
The Physiological and Pathological Role of Acyl-CoA Oxidation
 , , , and
, , , and
Abstract
:1. Introduction
1.1. Uptake and Activation of Fatty Acids
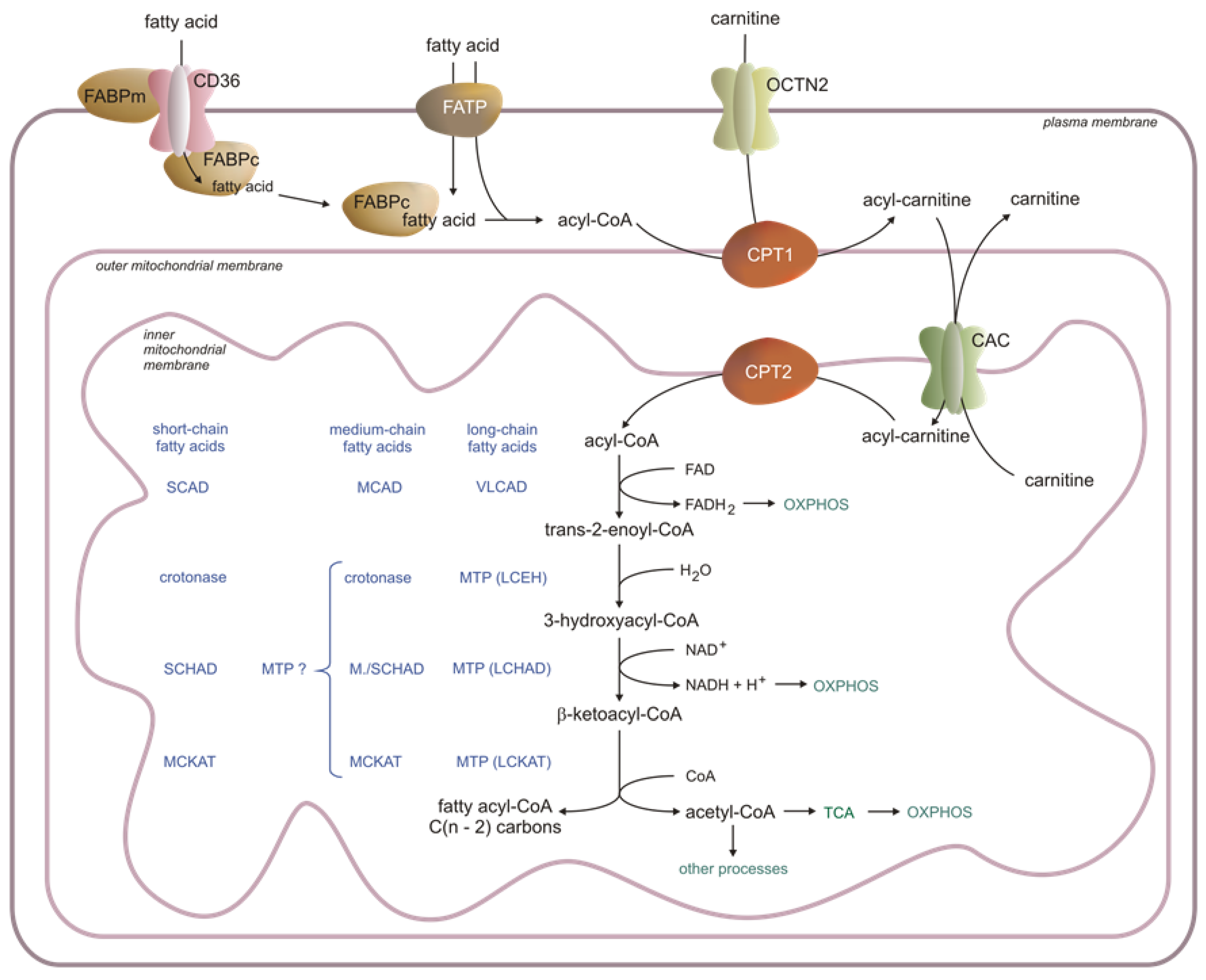
1.2. Carnitine Shuttle
1.2.1. Carnitine Palmitoyltransferase 1 (CPT1)
1.2.2. Carnitine Palmitoyltransferase 2 (CPT2) and Acylcarnitine Translocase CAC (SLC25A20)
1.3. Mitochondrial β-Oxidation
1.3.1. Oxidation of Long-Chain Acyl-CoA
1.3.2. Oxidation of Monounsaturated and Polyunsaturated Long-Chain Acyl-CoA
1.3.3. Oxidation of Medium-Chain Fatty Acids
1.3.4. Oxidation of Short-Chain Fatty Acids
1.4. Peroxisomal FAO
1.4.1. Peroxisomal α-Oxidation—Role in Phytol and Phytanic Acid Metabolism
1.4.2. Peroxisomes Are Essential for the Degradation of Dicarboxylic Acid Formed during ω-Oxidation in Microsomes
1.4.3. Peroxisomal FAO—Potential Role in the Utilization of Toxic FFAs
1.4.4. Peroxisomal FAO Related to the Synthesis of Cholesterol and Phospholipids
1.4.5. Peroxisomal FAO—Inhibition of Lipophagy
1.4.6. Peroxisomal FAO—Regulation of Mitochondrial β-Oxidation
1.4.7. Peroxisomal FAO As a Process Associated with the Production of H2O2—An Important Signaling Molecule and Toxic Substance
1.4.8. Microsomal Fatty Acid ω-Oxidation
2. The Function of FAO in Selected Organs
2.1. Liver
2.1.1. Mitochondrial FAO As a Regulator of Gluconeogenesis
2.1.2. Mitochondrial FAO As a Source of Acetyl-CoA for Protein Acetylation
2.1.3. The Potential Role of Mitochondrial FAO in the Regulation of Ureagenesis
2.1.4. The Potential Role of Mitochondrial FAO in Phase II Detoxication
2.1.5. Hepatic Manifestations of FAO Disorders (FAOD) Caused by Genetic Defects
2.2. Heart and Skeletal Muscles
2.3. Kidney
2.4. Lungs
2.5. Enterocytes and Colonocytes
2.6. βOX in Other Organs/Tissues/Cells
2.6.1. Adipocytes
2.6.2. Brain
2.6.3. Endothelium
2.6.4. Placenta
2.6.5. Peripheral White Blood Cells
2.6.6. Steroidogenic Cells
2.6.7. Osteoclast
2.6.8. Pancreatic β-Cell
3. FAO in Cancer
4. The Pathogenic Genetic Make-Up of FAO Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, Q.; Chen, Y.; Wang, Z.; He, H.; Yu, P. Cellular Uptake, Metabolism and Sensing of Long-Chain Fatty Acids. Front. Biosci. Landmark 2023, 28, 10. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Komen, J.; Ferdinandusse, S. Phytanic acid metabolism in health and disease. Biochim. Biophys. Acta 2011, 1811, 498–507. [Google Scholar] [CrossRef]
- Sanders, R.-J.; Ofman, R.; Valianpour, F.; Kemp, S.; Wanders, R.J.A. Evidence for two enzymatic pathways for omega-oxidation of docosanoic acid in rat liver microsomes. J. Lipid Res. 2005, 46, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.M.; Djouadi, F.; Kelly, D.P. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 1998, 273, 23786–23792. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef]
- Tahri-Joutey, M.; Andreoletti, P.; Surapureddi, S.; Nasser, B.; Cherkaoui-Malki, M.; Latruffe, N. Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by PPARα. Int. J. Mol. Sci. 2021, 22, 8969. [Google Scholar] [CrossRef]
- Maciejewska-Skrendo, A.; Buryta, M.; Czarny, W.; Król, P.; Stastny, P.; Petr, M.; Safranow, K.; Sawczuk, M. The Polymorphisms of the Peroxisome-Proliferator Activated Receptors’ Alfa Gene Modify the Aerobic Training Induced Changes of Cholesterol and Glucose. J. Clin. Med. 2019, 8, 1043. [Google Scholar] [CrossRef]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef]
- Ellinghaus, P.; Wolfrum, C.; Assmann, G.; Spener, F.; Seedorf, U. Phytanic acid activates the peroxisome proliferator-activated receptor α (PPARα) in sterol carrier protein 2-/sterol carrier protein x-deficient mice. J. Biol. Chem. 1999, 274, 2766–2772. [Google Scholar] [CrossRef]
- Duszka, K.; Gregor, A.; Guillou, H.; König, J.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Caloric Restriction-Common Pathways Affecting Metabolism, Health, and Longevity. Cells 2020, 9, 1708. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Muoio, D.M.; MacLean, P.S.; Lang, D.B.; Li, S.; Houmard, J.A.; Way, J.M.; Winegar, D.A.; Corton, J.C.; Dohm, G.L.; Kraus, W.E. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) alpha knock-out mice. Evidence for compensatory regulation by PPAR delta. J. Biol. Chem. 2002, 277, 26089–26097. [Google Scholar] [CrossRef]
- de Lange, P.; Farina, P.; Moreno, M.; Ragni, M.; Lombardi, A.; Silvestri, E.; Burrone, L.; Lanni, A.; Goglia, F. Sequential changes in the signal transduction responses of skeletal muscle following food deprivation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 2579–2581. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 2299–2301. [Google Scholar] [CrossRef]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor β/δ in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Neels, J.G.; Grimaldi, P.A. Physiological functions of peroxisome proliferator-activated receptor β. Physiol. Rev. 2014, 94, 795–858. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Abdelmagid, S.A.; Clarke, S.E.; Nielsen, D.E.; Badawi, A.; El-Sohemy, A.; Mutch, D.M.; Ma, D.W.L. Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS ONE 2015, 10, e0116195. [Google Scholar] [CrossRef]
- Janczy, A.; Szymanski, M.; Stankiewicz, M.; Kaska, L.; Waleron, K.; Stelmanska, E.; Sledzinski, T.; Mika, A. Increased Amount of Polyunsaturated Fatty Acids in the Intestinal Contents of Patients with Morbid Obesity. Obes. Surg. 2023, 33, 1228–1236. [Google Scholar] [CrossRef]
- Huber, A.H.; Kleinfeld, A.M. Unbound free fatty acid profiles in human plasma and the unexpected absence of unbound palmitoleate. J. Lipid Res. 2017, 58, 578–585. [Google Scholar] [CrossRef]
- Rojek, L.; Hebanowska, A.; Stojek, M.; Jagielski, M.; Goyke, E.; Szrok-Jurga, S.; Smoczynski, M.; Swierczynski, J.; Sledzinski, T.; Adrych, K. High levels of reactive oxygen species in pancreatic necrotic fluid of patients with walled-off pancreatic necrosis. Gastroenterol. Rev. Gastroenterol. 2020, 16, 56–61. [Google Scholar] [CrossRef]
- Jupin, M.; Michiels, P.J.; Girard, F.C.; Spraul, M.; Wijmenga, S.S. NMR identification of endogenous metabolites interacting with fatted and non-fatted human serum albumin in blood plasma: Fatty acids influence the HSA-metabolite interaction. J. Magn. Reson. 2013, 228, 81–94. [Google Scholar] [CrossRef]
- Roden, M.; Stingl, H.; Chandramouli, V.; Schumann, W.C.; Hofer, A.; Landau, B.R.; Nowotny, P.; Waldhäusl, W.; Shulman, G.I. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes 2000, 49, 701–707. [Google Scholar] [CrossRef]
- Pavićević, I.D.; Jovanović, V.B.; Takić, M.M.; Aćimović, J.M.; Penezić, A.Z.; Mandić, L.M. Quantification of total content of non-esterified fatty acids bound to human serum albumin. J. Pharm. Biomed. Anal. 2016, 129, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.F.P.; Bonen, A.; Glatz, J.F.C. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fat. Acids 2010, 82, 149–154. [Google Scholar] [CrossRef]
- De Leeuw, A.M.; Brouwer, A.; Knook, D.L. Sinusoidal endothelial cells of the liver: Fine structure and function in relation to age. J. Electron Microsc. Tech. 1990, 14, 218–236. [Google Scholar] [CrossRef] [PubMed]
- Arts, T.; Reneman, R.S.; Bassingthwaighte, J.B.; van der Vusse, G.J. Modeling Fatty Acid Transfer from Artery to Cardiomyocyte. PLoS Comput. Biol. 2015, 11, e1004666. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.C.; Nabben, M.; Luiken, J.J.F.P. CD36 (SR-B2) as master regulator of cellular fatty acid homeostasis. Curr. Opin. Lipidol. 2022, 33, 103. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Nenkov, M.; Chen, Y.; Press, A.T.; Kaemmerer, E.; Gassler, N. Fatty acid metabolism and acyl-CoA synthetases in the liver-gut axis. World J. Hepatol. 2021, 13, 1512. [Google Scholar] [CrossRef]
- Heden, T.D.; Franklin, M.P.; Dailey, C.; Mashek, M.T.; Chen, C.; Mashek, D.G. ACOT1 deficiency attenuates high-fat diet induced fat mass gain by increasing energy expenditure. J. Clin. Investig. 2023, 8, e160987. [Google Scholar] [CrossRef]
- Angelini, A.; Saha, P.K.; Jain, A.; Jung, S.Y.; Mynatt, R.L.; Pi, X.; Xie, L. PHDs/CPT1B/VDAC1 axis regulates long-chain fatty acid oxidation in cardiomyocytes. Cell Rep. 2021, 37, 109767. [Google Scholar] [CrossRef]
- Mashek, D.G.; Bornfeldt, K.E.; Coleman, R.A.; Berger, J.; Bernlohr, D.A.; Black, P.; DiRusso, C.C.; Farber, S.A.; Guo, W.; Hashimoto, N.; et al. Revised nomenclature for the mammalian long-chain acyl-CoA synthetase gene family. J. Lipid Res. 2004, 45, 1958–1961. [Google Scholar] [CrossRef]
- Pei, Z.; Fraisl, P.; Berger, J.; Jia, Z.; Forss-Petter, S.; Watkins, P.A. Mouse very long-chain Acyl-CoA synthetase 3/fatty acid transport protein 3 catalyzes fatty acid activation but not fatty acid transport in MA-10 cells. J. Biol. Chem. 2004, 279, 54454–54462. [Google Scholar] [CrossRef]
- Gimeno, R.E.; Ortegon, A.M.; Patel, S.; Punreddy, S.; Ge, P.; Sun, Y.; Lodish, H.F.; Stahl, A. Characterization of a heart-specific fatty acid transport protein. J. Biol. Chem. 2003, 278, 16039–16044. [Google Scholar] [CrossRef]
- Richards, M.R.; Harp, J.D.; Ory, D.S.; Schaffer, J.E. Fatty acid transport protein 1 and long-chain acyl coenzyme A synthetase 1 interact in adipocytes. J. Lipid Res. 2006, 47, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, R.E. Fatty acid transport proteins. Curr. Opin. Lipidol. 2007, 18, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Doege, H.; Baillie, R.A.; Ortegon, A.M.; Tsang, B.; Wu, Q.; Punreddy, S.; Hirsch, D.; Watson, N.; Gimeno, R.E.; Stahl, A. Targeted Deletion of FATP5 Reveals Multiple Functions in Liver Metabolism: Alterations in Hepatic Lipid Homeostasis. Gastroenterology 2006, 130, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Lewin, T.M.; Kim, J.H.; Granger, D.A.; Vance, J.E.; Coleman, R.A. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J. Biol. Chem. 2001, 276, 24674–24679. [Google Scholar] [CrossRef]
- Poppelreuther, M.; Rudolph, B.; Du, C.; Großmann, R.; Becker, M.; Thiele, C.; Ehehalt, R.; Füllekrug, J. The N-terminal region of acyl-CoA synthetase 3 is essential for both the localization on lipid droplets and the function in fatty acid uptake. J. Lipid Res. 2012, 53, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, H.; Hara, S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 2019, 144, 106363. [Google Scholar] [CrossRef]
- Bu, S.Y.; Mashek, D.G. Hepatic long-chain acyl-CoA synthetase 5 mediates fatty acid channeling between anabolic and catabolic pathways. J. Lipid Res. 2010, 51, 3270–3280. [Google Scholar] [CrossRef]
- Marszalek, J.R.; Kitidis, C.; DiRusso, C.C.; Lodish, H.F. Long-chain acyl-CoA synthetase 6 preferentially promotes DHA metabolism. J. Biol. Chem. 2005, 280, 10817–10826. [Google Scholar] [CrossRef]
- Vessey, D.A.; Kelley, M.; Warren, R.S. Characterization of the CoA ligases of human liver mitochondria catalyzing the activation of short- and medium-chain fatty acids and xenobiotic carboxylic acids. Biochim. Biophys. Acta Gen. Subj. 1999, 1428, 455–462. [Google Scholar] [CrossRef]
- Miyagawa, Y.; Mori, T.; Goto, K.; Kawahara, I.; Fujiwara-Tani, R.; Kishi, S.; Sasaki, T.; Fujii, K.; Ohmori, H.; Kuniyasu, H. Intake of medium-chain fatty acids induces myocardial oxidative stress and atrophy. Lipids Health Dis. 2018, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Puthillathu, N.; Vengilote, R.; Jaworski, D.M.; Namboodiri, A.M. Acetate Revisited: A Key Biomolecule at the Nexus of Metabolism, Epigenetics and Oncogenesis—Part 1: Acetyl-CoA, Acetogenesis and Acyl-CoA Short-Chain Synthetases. Front. Physiol. 2020, 11, 580167. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Araki, A.; Maruta, H.; Takahashi, Y.; Yamashita, H. Molecular cloning of rat acss3 and characterization of mammalian propionyl-CoA synthetase in the liver mitochondrial matrix. J. Biochem. 2017, 161, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Osborne, B.; Brown, S.H.J.; Small, L.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Contrasting metabolic effects of medium-versus long-chain fatty acids in skeletal muscle. J. Lipid Res. 2013, 54, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Faye, A.; Esnous, C.; Price, N.T.; Onfray, M.A.; Girard, J.; Prip-Buus, C. Rat liver carnitine palmitoyltransferase 1 forms an oligomeric complex within the outer mitochondrial membrane. J. Biol. Chem. 2007, 282, 26908–26916. [Google Scholar] [CrossRef]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef] [PubMed]
- Rufer, A.C.; Thoma, R.; Hennig, M. Structural insight into function and regulation of carnitine palmitoyltransferase. Cell. Mol. Life Sci. 2009, 66, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, M.J.; Kurama, T.; Dai, Y.; Suwa, A.; Asaumi, M.; Matsumoto, S.I.; Cha, S.H.; Shimokawa, T.; Lane, M.D. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 7282–7287. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, P.; Sahún, I.; McDonald, J.; Ramírez, S.; Jacas, J.; Gratacós, E.; Sierra, A.Y.; Serra, D.; Herrero, L.; Acker-Palmer, A.; et al. Ceramide levels regulated by carnitine palmitoyltransferase 1C control dendritic spine maturation and cognition. J. Biol. Chem. 2012, 287, 21224–21232. [Google Scholar] [CrossRef] [PubMed]
- Taïb, B.; Bouyakdan, K.; Hryhorczuk, C.; Rodaros, D.; Fulton, S.; Alquier, T. Glucose regulates hypothalamic long-chain fatty acid metabolism via AMP-activated kinase (AMPK) in neurons and astrocytes. J. Biol. Chem. 2013, 288, 37216–37229. [Google Scholar] [CrossRef] [PubMed]
- Van Weeghel, M.; Abdurrachim, D.; Nederlof, R.; Argmann, C.A.; Houtkooper, R.H.; Hagen, J.; Nabben, M.; Denis, S.; Ciapaite, J.; Kolwicz, S.C.; et al. Increased cardiac fatty acid oxidation in a mouse model with decreased malonyl-CoA sensitivity of CPT1B. Cardiovasc. Res. 2018, 114, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Louet, J.-F.F.; Le May, C.; Pégorier, J.-P.P.; Decaux, J.-F.F.; Girard, J. Regulation of liver carnitine palmitoyltransferase I gene expression by hormones and fatty acids. Biochem. Soc. Trans. 2001, 29, 310–316. [Google Scholar] [CrossRef]
- Bruce, C.R.; Hoy, A.J.; Turner, N.; Watt, M.J.; Allen, T.L.; Carpenter, K.; Cooney, G.J.; Febbraio, M.A.; Kraegen, E.W. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes 2009, 58, 550–558. [Google Scholar] [CrossRef]
- Park, E.A.; Mynatt, R.L.; Cook, G.A.; Kashfi, K. Insulin regulates enzyme activity, malonyl-CoA sensitivity and mRNA abundance of hepatic carnitine palmitoyltransferase-I. Biochem. J. 1995, 310, 853–858. [Google Scholar] [CrossRef]
- Faye, A.; Borthwick, K.; Esnous, C.; Price, N.T.; Gobin, S.; Jackson, V.N.; Zammit, V.A.; Girard, J.; Prip-Buus, C. Demonstration of N- and C-terminal domain intramolecular interactions in rat liver carnitine palmitoyltransferase 1 that determine its degree of malonyl-CoA sensitivity. Biochem. J. 2005, 387, 67. [Google Scholar] [CrossRef]
- Akkaoui, M.; Cohen, I.; Esnous, C.; Lenoir, V.; Sournac, M.; Girard, J.; Prip-Buus, C. Modulation of the hepatic malonyl-CoA-carnitine palmitoyltransferase 1A partnership creates a metabolic switch allowing oxidation of de novo fatty acids. Biochem. J. 2009, 420, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shi, J.; De Vries, Y.; Arvidson, D.N.; Cregg, J.M.; Woldegiorgis, G. Functional Studies of Yeast-Expressed Human Heart Muscle Carnitine Palmitoyltransferase I. Arch. Biochem. Biophys. 1997, 347, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, C.; Halberg, N.; Hillig, T.; Saha, A.K.; Ruderman, N.B.; Wojtaszewski, J.F.P.; Richter, E.A.; Kiens, B. Malonyl-CoA and carnitine in regulation of fat oxidation in human skeletal muscle during exercise. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Lefort, N.; Glancy, B.; Bowen, B.; Willis, W.T.; Bailowitz, Z.; De Filippis, E.A.; Brophy, C.; Meyer, C.; Højlund, K.; Yi, Z.; et al. Increased Reactive Oxygen Species Production and Lower Abundance of Complex I Subunits and Carnitine Palmitoyltransferase 1B Protein Despite Normal Mitochondrial Respiration in Insulin-Resistant Human Skeletal Muscle. Diabetes 2010, 59, 2444–2452. [Google Scholar] [CrossRef]
- Maples, J.M.; Brault, J.J.; Witczak, C.A.; Park, S.; Hubal, M.J.; Weber, T.M.; Houmard, J.A.; Shewchuk, B.M. Differential epigenetic and transcriptional response of the skeletal muscle carnitine palmitoyltransferase 1B (CPT1B) gene to lipid exposure with obesity. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E345. [Google Scholar] [CrossRef]
- Song, S.; Attia, R.R.; Connaughton, S.; Niesen, M.I.; Ness, G.C.; Elam, M.B.; Hori, R.T.; Cook, G.A.; Park, E.A. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol. Cell. Endocrinol. 2010, 325, 54–63. [Google Scholar] [CrossRef]
- Longo, N.; Amat Di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef]
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases caused by mutations in mitochondrial carrier genes SLC25: A review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef]
- Pochini, L.; Galluccio, M.; Scumaci, D.; Giangregorio, N.; Tonazzi, A.; Palmieri, F.; Indiveri, C. Interaction of β-lactam antibiotics with the mitochondrial carnitine/acylcarnitine transporter. Chem. Biol. Interact. 2008, 173, 187–194. [Google Scholar] [CrossRef]
- Doulias, P.T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef]
- Tonazzi, A.; Eberini, I.; Indiveri, C. Molecular mechanism of inhibition of the mitochondrial carnitine/acylcarnitine transporter by omeprazole revealed by proteoliposome assay, mutagenesis and bioinformatics. PLoS ONE 2013, 8, e82286. [Google Scholar] [CrossRef]
- Branco, V.; Godinho-Santos, A.; Gonçalves, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mitochondrial thioredoxin reductase inhibition, selenium status, and Nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic. Biol. Med. 2014, 73, 95–105. [Google Scholar] [CrossRef]
- Soni, M.S.; Rabaglia, M.E.; Bhatnagar, S.; Shang, J.; Ilkayeva, O.; Mynatt, R.; Zhou, Y.P.; Schadt, E.E.; Thornberry, N.A.; Muoio, D.M.; et al. Downregulation of carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion. Diabetes 2014, 63, 3805–3814. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Scalise, M.; La Russa, D.; Notaristefano, C.; Brunelli, E.; Barca, D.; Indiveri, C. Mitochondrial Carnitine/Acylcarnitine Transporter, a Novel Target of Mercury Toxicity. Chem. Res. Toxicol. 2015, 28, 1015–1022. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Lorusso, I.; De Palma, A.; Indiveri, C. The mitochondrial carnitine/acylcarnitine carrier is regulated by hydrogen sulfide via interaction with C136 and C155. Biochim. Biophys. Acta 2016, 1860, 20–27. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Indiveri, C. Post-translational modification by acetylation regulates the mitochondrial carnitine/acylcarnitine transport protein. Mol. Cell. Biochem. 2017, 426, 65–73. [Google Scholar] [CrossRef]
- Huizing, M.; Ruitenbeek, W.; Van den Heuvel, L.P.; Dolce, V.; Iacobazzi, V.; Smeitink, J.A.M.; Palmieri, F.; Frans Trijbels, J.M. Human mitochondrial transmembrane metabolite carriers: Tissue distribution and its implication for mitochondrial disorders. J. Bioenerg. Biomembr. 1998, 30, 277–284. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Convertini, P.; Infantino, V.; Scarcia, P.; Todisco, S.; Palmieri, F. Statins, fibrates and retinoic acid upregulate mitochondrial acylcarnitine carrier gene expression. Biochem. Biophys. Res. Commun. 2009, 388, 643–647. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Infantino, V.; Palmieri, F. Transcriptional regulation of the mitochondrial citrate and carnitine/acylcarnitine transporters: Two genes involved in fatty acid biosynthesis and β-oxidation. Biology 2013, 2, 284–303. [Google Scholar] [CrossRef]
- Lara, C.; Nicola, G.; Saverio, C.; Isabella, B.; Marino, P.; Cesare, I.; Giovanna, I.; Sabrina, C.; Annamaria, T. Human mitochondrial carnitine acylcarnitine carrier: Molecular target of dietary bioactive polyphenols from sweet cherry (Prunus avium L.). Chem. Biol. Interact. 2019, 307, 179–185. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
- Czumaj, A.; Szrok-Jurga, S.; Hebanowska, A.; Turyn, J.; Swierczynski, J.; Sledzinski, T.; Stelmanska, E. The pathophysiological role of CoA. Int. J. Mol. Sci. 2020, 21, 9057. [Google Scholar] [CrossRef]
- Aoyama, T.; Souri, M.; Ushikubo, S.; Kamijo, T.; Yamaguchi, S.; Kelley, R.I.; Rhead, W.J.; Uetake, K.; Tanaka, K.; Hashimoto, T. Purification of human very-long-chain acyl-coenzyme A dehydrogenase and characterization of its deficiency in seven patients. J. Clin. Investig. 1995, 95, 2465–2473. [Google Scholar] [CrossRef]
- Sinsheimer, A.; Mohsen, A.W.; Bloom, K.; Karunanidhi, A.; Bharathi, S.; Wu, Y.L.; Schiff, M.; Wang, Y.; Goetzman, E.S.; Ghaloul-Gonzalez, L.; et al. Development and Characterization of a Mouse Model for Acad9 deficiency. Mol. Genet. Metab. 2021, 134, 156. [Google Scholar] [CrossRef]
- Goetzman, E.S.; Alcorn, J.F.; Bharathi, S.S.; Uppala, R.; McHugh, K.J.; Kosmider, B.; Chen, R.; Zuo, Y.Y.; Beck, M.E.; McKinney, R.W.; et al. Long-chain Acyl-CoA dehydrogenase deficiency as a cause of pulmonary surfactant dysfunction. J. Biol. Chem. 2014, 289, 10668–10679. [Google Scholar] [CrossRef]
- Horowitz, J.F.; Klein, S. Lipid metabolism during endurance exercise. Am. J. Clin. Nutr. 2000, 72, 558S–563S. [Google Scholar] [CrossRef]
- Nochi, Z.; Olsen, R.K.J.; Gregersen, N. Short-chain acyl-CoA dehydrogenase deficiency: From gene to cell pathology and possible disease mechanisms. J. Inherit. Metab. Dis. 2017, 40, 641–655. [Google Scholar] [CrossRef]
- Xia, C.; Lou, B.; Fu, Z.; Mohsen, A.W.; Shen, A.L.; Vockley, J.; Kim, J.J.P. Molecular mechanism of interactions between ACAD9 and binding partners in mitochondrial respiratory complex I assembly. iScience 2021, 24, 103153. [Google Scholar] [CrossRef]
- Beck, M.E.; Zhang, Y.; Bharathi, S.S.; Kosmider, B.; Bahmed, K.; Dahmer, M.K.; Nogee, L.M.; Goetzman, E.S. The common K333Q polymorphism in long-chain acyl-CoA dehydrogenase (LCAD) reduces enzyme stability and function. Mol. Genet. Metab. 2020, 131, 83–89. [Google Scholar] [CrossRef]
- Henriques, B.J.; Katrine Jentoft Olsen, R.; Gomes, C.M.; Bross, P. Electron transfer flavoprotein and its role in mitochondrial energy metabolism in health and disease. Gene 2021, 776, 145407. [Google Scholar] [CrossRef]
- Salerno, K.M.; Domenico, J.; Le, N.Q.; Stiles, C.D.; Solov’Yov, I.A.; Martino, C.F. Long-Time Oxygen Localization in Electron Transfer Flavoprotein. J. Chem. Inf. Model. 2022, 62, 4191–4199. [Google Scholar] [CrossRef]
- Fould, B.; Garlatti, V.; Neumann, E.; Fenel, D.; Gaboriaud, C.; Arlaud, G.J. Structural and functional characterization of the recombinant human mitochondrial trifunctional protein. Biochemistry 2010, 49, 8608–8617. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Fu, Z.; Battaile, K.P.; Kim, J.J.P. Crystal structure of human mitochondrial trifunctional protein, a fatty acid β-oxidation metabolon. Proc. Natl. Acad. Sci. USA 2019, 116, 6069–6074. [Google Scholar] [CrossRef] [PubMed]
- Dagher, R.; Massie, R.; Gentil, B.J. MTP deficiency caused by HADHB mutations: Pathophysiology and clinical manifestations. Mol. Genet. Metab. 2021, 133, 1–7. [Google Scholar] [CrossRef]
- Zhang, D.; Yu, W.; Geisbrecht, B.V.; Gould, S.J.; Sprecher, H.; Schulz, H. Functional characterization of Delta3,Delta2-enoyl-CoA isomerases from rat liver. J. Biol. Chem. 2002, 277, 9127–9132. [Google Scholar] [CrossRef] [PubMed]
- Van Weeghel, M.; Te Brinke, H.; Van Lenthe, H.; Kulik, W.; Minkler, P.E.; Stoll, M.S.K.; Sass, J.O.; Janssen, U.; Stoffel, W.; Schwab, K.O.; et al. Functional redundancy of mitochondrial enoyl-CoA isomerases in the oxidation of unsaturated fatty acids. FASEB J. 2012, 26, 4316–4326. [Google Scholar] [CrossRef]
- Onwukwe, G.U.; Kursula, P.; Koski, M.K.; Schmitz, W.; Wierenga, R.K. Human Δ3,Δ2-enoyl-CoA isomerase, type 2: A structural enzymology study on the catalytic role of its ACBP domain and helix-10. FEBS J. 2015, 282, 746–768. [Google Scholar] [CrossRef]
- Horowitz, J.F.; Leone, T.C.; Feng, W.; Kelly, D.P.; Klein, S. Effect of endurance training on lipid metabolism in women: A potential role for PPARalpha in the metabolic response to training. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E348–E355. [Google Scholar] [CrossRef]
- Toogood, H.S.; Van Thiel, A.; Basran, J.; Sutcliffe, M.J.; Scrutton, N.S.; Leys, D. Extensive domain motion and electron transfer in the human electron transferring flavoprotein.medium chain Acyl-CoA dehydrogenase complex. J. Biol. Chem. 2004, 279, 32904–32912. [Google Scholar] [CrossRef]
- Jones, P.M.; Butt, Y.; Messmer, B.; Boriak, R.; Bennett, M.J. Medium-chain fatty acids undergo elongation before beta-oxidation in fibroblasts. Biochem. Biophys. Res. Commun. 2006, 346, 193–197. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J.A.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Vanhove, G.; Veldhoven, P.P.V.; Eyssen, H.J.; Mannaerts, G.P. Mitochondrial short-chain acyl-CoA dehydrogenase of human liver and kidney can function as an oxidase. Biochem. J. 1993, 292 Pt 1, 23–30. [Google Scholar] [CrossRef]
- Corydon, T.J.; Bross, P.; Jensen, T.G.; Corydon, M.J.; Lund, T.B.; Jensen, U.B.; Kim, J.J.P.; Gregersen, N.; Bolund, L. Rapid degradation of short-chain acyl-CoA dehydrogenase variants with temperature-sensitive folding defects occurs after import into mitochondria. J. Biol. Chem. 1998, 273, 13065–13071. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ohtake, A.; Abe, H.; Yamamoto, S.; Satoh, Y.; Takayanagi, M.; Niimi, H.; Mori, M.; Hashimoto, T. Molecular cloning and sequence analysis of the cDNA for human mitochondrial short-chain enoyl-CoA hydratase. Enzyme Protein 1993, 47, 9–13. [Google Scholar] [CrossRef]
- Nakagawa, J.; Waldner, H.; Meyer-Monard, S.; Hofsteenge, J.; Jenö, P.; Moroni, C. AUH, a gene encoding an AU-specific RNA binding protein with intrinsic enoyl-CoA hydratase activity. Proc. Natl. Acad. Sci. USA 1995, 92, 2051–2055. [Google Scholar] [CrossRef] [PubMed]
- Vredendaal, P.J.C.M.; Van Den Berg, I.E.T.; Malingré, H.E.M.; Stroobants, A.K.; OldeWeghuis, D.E.M.; Berger, R. Human short-chain L-3-hydroxyacyl-CoA dehydrogenase: Cloning and characterization of the coding sequence. Biochem. Biophys. Res. Commun. 1996, 223, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, G.P.; Debeer, L.J.; Thomas, J.; De Schepper, P.J. Mitochondrial and peroxisomal fatty acid oxidation in liver homogenates and isolated hepatocytes from control and clofibrate-treated rats. J. Biol. Chem. 1979, 254, 4585–4595. [Google Scholar] [CrossRef] [PubMed]
- Lazarow, P.B.; De Duve, C. A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc. Natl. Acad. Sci. USA 1976, 73, 2043–2046. [Google Scholar] [CrossRef] [PubMed]
- Kleiboeker, B.; Lodhi, I.J. Peroxisomal regulation of energy homeostasis: Effect on obesity and related metabolic disorders. Mol. Metab. 2022, 65, 101577. [Google Scholar] [CrossRef] [PubMed]
- Vilarinho, S.; Sari, S.; Mazzacuva, F.; Bilgüvar, K.; Esendagli-Yilmaz, G.; Jain, D.; Akyol, G.; Dalgiç, B.; Günel, M.; Clayton, P.T.; et al. ACOX2 deficiency: A disorder of bile acid synthesis with transaminase elevation, liver fibrosis, ataxia, and cognitive impairment. Proc. Natl. Acad. Sci. USA 2016, 113, 11289–11293. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; van Roermund, C.W.T.; Preece, M.A.; Koster, J.; Ebberink, M.S.; Waterham, H.R.; Wanders, R.J.A. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 952–958. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Baes, M.; Ribeiro, D.; Ferdinandusse, S.; Waterham, H.R. The physiological functions of human peroxisomes. Physiol. Rev. 2023, 103, 957–1024. [Google Scholar] [CrossRef] [PubMed]
- Westin, M.A.K.; Hunt, M.C.; Alexson, S.E.H. Short- and medium-chain carnitine acyltransferases and acyl-CoA thioesterases in mouse provide complementary systems for transport of beta-oxidation products out of peroxisomes. Cell. Mol. Life Sci. 2008, 65, 982–990. [Google Scholar] [CrossRef]
- Tawbeh, A.; Gondcaille, C.; Trompier, D.; Savary, S. Peroxisomal ABC Transporters: An Update. Int. J. Mol. Sci. 2021, 22, 6093. [Google Scholar] [CrossRef]
- Wang, M.; Wang, K.; Liao, X.; Hu, H.; Chen, L.; Meng, L.; Gao, W.; Li, Q. Carnitine Palmitoyltransferase System: A New Target for Anti-Inflammatory and Anticancer Therapy? Front. Pharmacol. 2021, 12, 76058. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Morita, M. ABC Transporter Subfamily D: Distinct Differences in Behavior between ABCD1-3 and ABCD4 in Subcellular Localization, Function, and Human Disease. Biomed Res. Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Wang, Y.; Palmfeldt, J.; Gregersen, N.; Makhov, A.M.; Conway, J.F.; Wang, M.; McCalley, S.P.; Basu, S.; Alharbi, H.S.; Croix, C. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J. Biol. Chem. 2019, 294, 12380–12391. [Google Scholar] [CrossRef] [PubMed]
- Roca-Saavedra, P.; Mariño-Lorenzo, P.; Miranda, J.M.; Porto-Arias, J.J.; Lamas, A.; Vazquez, B.I.; Franco, C.M.; Cepeda, A. Phytanic acid consumption and human health, risks, benefits and future trends: A review. Food Chem. 2017, 221, 237–247. [Google Scholar] [CrossRef]
- Steinberg, D.; Vroom, F.Q.; Engel, W.K.; Cammermeyer, J.; Mize, C.E.; Avigan, J. Refsum’s disease--a recently characterized lipidosis involving the nervous system. Combined clinical staff conference at the National Institutes of Health. Ann. Intern. Med. 1967, 66, 365–395. [Google Scholar] [CrossRef]
- Durrett, T.P.; Welti, R. The tail of chlorophyll: Fates for phytol. J. Biol. Chem. 2021, 296, 100802. [Google Scholar] [CrossRef] [PubMed]
- Wills, A.J.; Manning, N.J.; Reilly, M.M. Refsum’s disease. QJM 2001, 94, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Krauß, S.; Vetter, W. Phytol and Phytyl Fatty Acid Esters: Occurrence, Concentrations, and Relevance. Eur. J. Lipid Sci. Technol. 2018, 120, 1700387. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Vreken, P.; Ferdinandusse, S.; Jansen, G.A.; Waterham, H.R.; van Roermund, C.W.T.; Van Grunsven, E.G. Peroxisomal fatty acid alpha- and beta-oxidation in humans: Enzymology, peroxisomal metabolite transporters and peroxisomal diseases. Biochem. Soc. Trans. 2001, 29, 250. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Komen, J.C. Peroxisomes, Refsum’s disease and the alpha- and omega-oxidation of phytanic acid. Biochem. Soc. Trans. 2007, 35, 865–869. [Google Scholar] [CrossRef]
- Chen, M.H.; Raffield, L.M.; Mousas, A.; Sakaue, S.; Huffman, J.E.; Moscati, A.; Trivedi, B.; Jiang, T.; Akbari, P.; Vuckovic, D.; et al. Trans-ethnic and Ancestry-Specific Blood-Cell Genetics in 746,667 Individuals from 5 Global Populations. Cell 2020, 182, 1198–1213. [Google Scholar] [CrossRef]
- Goldfischer, S.; Johnson, A.B.; Essner, E.; Moore, C.; Ritch, R.H. Peroxisomal abnormalities in metabolic diseases. J. Histochem. Cytochem. 1973, 21, 972–977. [Google Scholar] [CrossRef]
- Monnens, L.; Bakkeren, J.; Parmentier, G.; Janssen, G.; van Haelst, U.; Trijbels, F.; Eyssen, H. Disturbances in bile acid metabolism of infants with the Zellweger (cerebro-hepato-renal) syndrome. Eur. J. Pediatr. 1980, 133, 31–35. [Google Scholar] [CrossRef]
- Cheillan, D. Zellweger Syndrome Disorders: From Severe Neonatal Disease to Atypical Adult Presentation. Adv. Exp. Med. Biol. 2020, 1299, 71–80. [Google Scholar] [CrossRef]
- Alam, A.; Locher, K.P. Structure and Mechanism of Human ABC Transporters. Annu. Rev. Biophys. 2023, 52, 275–300. [Google Scholar] [CrossRef]
- Chen, Z.-P.; Xu, D.; Wang, L.; Mao, Y.-X.; Li, Y.; Cheng, M.-T.; Zhou, C.-Z.; Hou, W.-T.; Chen, Y. Structural basis of substrate recognition and translocation by human very long-chain fatty acid transporter ABCD1. Nat. Commun. 2022, 13, 3299. [Google Scholar] [CrossRef]
- van Roermund, C.W.T.; IJlst, L.; Wagemans, T.; Wanders, R.J.A.; Waterham, H.R. A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 563–568. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Fourcade, S.; Savary, S.; Albet, S.; Gauthé, D.; Gondcaille, C.; Pineau, T.; Bellenger, J.; Bentejac, M.; Holzinger, A.; Berger, J.; et al. Fibrate induction of the adrenoleukodystrophy-related gene (ABCD2): Promoter analysis and role of the peroxisome proliferator-activated receptor PPARα. Eur. J. Biochem. 2001, 268, 3490–3500. [Google Scholar] [CrossRef]
- Leclercq, S.; Skrzypski, J.; Courvoisier, A.; Gondcaille, C.; Bonnetain, F.; André, A.; Chardigny, J.-M.; Bellenger, S.; Bellenger, J.; Narce, M.; et al. Effect of dietary polyunsaturated fatty acids on the expression of peroxisomal ABC transporters. Biochimie 2008, 90, 1602–1607. [Google Scholar] [CrossRef]
- Hayashi, H.; Takahata, S. Role of peroxisomal fatty acyl-CoA beta-oxidation in phospholipid biosynthesis. Arch. Biochem. Biophys. 1991, 284, 326–331. [Google Scholar] [CrossRef]
- Hayashi, H.; Oohashi, M. Incorporation of acetyl-CoA generated from peroxisomal beta-oxidation into ethanolamine plasmalogen of rat liver. Biochim. Biophys. Acta 1995, 1254, 319–325. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Yao, H.; Deng, S.; Gao, T.; Shang, L.; Chen, X.; Cui, X.; Zeng, J. Peroxisomal β-oxidation stimulates cholesterol biosynthesis in the liver in diabetic mice. J. Biol. Chem. 2022, 298, 101572. [Google Scholar] [CrossRef]
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of Autophagy by Cytosolic Acetyl-Coenzyme A. Mol. Cell 2014, 53, 710–725. [Google Scholar] [CrossRef]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1178–1187. [Google Scholar] [CrossRef]
- He, A.; Chen, X.; Tan, M.; Chen, Y.; Lu, D.; Zhang, X.; Dean, J.M.; Razani, B.; Lodhi, I.J. Acetyl-CoA Derived from Hepatic Peroxisomal β-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Mol. Cell 2020, 79, 30. [Google Scholar] [CrossRef]
- Chen, X.; Shang, L.; Deng, S.; Li, P.; Chen, K.; Gao, T.; Zhang, X.; Chen, Z.; Zeng, J. Peroxisomal oxidation of erucic acid suppresses mitochondrial fatty acid oxidation by stimulating malonyl-CoA formation in the rat liver. J. Biol. Chem. 2020, 295, 10168–10179. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C. Redox Signaling from and to Peroxisomes: Progress, Challenges, and Prospects. Antioxid. Redox Signal. 2019, 30, 95–112. [Google Scholar] [CrossRef]
- Terlecky, S.R.; Koepke, J.I.; Walton, P.A. Peroxisomes and aging. Biochim. Biophys. Acta 2006, 1763, 1749–1754. [Google Scholar] [CrossRef]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef]
- Vallejo, M.J.; Salazar, L.; Grijalva, M. Oxidative stress modulation and ROS-mediated toxicity in cancer: A review on in vitro models for plant-derived compounds. Oxid. Med. Cell. Longev. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Mahaseth, T.; Kuzminov, A. Potentiation of hydrogen peroxide toxicity: From catalase inhibition to stable DNA-iron complexes. Mutat. Res. Rev. Mutat. Res. 2017, 773, 274–281. [Google Scholar] [CrossRef]
- Feng, S.; Sun, Z.; Jia, X.; Li, L.; Wu, Y.; Wu, C.; Lin, L.; Liu, J.; Zeng, B. Lipophagy: Molecular Mechanisms and Implications in Hepatic Lipid Metabolism. Front. Biosci. 2023, 28, 6. [Google Scholar] [CrossRef]
- Sonoda, T.; Tatibana, M. Purification of N-acetyl-L-glutamate synthetase from rat liver mitochondria and substrate and activator specificity of the enzyme. J. Biol. Chem. 1983, 258, 9839–9844. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef]
- Dąbek, A.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients 2020, 12, 788. [Google Scholar] [CrossRef]
- Ramadhian, M.R. INHERITED VARIATIONS IN DRUGS EFFECT INDEPENDENT IN PHARMACOKINETIC: POLYMORPHISM IN PHASE II BIOTRANSFORMATION ENZYMES. JUKE Unila 2014, 4, 254–268. [Google Scholar]
- Hwang, C.Y.; Choe, W.; Yoon, K.S.; Ha, J.; Kim, S.S.; Yeo, E.J.; Kang, I. Molecular Mechanisms for Ketone Body Metabolism, Signaling Functions, and Therapeutic Potential in Cancer. Nutrients 2022, 14, 4932. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Metabolic and Signaling Roles of Ketone Bodies in Health and Disease. Annu. Rev. Nutr. 2021, 41, 49–77. [Google Scholar] [CrossRef]
- Lam, T.K.T.; Carpentier, A.; Lewis, G.F.; Van de Werve, G.; Fantus, I.G.; Giacca, A. Mechanisms of the free fatty acid-induced increase in hepatic glucose production. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E863–E873. [Google Scholar] [CrossRef]
- Batenburg, J.J.; Olson, M.S. Regulation of pyruvate dehydrogenase by fatty acid in isolated rat liver mitochondria. J. Biol. Chem. 1976, 251, 1364–1370. [Google Scholar] [CrossRef]
- Pougovkina, O.; Te Brinke, H.; Ofman, R.; Van Cruchten, A.G.; Kulik, W.; Wanders, R.J.A.; Houten, S.M.; De Boer, V.C.J. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet. 2014, 23, 3513–3522. [Google Scholar] [CrossRef]
- Sodji, Q.H.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. Chapter 15—In Vitro Histone Deacetylase Activity Screening: Making a Case for Better Assays; Zheng, Y.G., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 319–332. ISBN 978-0-12-801080-8. [Google Scholar]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Bharathi, S.S.; Zhang, Y.; Mohsen, A.W.; Uppala, R.; Balasubramani, M.; Schreiber, E.; Uechi, G.; Beck, M.E.; Vockley, J.; Rardin, M.J.; et al. Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J. Biol. Chem. 2013, 288, 33837–33847. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Hua, L.; Dittenhafer-Reed, K.E.; Schwer, B.; Lombard, D.B.; Li, Y.; Bunkenborg, J.; Alt, F.W.; Denu, J.M.; et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010, 12, 654–661. [Google Scholar] [CrossRef]
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 2009, 137, 560–570. [Google Scholar] [CrossRef]
- Yu, W.; Lin, Y.; Yao, J.; Huang, W.; Lei, Q.; Xiong, Y.; Zhao, S.; Guan, K.-L. Lysine 88 acetylation negatively regulates ornithine carbamoyltransferase activity in response to nutrient signals. J. Biol. Chem. 2009, 284, 13669–13675. [Google Scholar] [CrossRef]
- Hallows, W.C.; Yu, W.; Smith, B.C.; Devires, M.K.; Ellinger, J.J.; Someya, S.; Shortreed, M.R.; Prolla, T.; Markley, J.L.; Smith, L.M.; et al. Sirt3 Promotes the Urea Cycle and Fatty Acid Oxidation during Dietary Restriction. Mol. Cell 2011, 41, 139–149. [Google Scholar] [CrossRef]
- Walker, V. Ammonia metabolism and hyperammonemic disorders. Adv. Clin. Chem. 2014, 67, 73–150. [Google Scholar] [CrossRef] [PubMed]
- Fahien, L.A.; Schooler, J.M.; Gehred, G.A.; Cohen, P.P. Studies on the Mechanism of Action of Acetylglutamate as an Activator of Carbamyl Phosphate Synthetase. J. Biol. Chem. 1964, 239, 1935–1941. [Google Scholar] [CrossRef]
- Nissim, I.; Daikhin, Y.; Nissim, I.; Luhovyy, B.; Horyn, O.; Wehrli, S.L.; Yudkoff, M. Agmatine stimulates hepatic fatty acid oxidation: A possible mechanism for up-regulation of ureagenesis. J. Biol. Chem. 2006, 281, 8486–8496. [Google Scholar] [CrossRef]
- Ribas, G.S.; Lopes, F.F.; Deon, M.; Vargas, C.R. Hyperammonemia in Inherited Metabolic Diseases. Cell. Mol. Neurobiol. 2022, 42, 2593–2610. [Google Scholar] [CrossRef] [PubMed]
- Merritt, J.L.; MacLeod, E.; Jurecka, A.; Hainline, B. Clinical manifestations and management of fatty acid oxidation disorders. Rev. Endocr. Metab. Disord. 2020, 21, 479–493. [Google Scholar] [CrossRef]
- Ribas, G.S.; Vargas, C.R. Evidence that Oxidative Disbalance and Mitochondrial Dysfunction are Involved in the Pathophysiology of Fatty Acid Oxidation Disorders. Cell. Mol. Neurobiol. 2022, 42, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Pessayre, D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef]
- Amaral, A.U.; Cecatto, C.; Da Silva, J.C.; Wajner, A.; Godoy, K.D.S.; Ribeiro, R.T.; Wajner, M. cis-4-Decenoic and decanoic acids impair mitochondrial energy, redox and Ca(2+) homeostasis and induce mitochondrial permeability transition pore opening in rat brain and liver: Possible implications for the pathogenesis of MCAD deficiency. Biochim. Biophys. Acta 2016, 1857, 1363–1372. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Biswas, D.; Pulinilkunnil, T.; Lopaschuk, G.D. Myocardial Ketones Metabolism in Heart Failure. J. Card. Fail. 2020, 26, 998–1005. [Google Scholar] [CrossRef]
- Dong, S.; Qian, L.; Cheng, Z.; Chen, C.; Wang, K.; Hu, S.; Zhang, X.; Wu, T. Lactate and Myocadiac Energy Metabolism. Front. Physiol. 2021, 12, 715081. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- De Loof, M.; Renguet, E.; Ginion, A.; Bouzin, C.; Horman, S.; Beauloye, C.; Bertrand, L.; Bultot, L. Enhanced protein acetylation initiates fatty acid-mediated inhibition of cardiac glucose transport. Am. J. Physiol. Circ. Physiol. 2023, 324, H305–H317. [Google Scholar] [CrossRef] [PubMed]
- Olkowicz, M.; Tomczyk, M.; Debski, J.; Tyrankiewicz, U.; Przyborowski, K.; Borkowski, T.; Zabielska-Kaczorowska, M.; Szupryczynska, N.; Kochan, Z.; Smeda, M.; et al. Enhanced cardiac hypoxic injury in atherogenic dyslipidaemia results from alterations in the energy metabolism pattern. Metabolism 2021, 114, 154400. [Google Scholar] [CrossRef]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting fatty acid and carbohydrate oxidation—A novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1333–1350. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, L.; Battiprolu, P.K.; Fukushima, A.; Nguyen, K.; Milner, K.; Gupta, A.; Altamimi, T.; Byrne, N.; Mori, J.; et al. Malonyl CoA Decarboxylase Inhibition Improves Cardiac Function Post-Myocardial Infarction. JACC Basic Transl. Sci. 2019, 4, 385–400. [Google Scholar] [CrossRef]
- Shao, D.; Kolwicz, S.C.; Wang, P.; Roe, N.D.; Villet, O.; Nishi, K.; Hsu, Y.W.A.; Flint, G.V.; Caudal, A.; Wang, W.; et al. Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy. Circulation 2020, 142, 983–997. [Google Scholar] [CrossRef]
- Liu, Z.L.; Ding, J.; McMillen, T.S.; Villet, O.; Tian, R.; Shao, D. Enhancing fatty acid oxidation negatively regulates PPARs signaling in the heart. J. Mol. Cell. Cardiol. 2020, 146, 1–11. [Google Scholar] [CrossRef]
- Peterson, L.R.; Herrero, P.; Schechtman, K.B.; Racette, S.B.; Waggoner, A.D.; Kisrieva-Ware, Z.; Dence, C.; Klein, S.; Marsala, J.A.; Meyer, T.; et al. Effect of Obesity and Insulin Resistance on Myocardial Substrate Metabolism and Efficiency in Young Women. Circulation 2004, 109, 2191–2196. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, P.K.; O’Neill, B.T.; Roberts, M.W.; Buchanan, J.; Yun, U.J.; Cooksey, R.C.; Boudina, S.; Abel, E.D. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 2004, 53, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, J.R.; Carley, A.N.; Ji, R.; Zhang, X.; Fasano, M.; Schulze, P.C.; Lewandowski, E.D. Preservation of Acyl Coenzyme A Attenuates Pathological and Metabolic Cardiac Remodeling through Selective Lipid Trafficking. Circulation 2019, 139, 2765–2777. [Google Scholar] [CrossRef]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wüst, R.C.I.; Ferdinandusse, S.; IJlst, L.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Mayell, S.J.; Edwards, L.; Reynolds, F.E.; Chakrapani, A.B. Late presentation of medium-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2007, 30, 104. [Google Scholar] [CrossRef]
- El-Gharbawy, A.; Goldstein, A. Mitochondrial Fatty Acid Oxidation Disorders Associated with Cardiac Disease. Curr. Pathobiol. Rep. 2017, 5, 259–270. [Google Scholar] [CrossRef]
- Bonnet, D.; Martin, D.; De Lonlay, P.; Villain, E.; Jouvet, P.; Rabier, D.; Brivet, M.; Saudubray, J.M. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation 1999, 100, 2248–2253. [Google Scholar] [CrossRef]
- Sklirou, E.; Alodaib, A.N.; Dobrowolski, S.F.; Mohsen, A.W.A.; Vockley, J. Physiological Perspectives on the Use of Triheptanoin as Anaplerotic Therapy for Long Chain Fatty Acid Oxidation Disorders. Front. Genet. 2021, 11, 598760. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Charrow, J.; Ganesh, J.; Eswara, M.; Diaz, G.A.; McCracken, E.; Conway, R.; Enns, G.M.; Starr, J.; Wang, R.; et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol. Genet. Metab. 2016, 119, 223–231. [Google Scholar] [CrossRef]
- Vockley, J.; Burton, B.; Berry, G.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Chapman, K.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; et al. Effects of triheptanoin (UX007) in patients with long-chain fatty acid oxidation disorders: Results from an open-label, long-term extension study. J. Inherit. Metab. Dis. 2021, 44, 253–263. [Google Scholar] [CrossRef]
- Vockley, J.; Burton, B.; Berry, G.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Chapman, K.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; et al. OP017: Triheptanoin for the treatment of Long-Chain Fatty Acid Disorders (LC-FAOD): Final results of an open-label, long-term extension study. Genet. Med. 2022, 24, S349. [Google Scholar] [CrossRef]
- Hamilton-Craig, I.; Yudi, M.; Johnson, L.; Jayasinghe, R. Fenofibrate therapy in carnitine palmitoyl transferase type 2 deficiency. Case Rep. Med. 2012, 2012, 1–4. [Google Scholar] [CrossRef]
- Ørngreen, M.C.; Vissing, J.; Laforét, P. No effect of bezafibrate in patients with CPTII and VLCAD deficiencies. J. Inherit. Metab. Dis. 2015, 38, 373–374. [Google Scholar] [CrossRef]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.B.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef]
- Gavin, T.P.; Ernst, J.M.; Kwak, H.B.; Caudill, S.E.; Reed, M.A.; Garner, R.T.; Nie, Y.; Weiss, J.A.; Pories, W.J.; Dar, M.; et al. High incomplete skeletal muscle fatty acid oxidation explains low muscle insulin sensitivity in poorly controlled T2D. J. Clin. Endocrinol. Metab. 2018, 103, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Mengeste, A.M.; Rustan, A.C.; Lund, J. Skeletal muscle energy metabolism in obesity. Obesity 2021, 29, 1582–1595. [Google Scholar] [CrossRef] [PubMed]
- Fritzen, A.M.; Lundsgaard, A.M.; Kiens, B. Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise. Nat. Rev. Endocrinol. 2020, 16, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Simoneau, J.; Veerkamp, J.H.; Turcotte, L.P.; Kelley, D.E. Markers of capacity to utilize fatty acids in human skeletal muscle: Relation to insulin resistance and obesity and effects of weight loss. FASEB J. 1999, 13, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Rong, Q.; Han, B.; Li, Y.; Yin, H.; Li, J.; Hou, Y. Berberine Reduces Lipid Accumulation by Promoting Fatty Acid Oxidation in Renal Tubular Epithelial Cells of the Diabetic Kidney. Front. Pharmacol. 2022, 12, 729384. [Google Scholar] [CrossRef]
- Li, B.; Hao, J.; Zeng, J.; Sauter, E.R. SnapShot: FABP Functions. Cell 2020, 182, 1066.e1. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Li, Q.; Wei, S.; Zhou, Y.; Yu, W.; Xue, L.; Zhou, L.; Shen, L.; Lu, G.; et al. STAT6 contributes to renal fibrosis by modulating PPARα-mediated tubular fatty acid oxidation. Cell Death Dis. 2022, 13, 1–11. [Google Scholar] [CrossRef]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. 2015, 2, 52. [Google Scholar] [CrossRef]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 65. [Google Scholar] [CrossRef]
- Khan, S.; Gaivin, R.; Abramovich, C.; Boylan, M.; Calles, J.; Schelling, J.R. Fatty acid transport protein-2 regulates glycemic control and diabetic kidney disease progression. JCI Insight 2020, 5, e136845. [Google Scholar] [CrossRef]
- Miguel, V.; Tituaña, J.; Ignacio Herrero, J.; Herrero, L.; Serra, D.; Cuevas, P.; Barbas, C.; Puyol, D.R.; Márquez-Expósito, L.; Ruiz-Ortega, M.; et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Investig. 2021, 131, e140695. [Google Scholar] [CrossRef]
- Idrovo, J.P.; Yang, W.L.; Nicastro, J.; Coppa, G.F.; Wang, P. Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J. Surg. Res. 2012, 177, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, P.; Park, J.; Hurtado del Pozo, C.; Li, L.; Doke, T.; Huang, S.; Zhao, J.; Kang, H.M.; Shrestra, R.; Balzer, M.S.; et al. The Nuclear Receptor ESRRA Protects from Kidney Disease by Coupling Metabolism and Differentiation. Cell Metab. 2021, 33, 379–394.e8. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.J.; Ramkumar, N.; Summers, S.A. Gain of ‘FAOnction’, Loss of Fibrosis. Trends Endocrinol. Metab. 2021, 32, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Liu, Y. Understanding the mechanisms of kidney fibrosis. Nat. Rev. Nephrol. 2016, 12, 68–70. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Morel, J.D.; Sleiman, M.B.; Li, T.Y.; von Alvensleben, G.; Bachmann, A.M.; Hofer, D.; Broeckx, E.; Ma, J.Y.; Carreira, V.; Chen, T.; et al. Mitochondrial and NAD+ metabolism predict recovery from acute kidney injury in a diverse mouse population. JCI insight 2023, 8, e164626. [Google Scholar] [CrossRef]
- Gao, Z.; Chen, X. Fatty Acid β-Oxidation in Kidney Diseases: Perspectives on Pathophysiological Mechanisms and Therapeutic Opportunities. Front. Pharmacol. 2022, 13, 805281. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef]
- Li, S.; Wu, P.; Yarlagadda, P.; Vadjunec, N.M.; Proia, A.D.; Harris, R.A.; Portilla, D. PPARα ligand protects during cisplatin-induced acute renal failure by preventing inhibition of renal FAO and PDC activity. Am. J. Physiol. Ren. Physiol. 2004, 286, F572–F580. [Google Scholar] [CrossRef]
- Qiu, Y.; Hu, X.; Xu, C.; Lu, C.; Cao, R.; Xie, Y.; Yang, J. Ketogenic diet alleviates renal fibrosis in mice by enhancing fatty acid oxidation through the free fatty acid receptor 3 pathway. Front. Nutr. 2023, 10, 397. [Google Scholar] [CrossRef]
- Panov, A.V.; Mayorov, V.I.; Dikalova, A.E.; Dikalov, S.I. Long-Chain and Medium-Chain Fatty Acids in Energy Metabolism of Murine Kidney Mitochondria. Int. J. Mol. Sci. 2023, 24, 379. [Google Scholar] [CrossRef]
- Geng, J.; Liu, Y.; Dai, H.; Wang, C. Fatty Acid Metabolism and Idiopathic Pulmonary Fibrosis. Front. Physiol. 2022, 12, 794629. [Google Scholar] [CrossRef]
- Gu, L.; Larson Casey, J.L.; Andrabi, S.A.; Lee, J.H.; Meza-Perez, S.; Randall, T.D.; Carter, A.B. Mitochondrial calcium uniporter regulates PGC-1α expression to mediate metabolic reprogramming in pulmonary fibrosis. Redox Biol. 2019, 26, 101307. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, Q.; D’Souza, V.; Bartis, D.; Dancer, R.; Parekh, D.; Gao, F.; Lian, Q.; Jin, S.; Thickett, D.R. ResolvinD1 stimulates epithelial wound repair and inhibits TGF-β-induced EMT whilst reducing fibroproliferation and collagen production. Lab. Investig. 2018, 98, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Parks, B.W.; Black, L.L.; Zimmerman, K.A.; Metz, A.E.; Steele, C.; Murphy-Ullrich, J.E.; Kabarowski, J.H. CD36, but not G2A, modulates efferocytosis, infl ammation, and fibrosis following bleomycin-induced lung injury. J. Lipid Res. 2013, 54, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Langhans, W.; Leitner, C.; Arnold, M. Dietary fat sensing via fatty acid oxidation in enterocytes: Possible role in the control of eating. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, 554–565. [Google Scholar] [CrossRef]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 424615. [Google Scholar] [CrossRef]
- Roediger, W.E.W.; Millard, S. Selective inhibition of fatty acid oxidation in colonocytes by ibuprofen: A cause of colitis? Gut 1995, 36, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Malandrino, M.I.; Fucho, R.; Weber, M.; Calderon-Dominguez, M.; Mir, J.F.; Valcarcel, L.; Escoté, X.; Gómez-Serrano, M.; Peral, B.; Salvadó, L.; et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E756–E769. [Google Scholar] [CrossRef] [PubMed]
- Torchon, E.; Ray, R.; Hulver, M.W.; McMillan, R.P.; Voy, B.H. Fasting rapidly increases fatty acid oxidation in white adipose tissue of young broiler chickens. Adipocyte 2017, 6, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hurtado, E.; Lee, J.; Choi, J.; Wolfgang, M.J. Fatty acid oxidation is required for active and quiescent brown adipose tissue maintenance and thermogenic programing. Mol. Metab. 2018, 7, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J. Neurosci. 2003, 23, 5928–5935. [Google Scholar] [CrossRef] [PubMed]
- Dhopeshwarkar, G.A.; Subramanian, C.; Mead, J.F. Rapid uptke of [1-14C] acetate by the adult rat brain 15 seconds after carotid injection. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1971, 248, 41–47. [Google Scholar] [CrossRef]
- Gnaedinger, J.M.; Miller, J.C.; Latker, C.H.; Rapoport, S.I. Cerebral metabolism of plasma [14C]palmitate in awake, adult rat: Subcellular localization. Neurochem. Res. 1988, 13, 21–29. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. Biomed Res. Int. 2014, 2014, 1–22. [Google Scholar] [CrossRef]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef]
- Takahashi, S. Metabolic Compartmentalization between Astroglia and Neurons in Physiological and Pathophysiological Conditions of the Neurovascular Unit; Blackwell Publishing: Hoboken, NJ, USA, 2020; Volume 40, pp. 121–137. [Google Scholar]
- Ioannou, M.S. Current Insights into Fatty Acid Transport in the Brain. J. Membr. Biol. 2020, 253, 375–379. [Google Scholar] [CrossRef]
- Szrok-jurga, S.; Turyn, J.; Hebanowska, A.; Swierczynski, J.; Czumaj, A.; Sledzinski, T.; Stelmanska, E. The Role of Acyl-CoA β -Oxidation in Brain Metabolism and Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 13977. [Google Scholar] [CrossRef]
- Mallick, R.; Duttaroy, A.K. Modulation of endothelium function by fatty acids. Mol. Cell. Biochem. 2022, 477, 15–38. [Google Scholar] [CrossRef]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.S.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Brüning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Świerczyński, J.; Ścisłowski, P.; Aleksandrowicz, Z. Oxidation of palmitoyl-carnitine by mitochondria isolated from human term placenta. Biochem. Med. 1976, 16, 55–58. [Google Scholar] [CrossRef]
- Shekhawat, P.; Bennett, M.J.; Sadovsky, Y.; Nelson, D.M.; Rakheja, D.; Strauss, A.W. Human placenta metabolizes fatty acids: Implications for fetal fatty acid oxidation disorders and maternal liver diseases. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1098–E1105. [Google Scholar] [CrossRef] [PubMed]
- Rakheja, D.; Bennett, M.J.; Foster, B.M.; Domiati-Saad, R.; Rogers, B.B. Evidence for Fatty Acid Oxidation in Human Placenta, and the Relationship of Fatty Acid Oxidation Enzyme Activities with Gestational Age. Placenta 2002, 23, 447–450. [Google Scholar] [CrossRef]
- Oey, N.A.; den Boer, M.E.J.; Ruiter, J.P.N.; Wanders, R.J.A.; Duran, M.; Waterham, H.R.; Boer, K.; van der Post, J.A.M.; Wijburg, F.A. High activity of fatty acid oxidation enzymes in human placenta: Implications for fetal-maternal disease. J. Inherit. Metab. Dis. 2003, 26, 385–392. [Google Scholar] [CrossRef]
- Shin, E.K.; Kang, H.Y.; Yang, H.; Jung, E.M.; Jeung, E.B. The Regulation of Fatty Acid Oxidation in Human Preeclampsia. Reprod. Sci. 2016, 23, 1422–1433. [Google Scholar] [CrossRef]
- Mendez-Figueroa, H.; Chien, E.K.; Ji, H.; Nesbitt, N.L.; Bharathi, S.S.; Goetzman, E. Effects of labor on placental fatty acid β oxidation. J. Matern. Neonatal Med. 2013, 26, 150–154. [Google Scholar] [CrossRef]
- Powell, T.L.; Barner, K.; Madi, L.; Armstrong, M.; Manke, J.; Uhlson, C.; Jansson, T.; Ferchaud-Roucher, V. Sex-specific responses in placental fatty acid oxidation, esterification and transfer capacity to maternal obesity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158861. [Google Scholar] [CrossRef]
- Hulme, C.H.; Nicolaou, A.; Murphy, S.A.; Heazell, A.E.P.; Myers, J.E.; Westwood, M. The effect of high glucose on lipid metabolism in the human placenta. Sci. Rep. 2019, 9, 14114. [Google Scholar] [CrossRef]
- Pompéia, C.; Lopes, L.R.; Miyasaka, C.K.; Procópio, J.; Sannomiya, P.; Curi, R. Effect of fatty acids on leukocyte function. Brazilian J. Med. Biol. Res. = Rev. Bras. Pesqui. Med. Biol. 2000, 33, 1255–1268. [Google Scholar] [CrossRef]
- Pendergast, D.R.; Fisher, N.M.; Meksawan, K.; Doubrava, M.; Vladutiu, G.D. The distribution of white blood cell fat oxidation in health and disease. J. Inherit. Metab. Dis. 2004, 27, 89–99. [Google Scholar] [CrossRef]
- Schaefer, J.; Pourfarzam, M.; Bartlett, K.; Jackson, S.; Turnbull, D.M. Fatty acid oxidation in peripheral blood cells: Characterization and use for the diagnosis of defects of fatty acid oxidation. Pediatr. Res. 1995, 37, 354–360. [Google Scholar] [CrossRef]
- Stenlid, R.; Olsson, D.; Cen, J.; Manell, H.; Haglind, C.; Chowdhury, A.I.; Bergsten, P.; Nordenström, A.; Halldin, M. Altered mitochondrial metabolism in peripheral blood cells from patients with inborn errors of β-oxidation. Clin. Transl. Sci. 2022, 15, 182–194. [Google Scholar] [CrossRef]
- Tu, L.N.; Zhao, A.H.; Hussein, M.; Stocco, D.M.; Selvaraj, V. Translocator Protein (TSPO) Affects Mitochondrial Fatty Acid Oxidation in Steroidogenic Cells. Endocrinology 2016, 157, 1110–1121. [Google Scholar] [CrossRef]
- Park-Min, K.-H. Metabolic reprogramming in osteoclasts. Semin. Immunopathol. 2019, 41, 565–572. [Google Scholar] [CrossRef]
- Da, W.; Tao, L.; Zhu, Y. The Role of Osteoclast Energy Metabolism in the Occurrence and Development of Osteoporosis. Front. Endocrinol. 2021, 12, 675385. [Google Scholar] [CrossRef]
- Dodds, R.A.; Gowen, M.; Bradbeer, J.N. Microcytophotometric analysis of human osteoclast metabolism: Lack of activity in certain oxidative pathways indicates inability to sustain biosynthesis during resorption. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1994, 42, 599–606. [Google Scholar] [CrossRef]
- Lemma, S.; Sboarina, M.; Porporato, P.E.; Zini, N.; Sonveaux, P.; Di Pompo, G.; Baldini, N.; Avnet, S. Energy metabolism in osteoclast formation and activity. Int. J. Biochem. Cell Biol. 2016, 79, 168–180. [Google Scholar] [CrossRef]
- Koduru, S.V.; Sun, B.-H.; Walker, J.M.; Zhu, M.; Simpson, C.; Dhodapkar, M.; Insogna, K.L. The contribution of cross-talk between the cell-surface proteins CD36 and CD47-TSP-1 in osteoclast formation and function. J. Biol. Chem. 2018, 293, 15055–15069. [Google Scholar] [CrossRef] [PubMed]
- Dawodu, D.; Patecki, M.; Hegermann, J.; Dumler, I.; Haller, H.; Kiyan, Y. oxLDL inhibits differentiation and functional activity of osteoclasts via scavenger receptor-A mediated autophagy and cathepsin K secretion. Sci. Rep. 2018, 8, 11604. [Google Scholar] [CrossRef]
- Bellissimo, M.P.; Roberts, J.L.; Jones, D.P.; Liu, K.H.; Taibl, K.R.; Uppal, K.; Weitzmann, M.N.; Pacifici, R.; Drissi, H.; Ziegler, T.R.; et al. Metabolomic Associations with Serum Bone Turnover Markers. Nutrients 2020, 12, 3161. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, P.; Alekos, N.S.; Kim, S.P.; Li, Z.; Wolfgang, M.J.; Riddle, R.C. Mitochondrial fatty acid β-oxidation is important for normal osteoclast formation in growing female mice. Front. Physiol. 2022, 13, 997358. [Google Scholar] [CrossRef]
- Huang, Z.; Luo, R.; Yang, L.; Chen, H.; Zhang, X.; Han, J.; Wang, H.; Zhou, Z.; Wang, Z.; Shao, L. CPT1A-Mediated Fatty Acid Oxidation Promotes Precursor Osteoclast Fusion in Rheumatoid Arthritis. Front. Immunol. 2022, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yaney, G.C.; Corkey, B.E. Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 2003, 46, 1297–1312. [Google Scholar] [CrossRef]
- Berne, C. The metabolism of lipids in mouse pancreatic islets. The oxidation of fatty acids and ketone bodies. Biochem. J. 1975, 152, 661–666. [Google Scholar] [CrossRef]
- Malaisse, W.J. Insulin secretion: Multifactorial regulation for a single process of release. Diabetologia 1973, 9, 167–173. [Google Scholar] [CrossRef]
- Haber, E.P.; Ximenes, H.M.A.; Procópio, J.; Carvalho, C.R.O.; Curi, R.; Carpinelli, A.R. Pleiotropic effects of fatty acids on pancreatic beta-cells. J. Cell Physiol. 2003, 194, 1–12. [Google Scholar] [CrossRef]
- Nolan, C.J.; Madiraju, M.S.R.; Delghingaro-Augusto, V.; Peyot, M.-L.; Prentki, M. Fatty Acid Signaling in the β-Cell and Insulin Secretion. Diabetes 2006, 55, S16–S23. [Google Scholar] [CrossRef]
- El-Assaad, W.; Buteau, J.; Peyot, M.-L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated Fatty Acids Synergize with Elevated Glucose to Cause Pancreatic β-Cell Death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef]
- Gremlich, S.; Bonny, C.; Waeber, G.; Thorens, B. Fatty Acids Decrease IDX-1 Expression in Rat Pancreatic Islets and Reduce GLUT2, Glucokinase, Insulin, and Somatostatin Levels*. J. Biol. Chem. 1997, 272, 30261–30269. [Google Scholar] [CrossRef]
- Hellemans, K.; Kerckhofs, K.; Hannaert, J.C.; Martens, G.; Van Veldhoven, P.; Pipeleers, D. Peroxisome proliferator-activated receptor α-retinoid X receptor agonists induce beta-cell protection against palmitate toxicity. FEBS J. 2007, 274, 6094–6105. [Google Scholar] [CrossRef]
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-Generated Hydrogen Peroxide as Important Mediator of Lipotoxicity in Insulin-Producing Cells. Diabetes 2010, 60, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J. Gastroenterol. 2014, 20, 2279–2303. [Google Scholar] [CrossRef] [PubMed]
- Pakiet, A.; Kobiela, J.; Stepnowski, P.; Sledzinski, T.; Mika, A. Changes in lipids composition and metabolism in colorectal cancer: A review. Lipids Health Dis. 2019, 18, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, P.K.; Aneja, R.; Kapoor, S. Lipidomic landscape in cancer: Actionable insights for membrane-based therapy and diagnoses. Med. Res. Rev. 2022, 42, 983–1018. [Google Scholar] [CrossRef]
- Shi, J.; Fu, H.; Jia, Z.; He, K.; Fu, L.; Wang, W. High Expression of CPT1A Predicts Adverse Outcomes: A Potential Therapeutic Target for Acute Myeloid Leukemia. EBioMedicine 2016, 14, 55. [Google Scholar] [CrossRef]
- Liu, P.P.; Liu, J.; Jiang, W.Q.; Carew, J.S.; Ogasawara, M.A.; Pelicano, H.; Croce, C.M.; Estrov, Z.; Xu, R.H.; Keating, M.J.; et al. Elimination of Chronic Lymphocytic Leukemia Cells in Stromal Microenvironment by Targeting CPT with an Anti-Angina Drug Perhexiline. Oncogene 2016, 35, 5663. [Google Scholar] [CrossRef]
- Wu, Y.; Hurren, R.; MacLean, N.; Gronda, M.; Jitkova, Y.; Sukhai, M.A.; Minden, M.D.; Schimmer, A.D. Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells. Apoptosis 2015, 20, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef]
- Shao, H.; Mohamed, E.M.; Xu, G.G.; Waters, M.; Jing, K.; Ma, Y.; Zhang, Y.; Spiegel, S.; Idowu, M.O.; Fang, X. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget 2016, 7, 3832. [Google Scholar] [CrossRef] [PubMed]
- Mika, A.; Pakiet, A.; Czumaj, A.; Kaczynski, Z.; Liakh, I.; Kobiela, J.; Perdyan, A.; Adrych, K.; Makarewicz, W.; Sledzinski, T. Decreased Triacylglycerol Content and Elevated Contents of Cell Membrane Lipids in Colorectal Cancer Tissue: A Lipidomic Study. J. Clin. Med. 2020, 9, 1095. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, Z.; Lu, J.; Wang, Y.; Liu, Z.; He, M.; Zhao, Q.; Wang, Z.; Li, T.; Lu, Y.; et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 2018, 37, 6025–6040. [Google Scholar] [CrossRef]
- Jiang, N.; Xie, B.; Xiao, W.; Fan, M.; Xu, S.; Duan, Y.; Hamsafar, Y.; Evans, A.C.; Huang, J.; Zhou, W.; et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat. Commun. 2022, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Wang, C.Y.; Hung, Y.H.; Weng, T.Y.; Yen, M.C.; Lai, M.D. Systematic analysis of gene expression alterations and clinical outcomes for long-chain acyl-coenzyme A synthetase family in cancer. PLoS ONE 2016, 11, e0155660. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Jin, Y.; Yuan, Y.; Bai, C.; Wu, Y.; Zhu, H.; Lu, S. Validation and target gene screening of hsa-miR-205 in lung squamous cell carcinoma. Chin. Med. J. 2014, 127, 272–278. [Google Scholar] [PubMed]
- Sánchez-Martínez, R.; Cruz-Gil, S.; de Cedrón, M.G.; Álvarez-Fernández, M.; Vargas, T.; Molina, S.; García, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Wang, Y.; Sun, B.; Xiao, Z.; Ye, L.; Zhang, X. MiR-205 modulates abnormal lipid metabolism of hepatoma cells via targeting acyl-CoA synthetase long-chain family member 1 (ACSL1) mRNA. Biochem. Biophys. Res. Commun. 2014, 444, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Xiao, Z.; Wang, Y.; Zheng, M.; Song, T.; Cai, X.; Sun, B.; Ye, L.; Zhang, X. Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an mir-9-mediated RXRA signaling pathway. Cancer Res. 2015, 75, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Scholtens, D.; Holko, M.; Ivancic, D.; Lee, O.; Hu, H.; Chatterton, R.T.; Sullivan, M.E.; Hansen, N.; Bethke, K.; et al. Lipid metabolism genes in contralateral unaffected breast and estrogen receptor status of breast cancer. Cancer Prev. Res. 2013, 6, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Fraisl, P.; Shi, X.; Gabrielson, E.; Forss-Petter, S.; Berger, J.; Watkins, P.A. Very Long-Chain Acyl-CoA Synthetase 3: Overexpression and Growth Dependence in Lung Cancer. PLoS ONE 2013, 8, e69392. [Google Scholar] [CrossRef]
- Ye, X.; Zhang, Y.; Wang, X.; Li, Y.; Gao, Y. Tumor-suppressive functions of long-chain acyl-CoA synthetase 4 in gastric cancer. IUBMB Life 2016, 68, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.E.; Creighton, C.J.; Lee, P.; Zou, X.; Topham, M.K.; Stafforini, D.M. Expression of Long-chain Fatty Acyl-CoA Synthetase 4 in Breast and Prostate Cancers Is Associated with Sex Steroid Hormone Receptor Negativity 1. Transl. Oncol. 2010, 3, 91–98. [Google Scholar] [CrossRef]
- Cao, Y.; Pearman, A.T.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Intracellular unesterified arachidonic acid signals apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 11280–11285. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Wang, J.; Wen, X.; Marcus, M.T.; Daniels, G.; Zhang, D.Y.; Ye, F.; Wang, L.H.; Du, X.; et al. Long Chain Fatty Acyl-CoA Synthetase 4 Is a Biomarker for and Mediator of Hormone Resistance in Human Breast Cancer. PLoS ONE 2013, 8, e77060. [Google Scholar] [CrossRef]
- Hu, C.; Chen, L.; Jiang, Y.; Li, Y.; Wang, S. The effect of fatty acid-CoA ligase 4 on the growth of hepatic cancer cells. Cancer Biol. Ther. 2008, 7, 133–136. [Google Scholar] [CrossRef]
- Wu, X.; Deng, F.; Li, Y.; Daniels, G.; Du, X.; Ren, Q.; Wang, J.; Wang, L.H.; Yang, Y.; Zhang, V.; et al. ACSL4 promotes prostate cancer growth, invasion and hormonal resistance. Oncotarget 2015, 6, 44849–44863. [Google Scholar] [CrossRef]
- Cao, Y.; Dave, K.B.; Doan, T.P.; Prescott, S.M. Fatty acid CoA ligase 4 is up-regulated in colon adenocarcinoma. Cancer Res. 2001, 61, 8429–8434. [Google Scholar] [PubMed]
- Sung, Y.K.; Hwang, S.Y.; Park, M.K.; Bae, H.I.; Kim, W.H.; Kim, J.C.; Kim, M. Fatty acid-CoA ligase 4 is overexpressed in human hepatocellular carcinoma. Cancer Sci. 2003, 94, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Gassler, N.; Schneider, A.; Kopitz, J.; Schnölzer, M.; Obermüller, N.; Kartenbeck, J.; Otto, H.F.; Autschbach, F. Impaired Expression of Acyl-CoA-Synthetase 5 in Epithelial Tumors of the Small Intestine. Hum. Pathol. 2003, 34, 1048–1052. [Google Scholar] [CrossRef]
- Pitule, P.; Vycital, O.; Bruha, J.; Novak, P.; Hosek, P.; Treska, V.; Hlavata, I.; Soucek, P.; Kralickova, M.; Liska, V. Differential expression and prognostic role of selected genes in colorectal cancer patients. Anticancer Res. 2013, 33, 4855–4866. [Google Scholar]
- Gaisa, N.T.; Reinartz, A.; Schneider, U.; Klaus, C.; Heidenreich, A.; Jakse, G.; Kaemmerer, E.; Klinkhammer, B.M.; Knuechel, R.; Gassler, N. Levels of acyl-Coenzyme A synthetase 5 in urothelial cells and corresponding neoplasias reflect cellular differentiation. Histol. Histopathol. 2013, 28, 353–364. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Xiao, Q.; Li, Y.; Peng, Y.; Gan, Y.; Shu, G.; Yi, H.; Yin, G. Identification of CPT2 as a prognostic biomarker by integrating the metabolism-associated gene signature in colorectal cancer. BMC Cancer 2022, 22, 1038. [Google Scholar] [CrossRef]
- Wang, L.; Li, C.; Song, Y.; Yan, Z.K. Inhibition of carnitine palmitoyl transferase 1A-induced fatty acid oxidation suppresses cell progression in gastric cancer. Arch. Biochem. Biophys. 2020, 696, 108664. [Google Scholar] [CrossRef]
- Abudurexiti, M.; Zhu, W.; Wang, Y.; Wang, J.; Xu, W.; Huang, Y.; Zhu, Y.; Shi, G.; Zhang, H.; Zhu, Y.; et al. Targeting CPT1B as a potential therapeutic strategy in castration-resistant and enzalutamide-resistant prostate cancer. Prostate 2020, 80, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Vantaku, V.; Dong, J.; Ambati, C.R.; Perera, D.; Donepudi, S.R.; Amara, C.S.; Putluri, V.; Ravi, S.S.; Robertson, M.J.; Piyarathna, D.W.B.; et al. Multi-omics Integration Analysis Robustly Predicts High-Grade Patient Survival and Identifies CPT1B Effect on Fatty Acid Metabolism in Bladder Cancer. Clin. Cancer Res. 2019, 25, 3689–3701. [Google Scholar] [CrossRef]
- Chen, T.; Wu, G.; Hu, H.; Wu, C. Enhanced fatty acid oxidation mediated by CPT1C promotes gastric cancer progression. J. Gastrointest. Oncol. 2020, 11, 695–707. [Google Scholar] [CrossRef]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cheng, Y.; Su, D.; Gong, B.; He, X.; Zhou, X.; Pang, Z.; Cheng, L.; Chen, Y.; Yao, Z. Cpt1c regulated by AMPK promotes papillary thyroid carcinomas cells survival under metabolic stress conditions. J. Cancer 2017, 8, 3675–3681. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Lv, D.; Zheng, Y.; Wu, M.; Xu, C.; Zhang, Q.; Wu, L. Downregulation of CPT2 promotes tumorigenesis and chemoresistance to cisplatin in hepatocellular carcinoma. OncoTargets Ther. 2018, 11, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Liu, S.; Li, J.; Wu, L.; Lv, X.; Xu, J.; Chen, B.; Zhao, S.; Yang, H. CPT2 down-regulation promotes tumor growth and metastasis through inducing ROS/NFκB pathway in ovarian cancer. Transl. Oncol. 2021, 14, 101023. [Google Scholar] [CrossRef]
- Birkenkamp-Demtroder, K.; Christensen, L.L.; Olesen, S.H.; Frederiksen, C.M.; Laiho, P.; Aaltonen, L.A.; Laurberg, S.; Sørensen, F.B.; Hagemann, R.; Ørntoft, T.F. Gene expression in colorectal cancer. Cancer Res. 2002, 62, 4352–4363. [Google Scholar]
- Ren, H.; Li, W.; Liu, X.; Li, S.; Guo, H.; Wang, W.; Zhao, N. Identification and Validation of an 6-Metabolism-Related Gene Signature and Its Correlation With Immune Checkpoint in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 783934. [Google Scholar] [CrossRef]
- Cui, J.; Yi, G.; Li, J.; Li, Y.; Qian, D. Increased EHHADH Expression Predicting Poor Survival of Osteosarcoma by Integrating Weighted Gene Coexpression Network Analysis and Experimental Validation. Biomed Res. Int. 2021, 2021, 9917060. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Shen, Q.; Xie, H.; Zhou, X.; Li, J.; Feng, J.; Liu, H.; Wang, W.; Zhang, S.; Ni, S. Elevated expression of FABP3 and FABP4 cooperatively correlates with poor prognosis in non-small cell lung cancer (NSCLC). Oncotarget 2016, 7, 46253–46262. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Z.W.; Su, W.H.; Chen, J.; Wang, Y.L. Prognostic Value and Immune Infiltrates of ABCA8 and FABP4 in Stomach Adenocarcinoma. Biomed Res. Int. 2020, 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.Q.; Zhang, X.P.; Ma, N.; Zhang, E.B.; Li, J.J.; Jiang, Y.B.; Gao, Y.Z.; Yuan, Y.M.; Lan, S.Q.; Xie, D.; et al. FABP4 suppresses proliferation and invasion of hepatocellular carcinoma cells and predicts a poor prognosis for hepatocellular carcinoma. Cancer Med. 2018, 7, 2629–2640. [Google Scholar] [CrossRef]
- Kim, S.I.; Jung, M.; Dan, K.; Lee, S.; Lee, C.; Kim, H.S.; Chung, H.H.; Kim, J.W.; Park, N.H.; Song, Y.S.; et al. Proteomic discovery of biomarkers to predict prognosis of high-grade serous ovarian carcinoma. Cancers 2020, 12, 790. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, Z.; Li, D.; Liu, Z.; Yang, L.; Zou, Q.; Yuan, Y. LDHB and fabp4 are associated with progression and poor prognosis of pancreatic ductal adenocarcinomas. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Zhang, H.; Yuan, Y.; He, Q.; Zhou, J.; Li, S.; Sun, Y.; Li, D.Y.; Qiu, H.B.; Wang, W.; et al. Fatty acid oxidation controls CD8+Tissue-resident memory t-cell survival in gastric adenocarcinoma. Cancer Immunol. Res. 2020, 8, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Holzbeierlein, J.; Lal, P.; LaTulippe, E.; Smith, A.; Satagopan, J.; Zhang, L.; Ryan, C.; Smith, S.; Scher, H.; Scardino, P.; et al. Gene Expression Analysis of Human Prostate Carcinoma during Hormonal Therapy Identifies Androgen-Responsive Genes and Mechanisms of Therapy Resistance. Am. J. Pathol. 2004, 164, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Song, Y.H.; Xie, Y.; Wang, X.; Wang, Y.; Wang, C.; Liu, S.; Xue, S.L.; Li, Y.; Liu, B.; et al. Downregulation of HADH promotes gastric cancer progression via Akt signaling pathway. Oncotarget 2017, 8, 76279–76289. [Google Scholar] [CrossRef]
- Du, Z.; Zhang, X.; Gao, W.; Yang, J. Differentially expressed genes PCCA, ECHS1, and HADH are potential prognostic biomarkers for gastric cancer. Sci. Prog. 2021, 104, 1–14. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Wang, Z.; Xu, R.; Hu, X.; Sun, Y.; Wang, Q.; Ju, F.; Ren, S.; Zhang, C.; et al. The promising novel biomarkers and candidate small molecule drugs in kidney renal clear cell carcinoma: Evidence from bioinformatics analysis of high-throughput data. Mol. Genet. Genom. Med. 2019, 7, e607. [Google Scholar] [CrossRef]
- Jiang, H.; Chen, H.; Wan, P.; Chen, N. Decreased expression of HADH is related to poor prognosis and immune infiltration in kidney renal clear cell carcinoma. Genomics 2021, 113, 3556–3564. [Google Scholar] [CrossRef]
- Ren, J.; Feng, J.; Song, W.; Wang, C.; Ge, Y.; Fu, T. Development and validation of a metabolic gene signature for predicting overall survival in patients with colon cancer. Clin. Exp. Med. 2020, 20, 535–544. [Google Scholar] [CrossRef]
- Wei, J.; Xie, Q.; Liu, X.; Wan, C.; Wu, W.; Fang, K.; Yao, Y.; Cheng, P.; Deng, D.; Liu, Z. Identification the prognostic value of glutathione peroxidases expression levels in acute myeloid leukemia. Ann. Transl. Med. 2020, 8, 678. [Google Scholar] [CrossRef]
- Mamtani, M.; Kulkarni, H. Association of HADHA expression with the risk of breast cancer: Targeted subset analysis and meta-analysis of microarray data. BMC Res. Notes 2012, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef]
- Puca, F.; Yu, F.; Bartolacci, C.; Pettazzoni, P.; Carugo, A.; Huang-Hobbs, E.; Liu, J.; Zanca, C.; Carbone, F.; Del Poggetto, E.; et al. Medium-chain acyl-coa dehydrogenase protects mitochondria from lipid peroxidation in glioblastoma. Cancer Discov. 2021, 11, 2904–2923. [Google Scholar] [CrossRef]
- Su, Y.W.; Wu, P.S.; Lin, S.H.; Huang, W.Y.; Kuo, Y.S.; Lin, H.P. Prognostic value of the overexpression of fatty acid metabolism-related enzymes in Squamous cell Carcinoma of the head and neck. Int. J. Mol. Sci. 2020, 21, 6851. [Google Scholar] [CrossRef]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.J.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009, 69, 2163–2166. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92. [Google Scholar] [CrossRef]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-P.; Zhou, L.-S.; Zhao, Y.-Z.; Wang, S.-W.; Chen, L.-L.; Liu, L.-X.; Ling, Z.-Q.; Hu, F.-J.; Sun, Y.-P.; Zhang, J.-Y.; et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014, 33, 1304–1320. [Google Scholar] [CrossRef]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 2011, 1807, 726–734. [Google Scholar] [CrossRef]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; LeGonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S.; et al. Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef]
- Huang, J.; Duran, A.; Reina-Campos, M.; Valencia, T.; Castilla, E.A.; Müller, T.D.; Tschöp, M.H.; Moscat, J.; Diaz-Meco, M.T. Adipocyte p62/SQSTM1 Suppresses Tumorigenesis through Opposite Regulations of Metabolism in Adipose Tissue and Tumor. Cancer Cell 2018, 33, 770–784.e6. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Sebastiano, M.R.; Konstantinidou, G. Targeting long chain acyl-coa synthetases for cancer therapy. Int. J. Mol. Sci. 2019, 20, 3624. [Google Scholar] [CrossRef]
- Tung, S.; Shi, Y.; Wong, K.; Zhu, F.; Gorczynski, R.; Laister, R.C.; Minden, M.; Blechert, A.K.; Genzel, Y.; Reichl, U.; et al. PPARα and fatty acid oxidation mediate glucocorticoid resistance in chronic lymphocytic leukemia. Blood 2013, 122, 969–980. [Google Scholar] [CrossRef]
- Hermanova, I.; Arruabarrena-Aristorena, A.; Valis, K.; Nuskova, H.; Alberich-Jorda, M.; Fiser, K.; Fernandez-Ruiz, S.; Kavan, D.; Pecinova, A.; Niso-Santano, M.; et al. Pharmacological inhibition of fatty-acid oxidation synergistically enhances the effect of l-asparaginase in childhood ALL cells. Leukemia 2016, 30, 209–218. [Google Scholar] [CrossRef]
- Kitajima, S.; Yoshida, A.; Kohno, S.; Li, F.; Suzuki, S.; Nagatani, N.; Nishimoto, Y.; Sasaki, N.; Muranaka, H.; Wan, Y.; et al. The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 2017, 36, 5145–5157. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.P.Y.; Yeung, C.L.S.; Tey, S.K.; Mao, X.; Wong, S.W.K.; Ng, T.H.; Ko, F.C.F.; Kwong, E.M.L.; Tang, A.H.N.; Ng, I.O.L.; et al. Suppression of ACADM-Mediated Fatty Acid Oxidation Promotes Hepatocellular Carcinoma via Aberrant CAV1/SREBP1 Signaling. Cancer Res. 2021, 81, 3679–3692. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, R.; Cruz-Gil, S.; García-Álvarez, M.S.; Reglero, G.; De Molina, A.R. Complementary ACSL isoforms contribute to a non-Warburg advantageous energetic status characterizing invasive colon cancer cells. Sci. Rep. 2017, 7, 11143. [Google Scholar] [CrossRef] [PubMed]
- Orlando, U.D.; Castillo, A.F.; Dattilo, M.A.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Acyl-CoA synthetase-4, a new regulator of mTOR and a potential therapeutic target for enhanced estrogen receptor function in receptor-positive and -negative breast cancer. Oncotarget 2015, 6, 42632–42650. [Google Scholar] [CrossRef]
- Castillo, A.F.; Orlando, U.D.; Maloberti, P.M.; Prada, J.G.; Dattilo, M.A.; Solano, A.R.; Bigi, M.M.; Rios Medrano, M.A.; Torres, M.T.; Indo, S.; et al. New inhibitor targeting Acyl-CoA synthetase 4 reduces breast and prostate tumor growth, therapeutic resistance and steroidogenesis. Cell. Mol. Life Sci. 2020, 78, 2893–2910. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Sato, S.; Okabe, S.; Miyata, S.; Matsuura, M.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Acyl-CoA synthetase as a cancer survival factor: Its inhibition enhances the efficacy of etoposide. Cancer Sci. 2009, 100, 1556–1562. [Google Scholar] [CrossRef]
- Zhang, L.; Lv, J.; Chen, C.; Wang, X. Roles of acyl-CoA synthetase long-chain family member 5 and colony stimulating factor 2 in inhibition of palmitic or stearic acids in lung cancer cell proliferation and metabolism. Cell Biol. Toxicol. 2021, 37, 15–34. [Google Scholar] [CrossRef]
- Pei, Z.; Sun, P.; Huang, P.; Lal, B.; Laterra, J.; Watkins, P.A. Acyl-CoA synthetase VL3 knockdown inhibits human glioma cell proliferation and tumorigenicity. Cancer Res. 2009, 69, 9175–9182. [Google Scholar] [CrossRef]
- Lee, E.A.; Angka, L.; Rota, S.G.; Hanlon, T.; Mitchell, A.; Hurren, R.; Wang, X.M.; Gronda, M.; Boyaci, E.; Bojko, B.; et al. Targeting mitochondria with avocatin B induces selective leukemia cell death. Cancer Res. 2015, 75, 2478–2488. [Google Scholar] [CrossRef]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Del Valle, L.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Dheeraj, A.; Agarwal, C.; Schlaepfer, I.R.; Raben, D.; Singh, R.; Agarwal, R.; Deep, G. A novel approach to target hypoxic cancer cells via combining β-oxidation inhibitor etomoxir with radiation. Hypoxia 2018, 6, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Mascagna, D.; Ghanem, G.; Morandini, R.; D’Ischia, M.; Misuraca, G.; Lejeune, F.; Prota, G. Synthesis and cytotoxic properties of new N-substituted 4-am...: Melanoma Research. Melanoma Res. 1992, 2, 25–32. [Google Scholar] [CrossRef]
- Pucci, S.; Zonetti, M.J.; Fisco, T.; Polidoro, C.; Bocchinfuso, G.; Palleschi, A.; Novelli, G.; Spagnoli, L.G.; Mazzarelli, P. Carnitine palmitoyl transferase-1A (CPT1A): A new tumor specific target in human breast cancer. Oncotarget 2016, 7, 19982–19996. [Google Scholar] [CrossRef] [PubMed]
- Berge, K.; Tronstad, K.J.; Bohov, P.; Madsen, L.; Berge, R.K. Impact of mitochondrial β-oxidation in fatty acid-mediated inhibition of glioma cell proliferation. J. Lipid Res. 2003, 44, 118–127. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, J.H.; Wang, F.; Wang, Y.N.; He, M.M.; Wu, Q.N.; Lu, Y.X.; Yu, H.E.; Chen, Z.H.; Zhao, Q.; et al. Inhibition of fatty acid catabolism augments the efficacy of oxaliplatin-based chemotherapy in gastrointestinal cancers. Cancer Lett. 2020, 473, 74–89. [Google Scholar] [CrossRef]
- Liu, X.; Feng, R.; Du, L. The role of enoyl-CoA hydratase short chain 1 and peroxiredoxin 3 in PP2-induced apoptosis in human breast cancer MCF-7 cells. FEBS Lett. 2010, 584, 3185–3192. [Google Scholar] [CrossRef]
- Hernlund, E.; Ihrlund, L.S.; Khan, O.; Ates, Y.O.; Linder, S.; Panaretakis, T.; Shoshan, M.C. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-deoxyglucose and etomoxir. Int. J. Cancer 2008, 123, 476–483. [Google Scholar] [CrossRef]
- Yao, C.H.; Liu, G.Y.; Wang, R.; Moon, S.H.; Gross, R.W.; Patti, G.J. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase i is essential for cancer cell proliferation independent of β-oxidation. PLoS Biol. 2018, 16, e2003782. [Google Scholar] [CrossRef] [PubMed]
- Conti, R.; Mannucci, E.; Pessotto, P.; Tassoni, E.; Carminati, P.; Giannessi, F.; Arduini, A. Selective reversible inhibition of liver carnitine palmitoyl-transferase 1 by teglicar reduces gluconeogenesis and improves glucose homeostasis. Diabetes 2011, 60, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719. [Google Scholar] [CrossRef]
- Lien, E.C.; Westermark, A.M.; Zhang, Y.; Yuan, C.; Li, Z.; Lau, A.N.; Sapp, K.M.; Wolpin, B.M.; Vander Heiden, M.G. Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 2021, 599, 302–307. [Google Scholar] [CrossRef]
- Dmitrieva-Posocco, O.; Wong, A.C.; Lundgren, P.; Golos, A.M.; Descamps, H.C.; Dohnalová, L.; Cramer, Z.; Tian, Y.; Yueh, B.; Eskiocak, O.; et al. β-Hydroxybutyrate suppresses colorectal cancer. Nature 2022, 605, 160–165. [Google Scholar] [CrossRef]
- Abolhassani, R.; Berg, E.; Tenenbaum, G.; Israel, M. Inhibition of SCOT and Ketolysis Decreases Tumor Growth and Inflammation in the Lewis Cancer Model. Jap. J. Oncol. Clin. Res. 2022, 3, 1–12. [Google Scholar]
- Mahajan, A.; Spracklen, C.N.; Zhang, W.; Ng, M.C.Y.; Petty, L.E.; Kitajima, H.; Yu, G.Z.; Rüeger, S.; Speidel, L.; Kim, Y.J.; et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 2022, 54, 560–572. [Google Scholar] [CrossRef]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, Q.; Liu, Y.; Chen, S.; Zhang, Y.; Ma, Q.; Chen, X.; Liu, C.; Lei, H.; Chen, H.; et al. Genome-wide association study of metabolites in patients with coronary artery disease identified novel metabolite quantitative trait loci. Clin. Transl. Med. 2021, 11, e290. [Google Scholar] [CrossRef]
- Schlosser, P.; Li, Y.; Sekula, P.; Raffler, J.; Grundner-Culemann, F.; Pietzner, M.; Cheng, Y.; Wuttke, M.; Steinbrenner, I.; Schultheiss, U.T.; et al. Genetic studies of urinary metabolites illuminate mechanisms of detoxification and excretion in humans. Nat. Genet. 2020, 52, 167–176. [Google Scholar] [CrossRef]
- Li, Y.; Sekula, P.; Wuttke, M.; Wahrheit, J.; Hausknecht, B.; Schultheiss, U.T.; Gronwald, W.; Schlosser, P.; Tucci, S.; Ekici, A.B.; et al. Genome-Wide Association Studies of Metabolites in Patients with CKD Identify Multiple Loci and Illuminate Tubular Transport Mechanisms. J. Am. Soc. Nephrol. 2018, 29, 1513–1524. [Google Scholar] [CrossRef]
- Rhee, E.P.; Surapaneni, A.; Zheng, Z.; Zhou, L.; Dutta, D.; Arking, D.E.; Zhang, J.; Duong, T.V.; Chatterjee, N.; Luo, S.; et al. Trans-ethnic genome-wide association study of blood metabolites in the Chronic Renal Insufficiency Cohort (CRIC) study. Kidney Int. 2022, 101, 814–823. [Google Scholar] [CrossRef]
- Merritt, J.L.; Norris, M.; Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 2018, 6, 181–183. [Google Scholar] [CrossRef]
- Guerra, I.M.S.; Ferreira, H.B.; Melo, T.; Rocha, H.; Moreira, S.; Diogo, L.; Domingues, M.R.; Moreira, A.S.P. Mitochondrial Fatty Acid β-Oxidation Disorders: From Disease to Lipidomic Studies-A Critical Review. Int. J. Mol. Sci. 2022, 23, 13933. [Google Scholar] [CrossRef]
- Gregersen, N.; Winter, V.; Curtis, D.; Deufel, T.; Mack, M.; Hendrickx, J.; Willems, P.J.; Ponzone, A.; Parrella, T.; Ponzone, R.; et al. Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: The prevalent mutation G985 (K304E) is subject to a strong founder effect from northwestern Europe. Hum. Hered. 1993, 43, 342–350. [Google Scholar] [CrossRef]
- Morris, A.A.M. Disorders of mitochondrial fatty acid oxidation and related metabolic pathways. In Inborn Metabolic Diseases: Diagnosis and Treatment; Springer: Berlin/Heidelberg, Germany, 2012; pp. 201–216. ISBN 9783642157202. [Google Scholar]
- Nennstiel-Ratzel, U.; Arenz, S.; Maier, E.M.; Knerr, I.; Baumkötter, J.; Röschinger, W.; Liebl, B.; Hadorn, H.B.; Roscher, A.A.; Von Kries, R. Reduced incidence of severe metabolic crisis or death in children with medium chain acyl-CoA dehydrogenase deficiency homozygous for c.985A>G identified by neonatal screening. Mol. Genet. Metab. 2005, 85, 157–159. [Google Scholar] [CrossRef]
- Matsubara, Y.; Narisawa, K.; Ye-Qi, Y.; Tada, K.; Ikeda, H.; Danks, D.M.; Green, A.; McCabe, E.R.B. Prevalence of K329E mutation in medium-chain acyl-CoA dehydrogenase gene determined from Guthrie cards. Lancet 1991, 338, 552–553. [Google Scholar] [CrossRef]
- Von Muhlendahl, K.E.; Lehnert, W.; Monch, E. Medium-Chain-Acyl-CoA-Dehydrogenase(MCAD)-Defekt: Akute zerebrale Episoden und nicht-ketotische Hypoglykämien bei Kindern. DMW Dtsch. Medizinische Wochenschrift 1990, 115, 1235–1238. [Google Scholar] [CrossRef]
- Nagao, M. Frequency of 985A-to-G mutation in medium-chain acyl-CoA dehydrogenase gene among patients with sudden infant death syndrome, Reye syndrome, severe motor and intellectual disabilities and healthy newborns in Japan. Pediatr. Int. 1996, 38, 304–307. [Google Scholar] [CrossRef]
- Tyni, T.; Palotie, A.; Viinikka, L.; Valanne, L.; Salo, M.K.; Von Dobeln, U.; Jackson, S.; Wanders, R.; Venizelos, N.; Pihko, H. Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency with the G1528C mutation: Clinical presentation of thirteen patients. J. Pediatr. 1997, 130, 67–76. [Google Scholar] [CrossRef]
- Ijlst, L.; Ruiter, J.P.N.; Hoovers, J.M.N.; Jakobs, M.E.; Wanders, R.J.A. Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein α sub. J. Clin. Investig. 1996, 98, 1028–1033. [Google Scholar] [CrossRef]
- Jankowski, M.; Daca-Roszak, P.; Obracht-Prondzyński, C.; Płoski, R.; Lipska-Ziętkiewicz, B.S.; Ziętkiewicz, E. Genetic diversity in Kashubs: The regional increase in the frequency of several disease-causing variants. J. Appl. Genet. 2022, 63, 691–701. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Lindner, M.; Santer, R.; Grotzke, M.; Baumgartner, M.R.; Boehles, H.; Das, A.; Haase, C.; Hennermann, J.B.; Karall, D.; et al. Treatment recommendations in long-chain fatty acid oxidation defects: Consensus from a workshop. J. Inherit. Metab. Dis. 2009, 32, 498–505. [Google Scholar] [CrossRef]
- Wilcken, B. Fatty acid oxidation disorders: Outcome and long-term prognosis. J. Inherit. Metab. Dis. 2010, 33, 501–506. [Google Scholar] [CrossRef]
- Sperk, A.; Mueller, M.; Spiekerkoetter, U. Outcome in six patients with mitochondrial trifunctional protein disorders identified by newborn screening. Mol. Genet. Metab. 2010, 101, 205–207. [Google Scholar] [CrossRef]
- Karall, D.; Brunner-Krainz, M.; Kogelnig, K.; Konstantopoulou, V.; Maier, E.M.; Möslinger, D.; Plecko, B.; Sperl, W.; Volkmar, B.; Scholl-Bürgi, S. Clinical outcome, biochemical and therapeutic follow-up in 14 Austrian patients with Long-Chain 3-Hydroxy Acyl CoA Dehydrogenase Deficiency (LCHADD). Orphanet J. Rare Dis. 2015, 10, 1–11. [Google Scholar] [CrossRef]
- Kobayashi, T.; Minami, S.; Mitani, A.; Tanizaki, Y.; Booka, M.; Okutani, T.; Yamaguchi, S.; Ino, K. Acute fatty liver of pregnancy associated with fetal mitochondrial trifunctional protein deficiency. J. Obstet. Gynaecol. Res. 2015, 41, 799–802. [Google Scholar] [CrossRef]
- Brown, N.F.; Mullur, R.S.; Subramanian, I.; Esser, V.; Bennett, M.J.; Saudubray, J.M.; Feigenbaum, A.S.; Kobari, J.A.; Macleod, P.M.; McGarry, J.D.; et al. Molecular characterization of L-CPT I deficiency in six patients: Insights into function of the native enzyme. J. Lipid Res. 2001, 42, 1134–1142. [Google Scholar] [CrossRef]
- Gobin, S.; Bonnefont, J.P.; Prip-Buus, C.; Mugnier, C.; Ferrec, M.; Demaugre, F.; Saudubray, J.M.; Rostane, H.; Djouadi, F.; Wilcox, W.; et al. Organization of the human liver carnitine palmitoyltransferase 1 gene (CPT1A) and identification of novel mutations inn hyketotic hypoglycaemia. Hum. Genet. 2002, 111, 179–189. [Google Scholar] [CrossRef]
- Clemente, F.J.; Cardona, A.; Inchley, C.E.; Peter, B.M.; Jacobs, G.; Pagani, L.; Lawson, D.J.; Antão, T.; Vicente, M.; Mitt, M.; et al. A selective sweep on a deleterious mutation in CPT1A in Arctic populations. Am. J. Hum. Genet. 2014, 95, 584–589. [Google Scholar] [CrossRef]
- Collins, S.A.; Sinclair, G.; McIntosh, S.; Bamforth, F.; Thompson, R.; Sobol, I.; Osborne, G.; Corriveau, A.; Santos, M.; Hanley, B.; et al. Carnitine palmitoyltransferase 1A (CPT1A) P479L prevalence in live newborns in Yukon, Northwest Territories, and Nunavut. Mol. Genet. Metab. 2010, 101, 200–204. [Google Scholar] [CrossRef]
- Rinaldi, C.; Schmidt, T.; Situ, A.J.; Johnson, J.O.; Lee, P.R.; Chen, K.L.; Bott, L.C.; Fadó, R.; Harmison, G.H.; Parodi, S.; et al. Mutation in CPT1C associated with pure autosomal dominant spastic paraplegia. JAMA Neurol. 2015, 72, 561–570. [Google Scholar] [CrossRef]
- Hong, D.; Cong, L.; Zhong, S.; Liu, L.; Xu, Y.; Zhang, J. A novel CPT1C variant causes pure hereditary spastic paraplegia with benign clinical course. Ann. Clin. Transl. Neurol. 2019, 6, 610–614. [Google Scholar] [CrossRef]
- Boemer, F.; Deberg, M.; Schoos, R.; Caberg, J.H.; Gaillez, S.; Dugauquier, C.; Delbecque, K.; François, A.; Maton, P.; Demonceau, N.; et al. Diagnostic pitfall in antenatal manifestations of CPT II deficiency. Clin. Genet. 2016, 89, 193–197. [Google Scholar] [CrossRef]
- Anichini, A.; Fanin, M.; Vianey-Saban, C.; Cassandrini, D.; Fiorillo, C.; Bruno, C.; Angelini, C. Genotype—Phenotype correlations in a large series of patients with muscle type CPT II deficiency. Neurol. Res. 2011, 33, 24–32. [Google Scholar] [CrossRef]
- Ørngreen, M.C.; Dunø, M.; Ejstrup, R.; Christensen, E.; Schwartz, M.; Sacchetti, M.; Vissing, J. Fuel utilization in subjects with-carnitine palmitoyltransferase 2 gene mutations. Ann. Neurol. 2005, 57, 60–66. [Google Scholar] [CrossRef]
- Mak, C.M.; Lam, C.W.; Fong, N.C.; Siu, W.K.; Lee, H.C.H.; Siu, T.S.; Lai, C.K.; Law, C.Y.; Tong, S.F.; Poon, W.T.; et al. Fatal viral infection-associated encephalopathy in two Chinese boys: A genetically determined risk factor of thermolabile carnitine palmitoyltransferase II variants. J. Hum. Genet. 2011, 56, 617–621. [Google Scholar] [CrossRef]
- Kubota, M.; Chida, J.; Hoshino, H.; Ozawa, H.; Koide, A.; Kashii, H.; Koyama, A.; Mizuno, Y.; Hoshino, A.; Yamaguchi, M.; et al. Thermolabile CPT II variants and low blood ATP levels are closely related to severity of acute encephalopathy in Japanese children. Brain Dev. 2012, 34, 20–27. [Google Scholar] [CrossRef]
- Gallant, N.M.; Leydiker, K.; Tang, H.; Feuchtbaum, L.; Lorey, F.; Puckett, R.; Deignan, J.L.; Neidich, J.; Dorrani, N.; Chang, E.; et al. Biochemical, molecular, and clinical characteristics of children with short chain acyl-CoA dehydrogenase deficiency detected by newborn screening in California. Mol. Genet. Metab. 2012, 106, 55–61. [Google Scholar] [CrossRef]
- Pedersen, C.B.; Kølvraa, S.; Kølvraa, A.; Stenbroen, V.; Kjeldsen, M.; Ensenauer, R.; Tein, I.; Matern, D.; Rinaldo, P.; Vianey-Saban, C.; et al. The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Hum. Genet. 2008, 124, 43–56. [Google Scholar] [CrossRef]
- Van Maldegem, B.T.; Duran, M.; Wanders, R.J.A.; Niezen-Koning, K.E.; Hogeveen, M.; Ijlst, L.; Waterham, H.R.; Wijburg, F.A. Clinical, Biochemical, and Genetic Heterogeneity in Short-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. JAMA 2006, 296, 943–952. [Google Scholar] [CrossRef]
- Strauss, A.W.; Powell, C.K.; Hale, D.E.; Anderson, M.M.; Ahuja, A.; Brackett, J.C.; Sims, H.F. Molecular basis of human mitochondrial very-long-chain acyl-CoA dehydrogenase deficiency causing cardiomyopathy and sudden death in childhood. Proc. Natl. Acad. Sci. USA 1995, 92, 10496–10500. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Indo, Y.; Coates, P.M.; Hashimoto, T.; Tanaka, K. Identification of very-long-chain acyl-CoA dehydrogenase deficiency in three patients previously diagnosed with long-chain acyl-CoA dehydrogenase deficiency. Pediatr. Res. 1993, 34, 111–113. [Google Scholar] [CrossRef]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty acid oxidation disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Burrage, L.C.; Gibson, J.B.; Strenk, M.E.; Lose, E.J.; Bick, D.P.; Elsea, S.H.; Sutton, V.R.; Sun, Q.; Graham, B.H.; et al. Recurrent ACADVL molecular findings in individuals with a positive newborn screen for very long chain acyl-coA dehydrogenase (VLCAD) deficiency in the United States. Mol. Genet. Metab. 2015, 116, 139–145. [Google Scholar] [CrossRef]
- Burgin, H.J.; Murayama, K.; Ohtake, A.; McKenzie, M. Mitochondrial Short-Chain Enoyl-CoA Hydratase 1 Deficiency (ECHS1D). In Genetic Syndromes; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 1–5. [Google Scholar]
- Uesugi, M.; Mori, J.; Fukuhara, S.; Fujii, N.; Omae, T.; Sasai, H.; Ichimoto, K.; Murayama, K.; Osamura, T.; Hosoi, H. Short-chain enoyl-CoA hydratase deficiency causes prominent ketoacidosis with normal plasma lactate levels: A case report. Mol. Genet. Metab. Rep. 2020, 25, 100672. [Google Scholar] [CrossRef]
- Huffnagel, I.C.; Redeker, E.J.W.; Reneman, L.; Vaz, F.M.; Ferdinandusse, S.; Poll-The, B.T. Mitochondrial encephalopathy and transient 3-methylglutaconic aciduria in ECHS1 deficiency: Long-term follow-up. JIMD Rep. 2018, 39, 83–87. [Google Scholar] [PubMed]
- Fitzsimons, P.E.; Alston, C.L.; Bonnen, P.E.; Hughes, J.; Crushell, E.; Geraghty, M.T.; Tetreault, M.; O’Reilly, P.; Twomey, E.; Sheikh, Y.; et al. Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Am. J. Med. Genet. Part A 2018, 176, 1115–1127. [Google Scholar] [CrossRef]
- Peters, H.; Buck, N.; Wanders, R.; Ruiter, J.; Waterham, H.; Koster, J.; Yaplito-Lee, J.; Ferdinandusse, S.; Pitt, J. ECHS1 mutations in Leigh disease: A new inborn error of metabolism affecting valine metabolism. Brain 2014, 137, 2903–2908. [Google Scholar] [CrossRef]
- Sakai, C.; Yamaguchi, S.; Sasaki, M.; Miyamoto, Y.; Matsushima, Y.; Goto, Y. ichi ECHS1 mutations cause combined respiratory chain deficiency resulting in leigh syndrome. Hum. Mutat. 2015, 36, 232–239. [Google Scholar] [CrossRef]
- Sun, D.; Liu, Z.; Liu, Y.; Wu, M.; Fang, F.; Deng, X.; Liu, Z.; Song, L.; Murayama, K.; Zhang, C.; et al. Novel ECHS1 mutations in Leigh syndrome identified by whole-exome sequencing in five Chinese families: Case report. BMC Med. Genet. 2020, 21, 149. [Google Scholar] [CrossRef] [PubMed]
- Ganetzky, R.D.; Bloom, K.; Ahrens-Nicklas, R.; Edmondson, A.; Deardorff, M.A.; Bennett, M.J.; Ficicioglu, C. ECHS1 deficiency as a cause of severe neonatal lactic acidosis. JIMD Rep. 2016, 30, 33–37. [Google Scholar] [PubMed]
- Haack, T.B.; Jackson, C.B.; Murayama, K.; Kremer, L.S.; Schaller, A.; Kotzaeridou, U.; de Vries, M.C.; Schottmann, G.; Santra, S.; Büchner, B.; et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann. Clin. Transl. Neurol. 2015, 2, 492–509. [Google Scholar] [CrossRef]
- Olgiati, S.; Skorvanek, M.; Quadri, M.; Minneboo, M.; Graafland, J.; Breedveld, G.J.; Bonte, R.; Ozgur, Z.; van den Hout, M.C.G.N.; Schoonderwoerd, K.; et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov. Disord. 2016, 31, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Riley, L.G.; Bratkovic, D.; Ketteridge, D.; Manton, N.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Mohamed, M.; Gardeitchik, T.; et al. Unique presentation of cutis laxa with Leigh-like syndrome due to ECHS1 deficiency. J. Inherit. Metab. Dis. 2017, 40, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Klootwijk, E.D.; Reichold, M.; Helip-Wooley, A.; Tolaymat, A.; Broeker, C.; Robinette, S.L.; Reinders, J.; Peindl, D.; Renner, K.; Eberhart, K.; et al. Mistargeting of Peroxisomal EHHADH and Inherited Renal Fanconi’s Syndrome. N. Engl. J. Med. 2014, 370, 129–138. [Google Scholar] [CrossRef]
- Tolaymat, A.; Sakarcan, A.; Neiberger, R. Idiopathic Fanconi syndrome in a family. Part I. Clinical aspects. J. Am. Soc. Nephrol. 1992, 2, 1310–1317. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Theda, C.; Naidu, S.; Watkins, P.A.; Moser, A.B.; Moser, H.W. Neuronal migration abnormality in peroxisomal bifunctional enzyme defect. Ann. Neurol. 1996, 39, 268–271. [Google Scholar] [CrossRef]
- Fukuda, S.; Suzuki, Y.; Shimozawa, N.; Zhang, Z.; Orii, T.; Aoyama, T.; Hashimoto, T.; Kondo, N. Amino acid and nucleotide sequences of human peroxisomal enoyl-CoA hydratase: 3-hydroxyacyl-CoA dehydrogenase cDNA. J. Inherit. Metab. Dis. 1998, 21, 23–28. [Google Scholar] [CrossRef]
- Yang, S.Y.; He, X.Y.; Schulz, H. 3-Hydroxyacyl-CoA dehydrogenase and short chain 3-hydroxyacyl-CoA dehydrogenase in human health and disease. FEBS J. 2005, 272, 4874–4883. [Google Scholar] [CrossRef]
- Treacy, E.P.; Lambert, D.M.; Barnes, R.; Boriack, R.L.; Vockley, J.; O’Brien, L.K.; Jones, P.M.; Bennett, M.J. Short-chain hydroxyacyl-coenzyme A dehydrogenase deficiency presenting as unexpected infant death: A family study. J. Pediatr. 2000, 137, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J.; Weinberger, M.J.; Kobori, J.A.; Rinaldo, P.; Burlina, A.B. Mitochondrial Short-Chain L-3-Hydroxyacl-Coenzyme A Dehydrogenase Deficiency: A New Defect of Fatty Acid Oxidation. Pediatr. Res. 1996, 39, 185–188. [Google Scholar] [CrossRef]
- Bennett, M.J.; Spotswood, S.D.; Ross, K.F.; Comfort, S.; Koonce, R.; Boriack, R.L.; Ijlst, L.; Wanders, R.J.A. Fatal hepatic short-chain 1-3-hydroxyacyl-coenzyme dehydrogenase deficiency: Clinical, biochemical, and pathological studies on three subjects with this recently identified disorder of mitochondrial β-oxidation. Pediatr. Dev. Pathol. 1999, 2, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; Xie, W.; Caswell, R.; Damhuis, A.; Vianey-Saban, C.; Akcay, T.; Darendeliler, F.; Bas, F.; Guven, A.; Siklar, Z.; et al. Next-generation sequencing reveals deep intronic cryptic ABCC8 and HADH splicing founder mutations causing hyperinsulinism by pseudoexon activation. Am. J. Hum. Genet. 2013, 92, 131–136. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Patch, A.M.; Locke, J.M.; Akcay, T.; Simsek, E.; Alaei, M.; Yekta, Z.; Desai, M.; Kapoor, R.R.; Hussain, K.; et al. Genome-wide homozygosity analysis reveals HADH mutations as a common cause of diazoxide-responsive hyperinsulinemic-hypoglycemia in consanguineous pedigrees. J. Clin. Endocrinol. Metab. 2011, 96, 96. [Google Scholar] [CrossRef]
- Di Candia, S.; Gessi, A.; Pepe, G.; Valin, P.S.; Mangano, E.; Chiumello, G.; Gianolli, L.; Proverbio, M.C.; Mora, S. Identification of a diffuse form of hyperinsulinemic hypoglycemia by 18-fluoro-L-3, 4 dihydroxyphenylalanine positron emission tomography/CT in a patient carrying a novel mutation of the HADH gene. Eur. J. Endocrinol. 2009, 160, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Arora, C.; Padmanabha, H.; Christopher, R.; Mahale, R.; Bhat, M.; Arunachal, G.; Shekhar, R.; Mailankody, P.; Mathuranath, P.S. Pseudo-neonatal Adrenoleukodystrophy: A Rare Peroxisomal Disorder. Ann. Indian Acad. Neurol. 2022, 25, 275–278. [Google Scholar] [CrossRef]
- Aubourg, P.; Wanders, R. Peroxisomal disorders. Handb. Clin. Neurol. 2013, 113, 1593–1609. [Google Scholar] [CrossRef]
- Kemp, S.; Valianpour, F.; Mooyer, P.A.W.; Kulik, W.; Wanders, R.J.A. Method for measurement of peroxisomal very-long-chain fatty acid beta-oxidation in human skin fibroblasts using stable-isotope-labeled tetracosanoic acid. Clin. Chem. 2004, 50, 1824–1826. [Google Scholar] [CrossRef]
- Bezman, L.; Moser, H.W. Incidence of X-linked adrenoleukodystrophy and the relative frequency of its phenotypes. Am. J. Med. Genet. 1998, 76, 415–419. [Google Scholar] [CrossRef]
- Mosser, J.; Douar, A.M.; Sarde, C.O.; Kioschis, P.; Feil, R.; Moser, H.; Poustka, A.M.; Mandel, J.L.; Aubourg, P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 1993, 361, 726–730. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; De Visser, M.; Van Geel, B.M.; Wanders, R.J.A.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef]
- Kemp, S.; Theodoulou, F.L.; Wanders, R.J.A. Mammalian peroxisomal ABC transporters: From endogenous substrates to pathology and clinical significance. Br. J. Pharmacol. 2011, 164, 1753–1766. [Google Scholar] [CrossRef]
- Pugliese, A.; Beltramo, T.; Torre, D. Reye’s and Reye’s-like syndromes. Cell Biochem. Funct. 2008, 26, 741–746. [Google Scholar] [CrossRef]
- Schrör, K. Aspirin and Reye syndrome: A review of the evidence. Pediatr. Drugs 2007, 9, 195–204. [Google Scholar] [CrossRef]
- Chen, J.; Spracklen, C.N.; Marenne, G.; Varshney, A.; Corbin, L.J.; Luan, J.; Willems, S.M.; Wu, Y.; Zhang, X.; Horikoshi, M.; et al. The Trans-Ancestral Genomic Architecture of Glycemic Traits. Nat. Genet. 2021, 53, 840. [Google Scholar] [CrossRef] [PubMed]
- Xue, A.; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; Wu, Y.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 2941. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Lowe, J.K.; Kovvali, S.; Maller, J.B.; Salit, J.; Daly, M.J.; Stoffel, M.; Altshuler, D.M.; Friedman, J.M.; Breslow, J.L.; et al. Genome-wide association study of electrocardiographic conduction measures in an isolated founder population: Kosrae. Hear. Rhythm 2009, 6, 634–641. [Google Scholar] [CrossRef]
- Vuckovic, D.; Bao, E.L.; Akbari, P.; Lareau, C.A.; Mousas, A.; Jiang, T.; Chen, M.H.; Raffield, L.M.; Tardaguila, M.; Huffman, J.E.; et al. The Polygenic and Monogenic Basis of Blood Traits and Diseases. Cell 2020, 182, 1214–1231. [Google Scholar] [CrossRef] [PubMed]
- van Rheenen, W.; van der Spek, R.A.A.; Bakker, M.K.; van Vugt, J.J.F.A.; Hop, P.J.; Zwamborn, R.A.J.; de Klein, N.; Westra, H.J.; Bakker, O.B.; Deelen, P.; et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 2021, 53, 1636–1648. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Choi, J.E.; Park, B.; Cha, M.Y.; Hong, K.W.; Jung, D.H. Dyslipidaemia—Genotype Interactions with Nutrient Intake and Cerebro-Cardiovascular Disease. Biomedicines 2022, 10, 1615. [Google Scholar] [CrossRef] [PubMed]
- Sinnott-Armstrong, N.; Tanigawa, Y.; Amar, D.; Mars, N.; Benner, C.; Aguirre, M.; Venkataraman, G.R.; Wainberg, M.; Ollila, H.M.; Kiiskinen, T.; et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet. 2021, 53, 185–194. [Google Scholar] [CrossRef]
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A.; et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415–1429.e19. [Google Scholar] [CrossRef]
- Graham, S.E.; Clarke, S.L.; Wu, K.H.H.; Kanoni, S.; Zajac, G.J.M.; Ramdas, S.; Surakka, I.; Ntalla, I.; Vedantam, S.; Winkler, T.W.; et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 2021, 600, 675–679. [Google Scholar] [CrossRef]
- Morris, A.P.; Le, T.H.; Wu, H.; Akbarov, A.; van der Most, P.J.; Hemani, G.; Smith, G.D.; Mahajan, A.; Gaulton, K.J.; Nadkarni, G.N.; et al. Trans-ethnic kidney function association study reveals putative causal genes and effects on kidney-specific disease aetiologies. Nat. Commun. 2019, 10, 29. [Google Scholar] [CrossRef]
- Yin, X.; Chan, L.S.; Bose, D.; Jackson, A.U.; VandeHaar, P.; Locke, A.E.; Fuchsberger, C.; Stringham, H.M.; Welch, R.; Yu, K.; et al. Genome-wide association studies of metabolites in Finnish men identify disease-relevant loci. Nat. Commun. 2022, 13, 19. [Google Scholar] [CrossRef]
- Hysi, P.G.; Mangino, M.; Christofidou, P.; Falchi, M.; Karoly, E.D.; Mohney, R.P.; Valdes, A.M.; Spector, T.D.; Menni, C. Metabolome Genome-Wide Association Study Identifies 74 Novel Genomic Regions Influencing Plasma Metabolites Levels. Metabolites 2022, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef]
- Feofanova, E.V.; Chen, H.; Dai, Y.; Jia, P.; Grove, M.L.; Morrison, A.C.; Qi, Q.; Daviglus, M.; Cai, J.; North, K.E.; et al. A Genome-wide Association Study Discovers 46 Loci of the Human Metabolome in the Hispanic Community Health Study/Study of Latinos. Am. J. Hum. Genet. 2020, 107, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zheng, Y.; Alexander, D.; Morrison, A.C.; Coresh, J.; Boerwinkle, E. Genetic Determinants Influencing Human Serum Metabolome among African Americans. PLoS Genet. 2014, 10, e1004212. [Google Scholar] [CrossRef]
- Liu, H.; Doke, T.; Guo, D.; Sheng, X.; Ma, Z.; Park, J.; Vy, H.M.T.; Nadkarni, G.N.; Abedini, A.; Miao, Z.; et al. Epigenomic and transcriptomic analyses define core cell types, genes and targetable mechanisms for kidney disease. Nat. Genet. 2022, 54, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, Y.; Wedow, R.; Li, Y.; Brazel, D.M.; Chen, F.; Datta, G.; Davila-Velderrain, J.; McGuire, D.; Tian, C.; et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 2019, 51, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Karlsson Linnér, R.; Biroli, P.; Kong, E.; Meddens, S.F.W.; Wedow, R.; Fontana, M.A.; Lebreton, M.; Tino, S.P.; Abdellaoui, A.; Hammerschlag, A.R.; et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat. Genet. 2019, 51, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Ilkov, M.; Lamb, J.R.; Finkel, N.; Gudmundsson, E.F.; Pitts, R.; Hoover, H.; Gudmundsdottir, V.; Horman, S.R.; Aspelund, T.; et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 2018, 361, 769–773. [Google Scholar] [CrossRef]
- Thareja, G.; Belkadi, A.; Arnold, M.; Albagha, O.M.E.; Graumann, J.; Schmidt, F.; Grallert, H.; Peters, A.; Gieger, C.; Consortium, T.Q.G.P.R.; et al. Differences and commonalities in the genetic architecture of protein quantitative trait loci in European and Arab populations. Hum. Mol. Genet. 2022, 32, 907–916. [Google Scholar] [CrossRef]
- Hernandez Cordero, A.I.; Gonzales, N.M.; Parker, C.C.; Sokolof, G.; Vandenbergh, D.J.; Cheng, R.; Abney, M.; Sko, A.; Douglas, A.; Palmer, A.A.; et al. Genome-wide Associations Reveal Human-Mouse Genetic Convergence and Modifiers of Myogenesis, CPNE1 and STC2. Am. J. Hum. Genet. 2019, 105, 1222–1236. [Google Scholar] [CrossRef]
- Richardson, T.G.; Leyden, G.M.; Wang, Q.; Bell, J.A.; Elsworth, B.; Smith, G.D.; Holmes, M.V. Characterising metabolomic signatures of lipid-modifying therapies through drug target mendelian randomisation. PLoS Biol. 2022, 20, e3001547. [Google Scholar] [CrossRef]
- Christakoudi, S.; Evangelou, E.; Riboli, E.; Tsilidis, K.K. GWAS of allometric body-shape indices in UK Biobank identifies loci suggesting associations with morphogenesis, organogenesis, adrenal cell renewal and cancer. Sci. Rep. 2021, 11, 10688. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Verma, S.S.; Hall, M.A.; Goodloe, R.J.; Berg, R.L.; Carrell, D.S.; Carlson, C.S.; Chen, L.; Crosslin, D.R.; Denny, J.C.; et al. Electronic medical records and genomics (eMERGE) network exploration in cataract: Several new potential susceptibility loci. Mol. Vis. 2014, 20, 1281–1295. [Google Scholar]
- Timmins, I.R.; Zaccardi, F.; Nelson, C.P.; Franks, P.; Yates, T.; Dudbridge, F. Genome-wide association study of self-reported walking pace suggests beneficial effects of brisk walking on health and survival. Commun. Biol. 2020, 3, 634. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, E.; Gao, H.; Chu, C.; Ntritsos, G.; Blakeley, P.; Butts, A.R.; Pazoki, R.; Suzuki, H.; Koskeridis, F.; Yiorkas, A.M.; et al. New alcohol-related genes suggest shared genetic mechanisms with neuropsychiatric disorders. Nat. Hum. Behav. 2019, 3, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sealock, J.M.; Sanchez-Roige, S.; Clarke, T.K.; Levey, D.F.; Cheng, Z.; Li, B.; Polimanti, R.; Kember, R.L.; Smith, R.V.; et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat. Neurosci. 2020, 23, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Saunders, G.R.B.; Wang, X.; Chen, F.; Jang, S.K.; Liu, M.; Wang, C.; Gao, S.; Jiang, Y.; Khunsriraksakul, C.; Otto, J.M.; et al. Genetic diversity fuels gene discovery for tobacco and alcohol use. Nature 2022, 612, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Chouraki, V.; De Bruijn, R.F.A.G.; Chapuis, J.; Bis, J.C.; Reitz, C.; Schraen, S.; Ibrahim-Verbaas, C.A.; Grenier-Boley, B.; Delay, C.; Rogers, R.; et al. A genome-wide association meta-analysis of plasma Aβ peptides concentrations in the elderly. Mol. Psychiatry 2014, 19, 1326–1335. [Google Scholar] [CrossRef]
- Medina-Gomez, C.; Kemp, J.P.; Trajanoska, K.; Luan, J.; Chesi, A.; Ahluwalia, T.S.; Mook-Kanamori, D.O.; Ham, A.; Hartwig, F.P.; Evans, D.S.; et al. Life-Course Genome-wide Association Study Meta-analysis of Total Body BMD and Assessment of Age-Specific Effects. Am. J. Hum. Genet. 2018, 102, 88–102. [Google Scholar] [CrossRef]
- Richardson, T.G.; Sanderson, E.; Palmerid, T.M.; Korpelaid, M.A.; Ference, B.A.; Smith, G.D.; Holmes, M.V. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020, 17, e1003062. [Google Scholar] [CrossRef]
- Tin, A.; Marten, J.; Halperin Kuhns, V.L.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.B.; Qiu, C.; Gorski, M.; Yu, Z.; et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474. [Google Scholar] [CrossRef]
- Borges, M.C.; Haycock, P.C.; Zheng, J.; Hemani, G.; Holmes, M.V.; Davey Smith, G.; Hingorani, A.D.; Lawlor, D.A. Role of circulating polyunsaturated fatty acids on cardiovascular diseases risk: Analysis using Mendelian randomization and fatty acid genetic association data from over 114,000 UK Biobank participants. BMC Med. 2022, 20, 210. [Google Scholar] [CrossRef]
- Wyss, A.B.; Sofer, T.; Lee, M.K.; Terzikhan, N.; Nguyen, J.N.; Lahousse, L.; Latourelle, J.C.; Smith, A.V.; Bartz, T.M.; Feitosa, M.F.; et al. Multiethnic meta-analysis identifies ancestry-specific and cross-ancestry loci for pulmonary function. Nat. Commun. 2018, 9, 2976. [Google Scholar] [CrossRef]
- Chai, J.F.; Raichur, S.; Khor, I.W.; Torta, F.; Chew, W.S.; Herr, D.R.; Ching, J.; Kovalik, J.P.; Khoo, C.M.; Wenk, M.R.; et al. Associations with metabolites in Chinese suggest new metabolic roles in Alzheimer’s and Parkinson’s diseases. Hum. Mol. Genet. 2020, 29, 189–201. [Google Scholar] [CrossRef]
- Jia, Q.; Han, Y.; Huang, P.; Woodward, N.C.; Gukasyan, J.; Kettunen, J.; Ala-Korpela, M.; Anufrieva, O.; Wang, Q.; Perola, M.; et al. Genetic Determinants of Circulating Glycine Levels and Risk of Coronary Artery Disease. J. Am. Heart Assoc. 2019, 8. [Google Scholar] [CrossRef]
- Lotta, L.A.; Pietzner, M.; Stewart, I.D.; Wittemans, L.B.L.; Li, C.; Bonelli, R.; Raffler, J.; Biggs, E.K.; Oliver-Williams, C.; Auyeung, V.P.W.; et al. Cross-platform genetic discovery of small molecule products of metabolism and application to clinical outcomes. Nat. Genet. 2021, 53, 54. [Google Scholar] [CrossRef]
- Wittemans, L.B.L.; Lotta, L.A.; Oliver-Williams, C.; Stewart, I.D.; Surendran, P.; Karthikeyan, S.; Day, F.R.; Koulman, A.; Imamura, F.; Zeng, L.; et al. Assessing the causal association of glycine with risk of cardio-metabolic diseases. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Illig, T.; Gieger, C.; Zhai, G.; Römisch-Margl, W.; Wang-Sattler, R.; Prehn, C.; Altmaier, E.; Kastenmüller, G.; Kato, B.S.; Mewes, H.W.; et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 2010, 42, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Kachuri, L.; Jeon, S.; DeWan, A.T.; Metayer, C.; Ma, X.; Witte, J.S.; Chiang, C.W.K.; Wiemels, J.L.; de Smith, A.J. Genetic determinants of blood-cell traits influence susceptibility to childhood acute lymphoblastic leukemia. Am. J. Hum. Genet. 2021, 108, 1823–1835. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, M.H.; Bentley, A.R.; Leonard, H.; Meeks, K.A.C.; Ekoru, K.; Chen, G.; Nalls, M.A.; Simonsick, E.M.; Tarazona-Santos, E.; Lima-Costa, M.F.; et al. Trans-ethnic meta-analysis identifies new loci associated with longitudinal blood pressure traits. Sci. Rep. 2021, 11, 4075. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Bien, S.A.; Nishimura, K.K.; Haessler, J.; Hodonsky, C.J.; Baldassari, A.R.; Highland, H.M.; Wang, Z.; Preuss, M.; Sitlani, C.M.; et al. Multi-ethnic genome-wide association analyses of white blood cell and platelet traits in the Population Architecture using Genomics and Epidemiology (PAGE) study. BMC Genom. 2021, 22, 432. [Google Scholar] [CrossRef]
- Qayyum, R.; Snively, B.M.; Ziv, E.; Nalls, M.A.; Liu, Y.; Tang, W.; Yanek, L.R.; Lange, L.; Evans, M.K.; Ganesh, S.; et al. A meta-analysis and genome-wide association study of platelet count and mean platelet volume in African Americans. PLoS Genet. 2012, 8, e1002491. [Google Scholar] [CrossRef]
- Igarashi, M.; Nogawa, S.; Kawafune, K.; Hachiya, T.; Takahashi, S.; Jia, H.; Saito, K.; Kato, H. Identification of the 12q24 locus associated with fish intake frequency by genome-wide meta-analysis in Japanese populations. Genes Nutr. 2019, 14, 21. [Google Scholar] [CrossRef]
- Cho, S.K.; Kim, B.; Myung, W.; Chang, Y.; Ryu, S.; Kim, H.N.; Kim, H.L.; Kuo, P.H.; Winkler, C.A.; Won, H.H. Polygenic analysis of the effect of common and low-frequency genetic variants on serum uric acid levels in Korean individuals. Sci. Rep. 2020, 10, 9179. [Google Scholar] [CrossRef] [PubMed]
- Yasukochi, Y.; Sakuma, J.; Takeuchi, I.; Kato, K.; Oguri, M.; Fujimaki, T.; Horibe, H.; Yamada, Y. Identification of CDC42BPG as a novel susceptibility locus for hyperuricemia in a Japanese population. Mol. Genet. Genom. 2018, 293, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, L.; Feng, X.; Cheng, H.; Ge, X.; Bao, Y.; Huang, L.; Wang, F.; Liu, C.; Chen, X.; et al. Genome-wide association and Mendelian randomization study of blood copper levels and 213 deep phenotypes in humans. Commun. Biol. 2022, 5, 405. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, A.; Gudmundsdottir, V.; Axelsson, G.T.; Gudmundsson, E.F.; Jonsson, B.G.; Launer, L.J.; Lamb, J.R.; Jennings, L.L.; Aspelund, T.; Emilsson, V.; et al. A genome-wide association study of serum proteins reveals shared loci with common diseases. Nat. Commun. 2022, 13, 480. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, H.; Xu, H.; Geng, Q.; Mak, W.H.; Ling, F.; Su, Z.; Yang, F.; Zhang, T.; Chen, J.; et al. New genetic variants associated with major adverse cardiovascular events in patients with acute coronary syndromes and treated with clopidogrel and aspirin. Pharmacogenom. J. 2021, 21, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Jansen, P.R.; Savage, J.E.; Nandakumar, P.; Wang, X.; Agee, M.; Aslibekyan, S.; Auton, A.; Bell, R.K.; Bryc, K.; et al. Genome-wide meta-analysis of insomnia prioritizes genes associated with metabolic and psychiatric pathways. Nat. Genet. 2022, 54, 1125–1132. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name/Abbreviation | Organ/Tissue Localization | Subcellular Compartment | References |
|---|---|---|---|
| ACSVL [FATP2] | Liver, intestine, kidneys, brain | Peroxisomes, ER | [34] |
| ACSVL [FATP6] | Heart | Cytosol, plasma membrane | [35] |
| ACSVL [FATP3] | Lungs, gonads, adrenals | ER, mitochondrial membrane | [34] |
| ACSVL [FATP1] | Skeletal muscles, BAT, WAT, heart | Plasma membrane | [36] |
| ACSVL [FATP4] | Skeletal muscles, BAT, WAT, intestine, skin | Peroxisomes, ER, mitochondrial membrane | [37] |
| ACSVL [FATP5] | Liver | Plasma membrane | [38] |
| ACSL1 | Liver, heart, BAT, WAT, skeletal muscles | Mitochondria (outer mitochondrial membrane on the cytosolic side), lipid droplets, microsomes, plasma membrane | [39] |
| ACSL3 | Brain, gonads, small amounts in other tissues (liver) | Lipid droplets, the cytoplasmic face of ER, the outer mitochondrial membrane | [40] |
| ACSL4 | Adrenals, ovaries, testes, liver, skeletal muscles, small amounts in the brain | Endosomes, peroxisomes, plasma membrane, secretory vesicles, ER regions in close contact with mitochondria—mitochondrial-associated membranes | [41] |
| ACSL5 | BAT, the duodenal mucosa, liver, skeletal muscles, kidneys, lungs | The outer mitochondrial membrane on the cytosolic side | [42] |
| ACSL6 | Ovaries, testes, brain, skeletal muscles, small amounts in the WAT, kidneys, the duodenal mucosa | Plasma membrane | [43] |
| ACSM | Liver, skeletal muscles, cardiomyocytes, colonocytes, kidneys | Mitochondria. All ACSMs belong to a group of enzymes called XM-ligases (xenobiotic/medium-chain fatty acid-CoA ligases) | [44,45] |
| ACSS1 | Brain, blood, testes, intestine, heart, kidneys, skeletal muscles, BAT | Mitochondria. ACSS1 activates acetate | [46] |
| ACSS2 | Liver and kidneys | Cytosol, nucleus. ACSS2 activates acetate. ACSS2 is downregulated during fasting | [46,47] |
| ACSS3 | Liver | Mitochondria. ACSS3 has a higher affinity for propionate. ACSS3 is upregulated in the fasting state | [30,46] |
| Enzyme | Mitochondrial Compartment | Preferred Substrates (Acyl-CoAs) | Tissue/Organ/Cell | Reference |
|---|---|---|---|---|
| VLCAD | Inner mitochondrial membrane | LCFA (mainly palmitoyl-CoA) and VLCFA (C14–C22) | Muscles, heart, liver, skin fibroblasts | [84] |
| Acyl-CoA DH-9 (ACAD9) | Inner mitochondrial membrane | Unsaturated LCFA, VLCFA (C16:1, C18:1, C18:2; C22:6) | Brain, liver, heart, skeletal muscle | [85] |
| LCAD | Matrix | LCFA, unsaturated MCFA, SCFA, BCFA (in vitro) | Lungs—pulmonary surfactant | [86] |
| MCAD | Matrix | MCFA (C6:0–C12:0) | Heart, skeletal muscles, liver | [87] |
| SCAD | Matrix | SCFA (mainly butyryl-CoA); MCFA (C6:0–C12:0) | Liver, heart, skeletal muscle | [88] |
| Peroxisomal β-Oxidation | Mitochondrial β-Oxidation | References | |
|---|---|---|---|
| Proteins involved in the transport of FAs to peroxisomes/mitochondria | ABCD1, ABCD2, and ABCD3 | Carnitine transport system (CPT1, CPT2, CAC) | [115,116] |
| Substrates | VLCFAs (>C22), BCFAs (e.g., pristanic acid), PUFA, 2-hydroxy FAs, long-chain dicarboxylic acids, bile acid intermediates, and a number of prostanoids | VLCFAs (≤22), LCFAs, MCFAs, and SCFAs | [117,118] |
| Enzyme catalyzing the first reaction | ACOXs The transfer of electrons from FADH2 to oxygen results in the production of H2O2, which is subsequently cleaved by peroxisomal catalase | ACADs The electrons that originate from FADH2 are transported to ETF, the ETF dehydrogenase, and transferred to OXPHOS. Finally, they reduce oxygen to water, which results in the production of energy in the form of ATP | [82,110] |
| β-oxidation end products | Acetyl-CoA, NADH, MCFAs, and FADH2 | Acetyl-CoA, NADH, and FADH2 | [94,110] |
| Gene/Enzyme | Nature of Change | Type of Evaluation | Cancer Type | References |
|---|---|---|---|---|
| ACAD9 | Upregulated | mRNA level | Glioblastoma multiforme | [289] |
| ACSL1 | Downregulated | mRNA level | Lung cancer, breast cancer | [290,291] |
| Upregulated | mRNA level | Rectal adenocarcinoma, colon cancer, hepatocellular carcinoma | [290,292,293,294] | |
| ACSL3 | Downregulated | mRNA level | Ovarian cancer | [290] |
| Upregulated | mRNA level | Melanoma, ESR-negative breast cancer | [290,295] | |
| Protein level | Large-cell lung cancer, small-cell lung cancer | [296] | ||
| ACSL4 | Downregulated | mRNA and protein levels | Gastric cancer | [297] |
| mRNA level | Lung cancer | [290] | ||
| Upregulated | mRNA level | Colorectal cancer, ESR-negative breast cancer, triple-negative breast cancer, AR-negative prostate, hepatocellular carcinoma | [290,292,298,299,300,301] | |
| Protein level | Prostate cancer | [302] | ||
| mRNA and protein levels | Colon adenocarcinoma, hepatocellular carcinoma | [303,304] | ||
| ACSL5 | Downregulated | mRNA level | Breast cancer | [290] |
| mRNA and protein levels | Small intestine cancer | [305] | ||
| Upregulated | mRNA level | Bladder cancer, colorectal cancer | [290,306,307] | |
| ACSL6 | Downregulated | mRNA level | Leukemia | [290] |
| Upregulated | mRNA level | Colorectal cancer | [290,308] | |
| CPT1A | Upregulated | Protein level | Gastric cancer | [309] |
| mRNA level | Glioblastoma multiforme | [289] | ||
| CPT1B | Upregulated | mRNA and protein levels | Prostate cancer | [310] |
| mRNA level | High-grade bladder cancer | [311] | ||
| CPT1C | Upregulated | mRNA level | Gastric cancer, lung cancer, papillary thyroid carcinoma | [312,313,314] |
| CPT2 | Downregulated | mRNA level | Hepatocellular carcinoma, colorectal cancer, ovarian cancer | [308,315,316] |
| Upregulated | mRNA level | Glioblastoma multiforme | [289] | |
| ECH1 | Downregulated | mRNA level | Colorectal cancer | [317] |
| EHHADH | Downregulated | mRNA and protein levels | Hepatocellular carcinoma | [318] |
| Upregulated | mRNA level | Osteosarcoma | [319] | |
| FABP3 | Upregulated | mRNA and protein levels | Non-small-cell lung cancer | [320] |
| FABP4 | Downregulated | mRNA level | Stomach adenocarcinoma | [321] |
| mRNA and protein levels | Hepatocellular carcinoma | [322] | ||
| Upregulated | Protein level | High-grade serous ovarian carcinoma, pancreatic ductal adenocarcinoma, gastric adenocarcinoma | [323,324,325] | |
| mRNA and protein levels | Non-small-cell lung cancer, prostate cancer | [320,326] | ||
| FABP5 | Upregulated | Protein level | Gastric adenocarcinoma | [325] |
| HADH | Downregulated | Protein level | Gastric cancer | [327] |
| mRNA level | Gastric cancer, kidney renal clear cell carcinoma | [328,329,330] | ||
| Upregulated | mRNA level | Colon cancer, acute myeloid leukemia | [331,332] | |
| HADHA | Downregulated | mRNA level | Breast cancer | [333] |
| LCAD | Downregulated | mRNA level | Hepatocellular carcinoma | [334] |
| MCAD | Upregulated | Protein level | Glioblastoma, squamous cell carcinoma of the head and neck | [335,336] |
| SCAD | Downregulated | mRNA level | Colorectal cancer | [317] |
| Targeted Enzyme | Inhibitor/Interfering Compound | Experimental Models | Effects | References |
|---|---|---|---|---|
| ACSL4 | Rosiglitazone | Breast cancer cell lines | Inhibition of cancer cell growth | [356] |
| PRGL493 | Breast cancer cell lines, prostate cancer cell lines | Inhibition of cancer cell growth and sensitization to chemotherapy | [357] | |
| ACSL5 | Triacsin C | Glioma cell lines | Inhibition of cancer cell survival | [358] |
| Small interfering RNA | Lung cancer cell lines | Inhibition of cancer cell growth | [359] | |
| ACSVL3 | Small interfering RNA | Glioblastoma cell lines | Inhibition of cancer cell growth and tumourigenicity | [360] |
| CPT1 | Avocatin B | Primary myeloid leukemia cells | Inhibition of cancer cell survival | [361] |
| Etomoxir | Leukemia, breast, prostate, colorectal cancer cell lines, and the xenograft model | Inhibition of cancer cell growth, survival, and tumourigenicity | [288,362,363,364,365] | |
| Lung cell lines | Sensitization to radiation | [366] | ||
| Oxfenicine | Melanoma cell lines | Inhibition of cancer cell growth | [367] | |
| Small interfering RNAs | Brest cancer cell lines | Inhibition of cancer cell survival | [368] | |
| CPT2 | Aminocarnitine | Glioma cell lines | Inhibition of cancer cell growth | [369] |
| Perhexiline | Gastrointestinal cancer cell lines | Inhibition of cancer cell survival and sensitization to chemotherapy | [370] | |
| ECHS1 | Small interfering RNA, PP2 | Breast cancer cell lines | Inhibition of cancer cell survival | [371] |
| MCAD | Hairpin RNA interference | Glioblastoma cell lines | Inhibition of cancer cell survival | [335] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szrok-Jurga, S.; Czumaj, A.; Turyn, J.; Hebanowska, A.; Swierczynski, J.; Sledzinski, T.; Stelmanska, E. The Physiological and Pathological Role of Acyl-CoA Oxidation. Int. J. Mol. Sci. 2023, 24, 14857. https://doi.org/10.3390/ijms241914857
Szrok-Jurga S, Czumaj A, Turyn J, Hebanowska A, Swierczynski J, Sledzinski T, Stelmanska E. The Physiological and Pathological Role of Acyl-CoA Oxidation. International Journal of Molecular Sciences. 2023; 24(19):14857. https://doi.org/10.3390/ijms241914857
Chicago/Turabian StyleSzrok-Jurga, Sylwia, Aleksandra Czumaj, Jacek Turyn, Areta Hebanowska, Julian Swierczynski, Tomasz Sledzinski, and Ewa Stelmanska. 2023. "The Physiological and Pathological Role of Acyl-CoA Oxidation" International Journal of Molecular Sciences 24, no. 19: 14857. https://doi.org/10.3390/ijms241914857