


Treatment of Alzheimer’s Disease: Beyond Symptomatic Therapies
 , ,
, ,
Abstract
:1. Introduction
2. Immunotherapy Overview
3. Failure of Many Clinical Trials: Why?
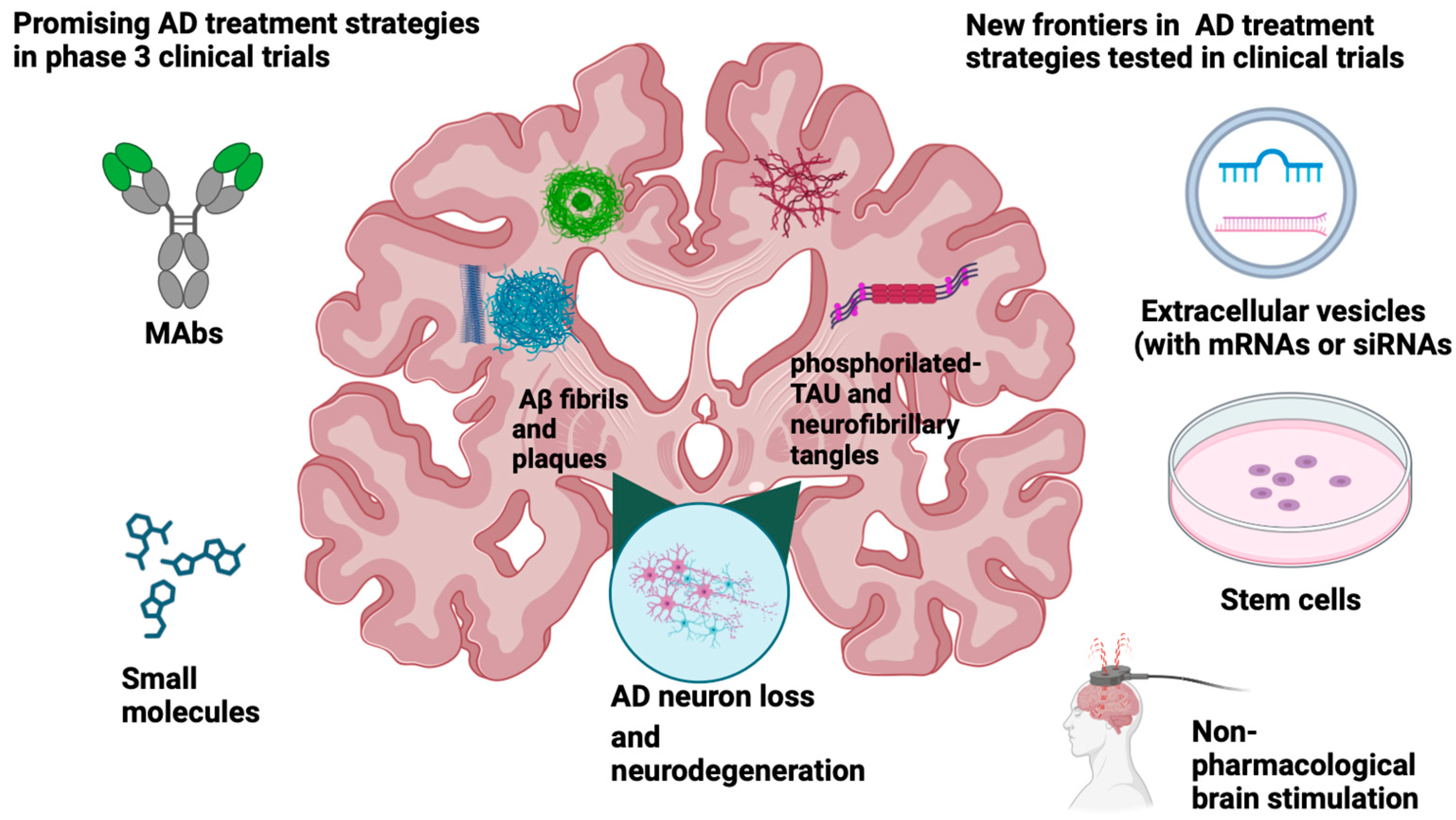
4. State of the Art of AD Disease-Modifying Treatments (DMT)s
4.1. mAbs Anti-Aβ and Anti-Tau
4.1.1. Aducanumab
4.1.2. Donanemab
4.1.3. Gantenerumab
4.1.4. Lecanemab
4.1.5. Remternetug
4.1.6. Solanezumab
4.1.7. E2814
4.1.8. Semaglutide
4.1.9. Tertomotide
4.2. Small Molecules
4.2.1. Hydralazine
4.2.2. Omega 3
4.2.3. Metformin
4.2.4. AGB101
4.2.5. Anavex2-73
4.2.6. Fosfogonimenton
4.2.7. Masitinib
4.2.8. NE3107
4.2.9. Nilotinib
4.2.10. Melatonin
4.2.11. Simufilam
4.2.12. ALZ-801
4.2.13. Hydromethylthionine Mesylate
5. Other Therapeutic Approaches: Stem Cells
6. Non-Pharmacological Interventions
7. Future of AD Diagnosis and Treatment
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prince, M.J. S3-01-01: Prevention report from Alzheimer’s disease international (ADI). Alzheimer’s Dement. 2015, 11, 210. [Google Scholar] [CrossRef]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2023. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2023, 9, e12385. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Fontana, I.C.; Zimmer, A.R.; Rocha, A.S.; Gosmann, G.; Souza, D.O.; Lourenco, M.V.; Ferreira, S.T.; Zimmer, E.R. Amyloid-β oligomers in cellular models of Alzheimer’s disease. J. Neurochem. 2020, 155, 348–369. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef]
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-β Receptors: The Good, the Bad, and the Prion Protein. J. Biol. Chem. 2016, 291, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef]
- O’Nuallain, B.; Freir, D.B.; Nicoll, A.J.; Risse, E.; Ferguson, N.; Herron, C.E.; Collinge, J.; Walsh, D.M. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J. Neurosci. 2010, 30, 14411–14419. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Borchelt, D.R.; Giasson, B.I.; Lewis, J. Thinking laterally about neurodegenerative proteinopathies. J. Clin. Investig. 2013, 123, 1847–1855. [Google Scholar] [CrossRef]
- Ramirez-Alvarado, M. Amyloid formation in light chain amyloidosis. Curr. Top. Med. Chem. 2012, 12, 2523–2533. [Google Scholar] [CrossRef] [PubMed]
- Limbocker, R.; Cremades, N.; Cascella, R.; Tessier, P.M.; Vendruscolo, M.; Chiti, F. Characterization of Pairs of Toxic and Nontoxic Misfolded Protein Oligomers Elucidates the Structural Determinants of Oligomer Toxicity in Protein Misfolding Diseases. Acc. Chem. Res. 2023, 56, 1395–1405. [Google Scholar] [CrossRef]
- Hipp, M.S.; Park, S.H.; Hartl, U.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabel, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Limbocker, R.; Errico, S.; Barbut, D.; Knowles, T.P.J.; Vendruscolo, M.; Chiti, F.; Zasloff, M. Squalamine and trodusquemine: Two natural products for neurodegenerative diseases, from physical chemistry to the clinic. Nat. Prod. Rep. 2022, 39, 742–753. [Google Scholar] [CrossRef]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Shao, E.; Mucke, L. Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 2021, 371, eabb8255. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Swift, I.J.; Sogorb-Esteve, A.; Heller, C.; Synofzik, M.; Otto, M.; Graff, C.; Galimberti, D.; Todd, E.; Heslegrave, A.J.; Van Der Ende, E.L.; et al. Fluid biomarkers in frontotemporal dementia: Past, present and future. J. Neurol. Neurosurg. Psychiatry 2021, 92, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Götz, J.; Chen, F.; Van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Wang, L.; Benzinger, T.L.; Su, Y.; Christensen, J.; Friedrichsen, K.; Aldea, P.; McConathy, J.; Cairns, N.J.; Fagan, A.M.; Morris, J.C.; et al. Evaluation of Tau Imaging in Staging Alzheimer Disease and Revealing Interactions between β-Amyloid and Tauopathy. JAMA Neurol. 2016, 73, 1070–1077. [Google Scholar] [CrossRef]
- Horie, K.; Barthelemy, N.R.; Sato, C.; Bateman, R.J. CSF tau microtubule binding region identifies tau tangle and clinical stages of Alzheimer’s disease. Brain 2021, 144, 515–527. [Google Scholar] [CrossRef]
- Hampel, H.; Blennow, K.; Shaw, L.M.; Hoessler, Y.C.; Zetterberg, H.; Trojanowski, J.Q. Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp. Gerontol. 2010, 45, 30–40. [Google Scholar] [CrossRef] [PubMed]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Golde, T.E. Disease modifying therapy for AD? J. Neurochem. 2006, 99, 689–707. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guldo, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef]
- Golde, T.E. Disease-Modifying Therapies for Alzheimer’s Disease: More Questions than Answers. Neurotherapeutics 2022, 19, 209–227. [Google Scholar] [CrossRef]
- Morgan, D. Immunotherapy for Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 425–432. [Google Scholar] [CrossRef]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Boada Rovira, M.; Forette, F.; et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Vellas, B.; Black, R.; Thal, L.; Fox, N.; Daniels, M.; McLennan, G.; Tompkins, C.; Leibman, C.; Pomfret, M.; Grundman, M. Long-term follow-up of patients immunized with AN1792: Reduced functional decline in antibody responders. Curr. Alzheimer Res. 2009, 6, 144–151. [Google Scholar] [CrossRef]
- Van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry. 2018, 83, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.S.; Cashman, N.R. Passive immunotherapies targeting Aβ and tau in Alzheimer’s disease. Neurobiol. Dis. 2020, 144, 105010. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; Van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Doggrell, S.A. Lessons that can be learnt from the failure of verubecestat in Alzheimer’s disease. Expert Opin. Pharmacother. 2019, 20, 2095–2099. [Google Scholar] [CrossRef]
- Hawkes, N. Pfizer abandons research into Alzheimer’s and Parkinson’s diseases. BMJ 2018, 360, k122. [Google Scholar] [CrossRef]
- Kim, C.K.; Lee, Y.R.; Ong, L.; Gold, M.; Kalali, A.; Sarkar, J. Alzheimer’s Disease: Key Insights from Two Decades of Clinical Trial Failures. J. Alzheimer’s Dis. 2022, 87, 83–100. [Google Scholar] [CrossRef]
- Cummings, J.; Rabinovici, G.D.; Atri, A.; Aisen, P.; Apostolova, L.G.; Hendrix, S.; Sabbagh, M.; Selkoe, D.; Weiner, M.; Salloway, S. Aducanumab: Appropriate Use Recommendations Update. J. Prev. Alzheimer’s Dis. 2022, 9, 221–230. [Google Scholar] [CrossRef]
- Canevelli, M.; Rossi, P.D.; Astrone, P.; Consorti, E.; Vanacore, N.; Cesari, M. “Real world” eligibility for aducanumab. J. Am. Geriatr. Soc. 2021, 69, 2995–2998. [Google Scholar] [CrossRef]
- European Medicines Agency. Clinical Investigation of Medicines for the Treatment of Alzheimer’s Disease—Scientific Guideline. Available online: https://www.ema.europa.eu/en/clinical-investigation-medicines-treatment-alzheimers-disease-scientific-guideline (accessed on 8 August 2023).
- FDA. Alzheimer’s Disease: Developing Drugs for Treatment Guidance for Industry; FDA: Silver Spring, MD, USA, 2018. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/alzheimers-disease-developing-drugs-treatment-guidance-industy (accessed on 8 August 2023).
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Mattsson-Carlgren, N.; Leuzy, A.; Janelidze, S.; Palmqvist, S.; Stomrud, E.; Strandberg, O.; Smith, R.; Hansson, O. The implications of different approaches to define AT(N) in Alzheimer disease. Neurology 2020, 94, E2233–E2244. [Google Scholar] [CrossRef]
- Hansson, O.; Blennow, K.; Zetterberg, H.; Dage, J. Blood biomarkers for Alzheimer’s disease in clinical practice and trials. Nat. Aging 2023, 3, 506–519. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Verberk, I.M.W.; Thijssen, E.H.; Vermunt, L.; Hansson, O.; Zetterberg, H.; van der Flier, W.M.; Mielke, M.M.; del Campo, M. Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; van der Kant, R.; Hansson, O. Tau biomarkers in Alzheimer’s disease: Towards implementation in clinical practice and trials. Lancet Neurol. 2022, 21, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, J.M.; Noviasky, J.; Britton, S. Aducanumab for Alzheimer’s Disease: Summarized Data From EMERGE, ENGAGE, and PRIME Studies. Sr. care Pharm. 2022, 37, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients with Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Vaz, M.; Silva, V.; Monteiro, C.; Silvestre, S. Role of Aducanumab in the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Clin. Interv. Aging 2022, 17, 797–810. [Google Scholar] [CrossRef]
- Biogen. Biogen and Eisai to Discontinue Phase 3 ENGAGE and EMERGE Trials of Aducanumab in Alzheimer’s Disease. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-and-eisai-discontinue-phase-3-engage-and-emerge-trials (accessed on 7 August 2023).
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef]
- Haeberlein, S.B.; Von Hehn, C.; Tian, Y.; Chalkias, S.; Muralidharan, K.K.; Chen, T.; Wu, S.; Li, J.; Skordos, L.; Nisenbaum, L.; et al. EMERGE and ENGAGE Topline Results: Two Phase 3 Studies to Evaluate Aducanumab in Patients with Early Alzheimer’s Disease. Alzheimer’s Dement. 2020, 16, e047259. [Google Scholar] [CrossRef]
- FDA. FDA Grants Accelerated Approval for Alzheimer’s Drug. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 7 August 2023).
- Sperling, R.A.; Jack, C.R.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dement. 2011, 7, 367–385. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; DeLong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.M.; Siemers, E.R.; Yu, P.; Andersen, S.W.; Holdridge, K.C.; Sims, J.R.; Sundell, K.; Stern, Y.; Rentz, D.M.; Dubois, B.; et al. A Combined Measure of Cognition and Function for Clinical Trials: The Integrated Alzheimer’s Disease Rating Scale (iADRS). J. Prev. Alzheimer’s Dis. 2015, 2, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, D.; Feligioni, M.; Scaccianoce, S.; Caruso, A.; Piccinin, S.; Schepisi, C.; Errico, F.; Mercuri, N.B.; Nicoletti, F.; Nisticò, R. Profile of gantenerumab and its potential in the treatment of Alzheimer’s disease. Drug Des. Devel. Ther. 2013, 7, 1359–1364. [Google Scholar] [CrossRef]
- Bateman, R.J.; Cummings, J.; Schobel, S.; Salloway, S.; Vellas, B.; Boada, M.; Black, S.E.; Blennow, K.; Fontoura, P.; Klein, G.; et al. Gantenerumab: An anti-amyloid monoclonal antibody with potential disease-modifying effects in early Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 178. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Deptula, D.; Thurfjell, L.; Barkhof, F.; Bohrmann, B.; Brooks, D.J.; Klunk, W.E.; Ashford, E.; Yoo, K.; Xu, Z.X.; et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch. Neurol. 2012, 69, 198–207. [Google Scholar] [CrossRef]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimer’s Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef]
- [Ad Hoc Announcement Pursuant to Art. 53 LR] Roche Provides Update on Phase III GRADUATE Programme Evaluating Gantenerumab in Early Alzheimer’s Disease. Available online: https://www.globenewswire.com/news-release/2022/11/14/2554515/0/en/Ad-hoc-announcement-pursuant-to-Art-53-LR-Roche-provides-update-on-Phase-III-GRADUATE-programme-evaluating-gantenerumab-in-early-Alzheimer-s-disease.html (accessed on 7 August 2023).
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Roberts, M.; Sevastou, I.; Imaizumi, Y.; Mistry, K.; Talma, S.; Dey, M.; Gartlon, J.; Ochiai, H.; Zhou, Z.; Akasofu, S.; et al. Pre-clinical characterisation of E2814, a high-affinity antibody targeting the microtubule-binding repeat domain of tau for passive immunotherapy in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 13. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 142–143. [Google Scholar] [CrossRef] [PubMed]
- A Study of Remternetug (LY3372993) in Participants with Alzheimer’s Disease (TRAILRUNNER-ALZ 1). Available online: https://trials.lilly.com/en-US/trial/351135 (accessed on 7 August 2023).
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Sperling, R.A.; Donohue, M.C.; Raman, R.; Rafii, M.S.; Johnson, K.; Masters, C.L.; van Dyck, C.H.; Iwatsubo, T.; Marshall, G.A.; Yaari, R.; et al. Trial of Solanezumab in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Younes, K.; Sha, S.J. The most valuable player or the tombstone: Is tau the correct target to treat Alzheimer’s disease? Brain 2023, 146, 2211–2213. [Google Scholar] [CrossRef]
- Buccellato, F.R.; D’Anca, M.; Galimberti, D.; Fenoglio, C.; Scarpini, E. Role of Oxidative Damage in Alzheimer’s Disease and Neurodegeneration: From Pathogenic Mechanisms to Biomarker Discovery. Antioxidants 2021, 10, 1353. [Google Scholar] [CrossRef]
- Neth, B.J.; Craft, S. Insulin Resistance and Alzheimer’s Disease: Bioenergetic Linkages. Front. Aging Neurosci. 2017, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.C.; Xing, X.; Wang, C.; Ma, D.; Nho, K.; Crane, P.K.; Elahi, F.; Ziegler, D.A.; Liang, G.; Cheng, Q.; et al. β-amyloid and tau drive early Alzheimer’s disease decline while glucose hypometabolism drives late decline. Commun. Biol. 2020, 3, 352. [Google Scholar] [CrossRef]
- Perry, T.; Haughey, N.J.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J. Pharmacol. Exp. Ther. 2002, 302, 881–888. [Google Scholar] [CrossRef] [PubMed]
- During, M.J.; Cao, L.; Zuzga, D.S.; Francis, J.S.; Fitzsimons, H.L.; Jiao, X.; Bland, R.J.; Klugmann, M.; Banks, W.A.; Drucker, D.J.; et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat. Med. 2003, 9, 1173–1179. [Google Scholar] [CrossRef]
- Gejl, M.; Brock, B.; Egefjord, L.; Vang, K.; Rungby, J.; Gjedde, A. Blood-Brain Glucose Transfer in Alzheimer’s disease: Effect of GLP-1 Analog Treatment. Sci. Rep. 2017, 7, 17490. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Schwartzentruber, D.J.; Hwu, P.; Marincola, F.M.; Topalian, S.L.; Restifo, N.P.; Dudley, M.E.; Schwarz, S.L.; Spiess, P.J.; et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998, 4, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Chiodi, I.; Mondello, C. Telomere-independent functions of telomerase in nuclei, cytoplasm, and mitochondria. Front. Oncol. 2012, 2, 133. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 2011, 11, 161–176. [Google Scholar] [CrossRef]
- Park, H.H.; Lee, K.Y.; Kim, S.; Lee, J.W.; Choi, N.Y.; Lee, E.H.; Lee, Y.J.; Lee, S.H.; Koh, S.H. Novel vaccine peptide GV1001 effectively blocks β-amyloid toxicity by mimicking the extra-telomeric functions of human telomerase reverse transcriptase. Neurobiol. Aging 2014, 35, 1255–1274. [Google Scholar] [CrossRef]
- Park, H.H.; Yu, H.J.; Kim, S.; Kim, G.; Choi, N.Y.; Lee, E.H.; Lee, Y.J.; Yoon, M.Y.; Lee, K.Y.; Koh, S.H. Neural stem cells injured by oxidative stress can be rejuvenated by GV1001, a novel peptide, through scavenging free radicals and enhancing survival signals. Neurotoxicology 2016, 55, 131–141. [Google Scholar] [CrossRef]
- Koh, S.H.; Kwon, H.S.; Choi, S.H.; Jeong, J.H.; Na, H.R.; Lee, C.N.; Yang, Y.S.; Lee, A.Y.; Lee, J.H.; Park, K.W.; et al. Efficacy and safety of GV1001 in patients with moderate-to-severe Alzheimer’s disease already receiving donepezil: A phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. Alzheimer’s Res. Ther. 2021, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, A.; Sun, H.; Han, Y.; Kong, L.; Wang, X. Two decades of new drug discovery and development for Alzheimer’s disease. RSC Adv. 2017, 7, 6046–6058. [Google Scholar] [CrossRef]
- Holmes, C. Review: Systemic inflammation and Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2013, 39, 51–68. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.; Lee, H.-g.; Perry, G.; Smith, M.; Zhu, X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 525–532. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Craig, L.A.; Hong, N.S.; McDonald, R.J. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2011, 35, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Wieckowska, A.; Panek, D.; Malawska, B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef] [PubMed]
- García-Morales, V.; González-Acedo, A.; Melguizo-Rodríguez, L.; Pardo-Moreno, T.; Costela-Ruiz, V.J.; Montiel-Troya, M.; Ramos-Rodríguez, J.J. Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease. Biomedicines 2021, 9, 1910. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, E.; Zhang, Y.; Saremi, B.; Yadavali, S.; Hakimi, A.; Dehghani, M.; Goodarzi, M.; Tu, X.; Robertson, S.; Lin, R.; et al. Hydralazine induces stress resistance and extends C. elegans lifespan by activating the NRF2/SKN-1 signalling pathway. Nat. Commun. 2017, 8, 2223. [Google Scholar] [CrossRef]
- Barnes, S.; Chowdhury, S.; Gatto, N.M.; Fraser, G.E.; Lee, G.J. Omega-3 fatty acids are associated with blood-brain barrier integrity in a healthy aging population. Brain Behav. 2021, 11, e2273. [Google Scholar] [CrossRef]
- Amen, D.G.; Harris, W.S.; Kidd, P.M.; Meysami, S.; Raji, C.A. Quantitative Erythrocyte Omega-3 EPA Plus DHA Levels are Related to Higher Regional Cerebral Blood Flow on Brain SPECT. J. Alzheimer’s Dis. 2017, 58, 1189–1199. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J. Alzheimer’s Dis. 2016, 51, 501–514. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Perry, G.; Zhu, X.; IMoreira, P.I.; Smith, M.A. Insulin-resistant brain state: The culprit in sporadic Alzheimer’s disease? Ageing Res. Rev. 2011, 10, 264–273. [Google Scholar] [CrossRef]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef]
- Ferrendelli, J.A.; French, J.; Leppik, I.; Morrell, M.J.; Herbeuval, A.; Han, J.; Magnus, L. Use of levetiracetam in a population of patients aged 65 years and older: A subset analysis of the KEEPER trial. Epilepsy Behav. 2003, 4, 702–709. [Google Scholar] [CrossRef]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Akinola, M.; Tai, X.Y.; Symmonds, M.; Davis Jones, G.; Mura, S.; Galloway, J.; Hallam, A.; Chan, J.Y.C.; Koychev, I.; et al. An Investigation of Levetiracetam in Alzheimer’s Disease (ILiAD): A double-blind, placebo-controlled, randomised crossover proof of concept study. Trials 2021, 22, 508. [Google Scholar] [CrossRef]
- Shi, M.; Chen, F.; Chen, Z.; Yang, W.; Yue, S.; Zhang, J.; Chen, X. Sigma-1 Receptor: A Potential Therapeutic Target for Traumatic Brain Injury. Front. Cell. Neurosci. 2021, 15, 685201. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Kimura, Y.; Tsukada, H.; Kobayashi, T.; Nishiyama, S.; Kakiuchi, T.; Ohba, H.; Harada, N.; Matsuno, K.; Ishii, K.; et al. An increase of sigma1 receptors in the aged monkey brain. Neurobiol. Aging 2003, 24, 745–752. [Google Scholar] [CrossRef]
- Ishiwata, K.; Kobayashi, T.; Kawamura, K.; Matsuno, K. Age-related changes of the binding of [3h]SA4503 to sigma1 receptors in the rat brain. Ann. Nucl. Med. 2003, 17, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Horsager, J.; Fedorova, T.D.; Berge, N.V.D.; Klinge, M.W.; Knudsen, K.; Hansen, A.K.; Alstrup, A.K.O.; Krogh, K.; Gormsen, L.; Borghammer, P. Cardiac 11C-Donepezil Binding Increases with Age in Healthy Humans: Potentially Signifying Sigma-1 Receptor Upregulation. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 365–370. [Google Scholar] [CrossRef]
- Mishina, M.; Ohyama, M.; Ishii, K.; Kitamura, S.; Kimura, Y.; Oda, K.I.; Kawamura, K.; Sasaki, T.; Kobayashi, S.; Katayama, Y.; et al. Low density of sigma1 receptors in early Alzheimer’s disease. Ann. Nucl. Med. 2008, 22, 151–156. [Google Scholar] [CrossRef]
- Minoshima, S.; Giordani, B.; Berent, S.; Frey, K.A.; Foster, N.L.; Kuhl, D.E. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 1997, 42, 85–94. [Google Scholar] [CrossRef]
- Tsai, S.Y.A.; Pokrass, M.J.; Klauer, N.R.; Nohara, H.; Su, T.P. Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc. Natl. Acad. Sci. USA 2015, 112, 6742–6747. [Google Scholar] [CrossRef] [PubMed]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (σ1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. 2011, 25, 1101–1117. [Google Scholar] [CrossRef]
- Funakoshi, H.; Nakamura, T. Hepatocyte Growth Factor (HGF): Neurotrophic Functions and Therapeutic Implications for Neuronal Injury/Diseases. Curr. Signal Transduct. Ther. 2012, 6, 156–167. [Google Scholar] [CrossRef]
- Koike, H.; Ishida, A.; Shimamura, M.; Mizuno, S.; Nakamura, T.; Ogihara, T.; Kaneda, Y.; Morishita, R. Prevention of onset of Parkinson’s disease by in vivo gene transfer of human hepatocyte growth factor in rodent model: A model of gene therapy for Parkinson’s disease. Gene Ther. 2006, 13, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, D.; Sato, N.; Shimamura, M.; Kurinami, H.; Takeda, S.; Shinohara, M.; Suzuki, S.; Kojima, M.; Ogihara, T.; Morishita, R. Alleviation of Abeta-induced cognitive impairment by ultrasound-mediated gene transfer of HGF in a mouse model. Gene Ther. 2008, 15, 561–571. [Google Scholar] [CrossRef]
- Hamasaki, H.; Honda, H.; Suzuki, S.O.; Hokama, M.; Kiyohara, Y.; Nakabeppu, Y.; Iwaki, T. Down-regulation of MET in hippocampal neurons of Alzheimer’s disease brains. Neuropathology 2014, 34, 284–290. [Google Scholar] [CrossRef]
- Liu, H.; Luo, K.; Luo, D. Guanosine monophosphate reductase 1 is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 2018, 8, 2759. [Google Scholar] [CrossRef]
- Johnston, J.L.; Reda, S.M.; Setti, S.E.; Taylor, R.W.; Berthiaume, A.A.; Walker, W.E.; Wu, W.; Moebius, H.J.; Church, K.J. Fosgonimeton, a Novel Positive Modulator of the HGF/MET System, Promotes Neurotrophic and Procognitive Effects in Models of Dementia. Neurotherapeutics 2023, 20, 431–451. [Google Scholar] [CrossRef]
- Li, J.-W.; Zong, Y.; Cao, X.-P.; Tan, L.; Tan, L. Microglial priming in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 176. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef]
- Vermersch, P.; Brieva-Ruiz, L.; Fox, R.J.; Paul, F.; Ramio-Torrenta, L.; Schwab, M.; Moussy, A.; Mansfield, C.; Hermine, O.; Maciejowski, M. Efficacy and Safety of Masitinib in Progressive Forms of Multiple Sclerosis: A Randomized, Phase 3, Clinical Trial. Neurol. Neuroimmunol. Neuroinflammation 2022, 9, 1148. [Google Scholar] [CrossRef]
- Li, T.; Martin, E.; Abada, Y.S.; Boucher, C.; Cès, A.; Youssef, I.; Fenaux, G.; Forand, Y.; Legrand, A.; Nachiket, N.; et al. Effects of Chronic Masitinib Treatment in APPswe/PSEN1dE9 Transgenic Mice Modeling Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 76, 1339–1345. [Google Scholar] [CrossRef]
- Dubois, B.; López-Arrieta, J.; Lipschitz, S.; Triantafyllos, D.; Spiru, L.; Moroz, S.; Venger, O.; Vermersch, P.; Moussy, A.; Mansfield, C.D.; et al. Masitinib for mild-to-moderate Alzheimer’s disease: Results from a randomized, placebo-controlled, phase 3, clinical trial. Alzheimer’s Res. Ther. 2023, 15. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Chen, M.J.; Ramesha, S.; Weinstock, L.D.; Gao, T.; Ping, L.; Xiao, H.; Dammer, E.B.; Duong, D.D.; Levey, A.I.; Lah, J.J.; et al. Extracellular signal-regulated kinase regulates microglial immune responses in Alzheimer’s disease. J. Neurosci. Res. 2021, 99, 1704–1721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guo, S.; Zhang, X.; Tang, S.; Wang, L.; Han, X.; Shao, W.; Cong, L.; Du, Y. Amyloid β oligomer-induced ERK1/2-dependent serine 636/639 phosphorylation of insulin receptor substrate-1 impairs insulin signaling and glycogen storage in human astrocytes. Gene 2015, 561, 76–81. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef]
- Reading, C.L.; Frincke, J.M.; White, S.K. Molecular targets for 17α-ethynyl-5-androstene-3β,7β,17β-triol, an anti-inflammatory agent derived from the human metabolome. PLoS ONE 2012, 7, e32147. [Google Scholar] [CrossRef]
- Lambert, W.S.; Carlson, B.J.; Formichella, C.R.; Sappington, R.M.; Ahlem, C.; Calkins, D.J. Oral Delivery of a Synthetic Sterol Reduces Axonopathy and Inflammation in a Rodent Model of Glaucoma. Front. Neurosci. 2017, 11, 45. [Google Scholar] [CrossRef]
- Khan, R.S.; Dine, K.; Luna, E.; Ahlem, C.; Shindler, K.S. HE3286 reduces axonal loss and preserves retinal ganglion cell function in experimental optic neuritis. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5744–5751. [Google Scholar] [CrossRef] [PubMed]
- BioVie Announces Positive Results for NE3107 in Parkinson’s. Available online: https://www.globenewswire.com/news-release/2022/12/05/2567854/0/en/BioVie-Announces-Positive-Results-for-NE3107-in-Parkinson-s-and-Alzheimer-s-Phase-2-Trials.html (accessed on 7 August 2023).
- Reading, C.L.; Ahlem, C.N.; Murphy, M.F. NM101 Phase III study of NE3107 in Alzheimer’s disease: Rationale, design and therapeutic modulation of neuroinflammation and insulin resistance. Neurodegener. Dis. Manag. 2021, 11, 289–298. [Google Scholar] [CrossRef]
- BioVie Presents Data Highlighting Role of Insulin. Available online: https://www.globenewswire.com/news-release/2023/06/26/2694368/0/en/BioVie-Presents-Data-Highlighting-Role-of-Insulin-Resistance-and-Neuroinflammation-in-the-Development-of-Mild-to-Moderate-in-Alzheimer-s-Disease.html (accessed on 7 August 2023).
- Lim, J.; Yue, Z. Neuronal aggregates: Formation, clearance, and spreading. Dev. Cell 2015, 32, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef]
- Sarkar, S.; Ravikumar, B.; Rubinsztein, D.C. Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzymol. 2009, 453, 83–110. [Google Scholar] [CrossRef]
- Cai, Z.; Zhou, Y.; Liu, Z.; Ke, Z.; Zhao, B. Autophagy dysfunction upregulates beta-amyloid peptides via enhancing the activity of γ-secretase complex. Neuropsychiatr. Dis. Treat. 2015, 11, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- DeRemer, D.L.; Ustun, C.; Natarajan, K. Nilotinib: A second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin. Ther. 2008, 30, 1956–1975. [Google Scholar] [CrossRef]
- La Barbera, L.; Vedele, F.; Nobili, A.; Krashia, P.; Spoleti, E.; Latagliata, E.C.; Cutuli, D.; Cauzzi, E.; Marino, R.; Viscomi, M.T.; et al. Nilotinib restores memory function by preventing dopaminergic neuron degeneration in a mouse model of Alzheimer’s Disease. Prog. Neurobiol. 2021, 202, 102031. [Google Scholar] [CrossRef] [PubMed]
- Nobili, A.; La Barbera, L.; D’Amelio, M. Targeting autophagy as a therapeutic strategy to prevent dopamine neuron loss in early stages of Alzheimer disease. Autophagy 2021, 17, 1278–1280. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Odero, G.G.; Glazner, G.; Turner, R.S.; Albensi, B.C. Nilotinib Improves Bioenergetic Profiling in Brain Astroglia in the 3xTg Mouse Model of Alzheimer’s Disease. Aging Dis. 2021, 12, 441–465. [Google Scholar] [CrossRef]
- Turner, R.S.; Hebron, M.L.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, J.N.; Anjum, M.; Pagan, F.; Torres-Yaghi, Y.; Shi, W.; et al. Nilotinib Effects on Safety, Tolerability, and Biomarkers in Alzheimer’s Disease. Ann. Neurol. 2020, 88, 183–194. [Google Scholar] [CrossRef]
- Liu, R.Y.; Zhou, J.N.; Van Heerikhuize, J.; Hofman, M.A.; Swaab, D.F. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-ε4/4 genotype. J. Clin. Endocrinol. Metab. 1999, 84, 323–327. [Google Scholar] [CrossRef]
- Ramírez-Rodríguez, G.; Vega-Rivera, N.M.; Benítez-King, G.; Castro-García, M.; Ortíz-López, L. Melatonin supplementation delays the decline of adult hippocampal neurogenesis during normal aging of mice. Neurosci. Lett. 2012, 530, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.A.; Sos, M.; Omar, R.A.; Bick, R.J.; Hickson-Bick, D.L.M.; Reiter, R.J.; Efthimiopoulos, S.; Robakis, N.K. Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J. Neurosci. 1997, 17, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, E.; Bryant-Thomas, T.; Quinto, J.P.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.J.; Smith, M.A.; Perry, G.; et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar] [CrossRef]
- He, P.; Ouyang, X.; Zhou, S.; Yin, W.; Tang, C.; Laudon, M.; Tian, S. A novel melatonin agonist Neu-P11 facilitates memory performance and improves cognitive impairment in a rat model of Alzheimer’ disease. Horm. Behav. 2013, 64, 1–7. [Google Scholar] [CrossRef]
- Benca, R.M.; Teodorescu, M. Sleep physiology and disorders in aging and dementia. Handb. Clin. Neurol. 2019, 167, 477–493. [Google Scholar] [CrossRef]
- Schneider, L.S.; Laudon, M.; Nir, T.; Caceres, J.; Ianniciello, G.; Capulli, M.; Zisapel, N. A Polymorphism Cluster at the 2q12 locus May Predict Response to Piromelatine in Patients with Mild Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 247–254. [Google Scholar] [CrossRef]
- Wang, H.Y.; Lee, D.H.S.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Li, W.; Benedetti, N.J.; Lee, D.H.S. Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J. Biol. Chem. 2003, 278, 31547–31553. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Bakshi, K.; Frankfurt, M.; Stucky, A.; Goberdhan, M.; Shah, S.M.; Burns, L.H. Reducing amyloid-related Alzheimer’s disease pathogenesis by a small molecule targeting filamin A. J. Neurosci. 2012, 32, 9773–9784. [Google Scholar] [CrossRef]
- Wang, H.Y.; Lee, K.C.; Pei, Z.; Khan, A.; Bakshi, K.; Burns, L.H. PTI-125 binds and reverses an altered conformation of filamin A to reduce Alzheimer’s disease pathogenesis. Neurobiol. Aging 2017, 55, 99–114. [Google Scholar] [CrossRef]
- Burns, L.H.; Pei, Z.; Wang, H.Y. Targeting α7 nicotinic acetylcholine receptors and their protein interactions in Alzheimer’s disease drug development. Drug Dev. Res. 2023, early view. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimer’s Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Hey, J.A.; Kocis, P.; Hort, J.; Abushakra, S.; Power, A.; Vyhnálek, M.; Yu, J.Y.; Tolar, M. Discovery and Identification of an Endogenous Metabolite of Tramiprosate and Its Prodrug ALZ-801 that Inhibits Beta Amyloid Oligomer Formation in the Human Brain. CNS Drugs 2018, 32, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Abushakra, S.; Porsteinsson, A.; Vellas, B.; Cummings, J.; Gauthier, S.; Hey, J.A.; Power, A.; Hendrix, S.; Wang, P.; Shen, L.; et al. Clinical Benefits of Tramiprosate in Alzheimer’s Disease Are Associated with Higher Number of APOE4 Alleles: The “APOE4 Gene-Dose Effect”. J. Prev. Alzheimer’s Dis. 2016, 3, 219–228. [Google Scholar] [CrossRef]
- Abushakra, S.; Porsteinsson, A.; Scheltens, P.; Sadowsky, C.; Vellas, B.; Cummings, J.; Gauthier, S.; Hey, J.A.; Power, A.; Wang, P.; et al. Clinical Effects of Tramiprosate in APOE4/4 Homozygous Patients with Mild Alzheimer’s Disease Suggest Disease Modification Potential. J. Prev. Alzheimer’s Dis. 2017, 4, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Frontzkowski, L.; Ewers, M.; Brendel, M.; Biel, D.; Ossenkoppele, R.; Hager, P.; Steward, A.; Dewenter, A.; Römer, S.; Rubinski, A.; et al. Earlier Alzheimer’s disease onset is associated with tau pathology in brain hub regions and facilitated tau spreading. Nat. Commun. 2022, 13, 4899. [Google Scholar] [CrossRef]
- Wischik, C.M.; Bentham, P.; Gauthier, S.; Miller, S.; Kook, K.; Schelter, B.O. Oral Tau Aggregation Inhibitor for Alzheimer’s Disease: Design, Progress and Basis for Selection of the 16 mg/day Dose in a Phase 3, Randomized, Placebo-Controlled Trial of Hydromethylthionine Mesylate. J. Prev. Alzheimer’s Dis. 2022, 9, 780–790. [Google Scholar] [CrossRef]
- Kim, J.; Lee, Y.; Lee, S.; Kim, K.; Song, M.; Lee, J. Mesenchymal Stem Cell Therapy and Alzheimer’s Disease: Current Status and Future Perspectives. J. Alzheimer’s Dis. 2020, 77, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rasmusson, I. Immune modulation by mesenchymal stem cells. Exp. Cell Res. 2006, 312, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jin, H.K.; Bae, J.-s. Bone marrow-derived mesenchymal stem cells reduce brain amyloid-beta deposition and accelerate the activation of microglia in an acutely induced Alzheimer’s disease mouse model. Neurosci. Lett. 2009, 450, 136–141. [Google Scholar] [CrossRef]
- Cooney, D.S.; Wimmers, E.G.; Ibrahim, Z.; Grahammer, J.; Christensen, J.M.; Brat, G.A.; Wu, L.W.; Sarhane, K.A.; Lopez, J.; Wallner, C.; et al. Mesenchymal Stem Cells Enhance Nerve Regeneration in a Rat Sciatic Nerve Repair and Hindlimb Transplant Model. Sci. Rep. 2016, 6, 31306. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Wang, I.F.; Chiang, P.M.; Wang, L.C.; Shen, C.K.J.; Tsai, K.J. G-CSF-mobilized Bone Marrow Mesenchymal Stem Cells Replenish Neural Lineages in Alzheimer’s Disease Mice via CXCR4/SDF-1 Chemotaxis. Mol. Neurobiol. 2017, 54, 6198–6212. [Google Scholar] [CrossRef]
- Sanooghi, D.; Amini, N.; Azedi, F.; Bagher, Z.; Parvishan, A.; Lotfi, A.; Rashidi, N.; Lotfi, E.; Sayahpour, F.A.; Faghihi, F. Differentiation of Mesenchymal Stem Cells Derived from Human Adipose Tissue into Cholinergic-like Cells: An in Vitro Study. Basic. Clin. Neurosci. 2021, 12, 315–324. [Google Scholar] [CrossRef]
- Nasiri, E.; Alizadeh, A.; Roushandeh, A.M.; Gazor, R.; Hashemi-Firouzi, N.; Golipoor, Z. Melatonin-pretreated adipose-derived mesenchymal stem cells efficeintly improved learning, memory, and cognition in an animal model of Alzheimer’s disease. Metab. Brain Dis. 2019, 34, 1131–1143. [Google Scholar] [CrossRef]
- Hernández, A.E.; García, E. Mesenchymal Stem Cell Therapy for Alzheimer’s Disease. Stem Cells Int. 2021, 2021, 7834421. [Google Scholar] [CrossRef]
- Biglari, N.; Mehdizadeh, A.; Vafaei Mastanabad, M.; Gharaeikhezri, M.H.; Gol Mohammad Pour Afrakoti, L.; Pourbala, H.; Yousefi, M.; Soltani-Zangbar, M.S. Application of mesenchymal stem cells (MSCs) in neurodegenerative disorders: History, findings, and prospective challenges. Pathol. Res. Pract. 2023, 247, 154541. [Google Scholar] [CrossRef]
- Chen, H.X.; Liang, F.C.; Gu, P.; Xu, B.L.; Xu, H.J.; Wang, W.T.; Hou, J.Y.; Xie, D.X.; Chai, X.Q.; An, S.J. Exosomes derived from mesenchymal stem cells repair a Parkinson’s disease model by inducing autophagy. Cell Death Dis. 2020, 11, 288. [Google Scholar] [CrossRef]
- D’anca, M.; Fenoglio, C.; Buccellato, F.R.; Visconte, C.; Galimberti, D.; Scarpini, E. Extracellular Vesicles in Multiple Sclerosis: Role in the Pathogenesis and Potential Usefulness as Biomarkers and Therapeutic Tools. Cells 2021, 10, 1733. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; Wang, S.; Zhao, R.C. Exosomes from mesenchymal stem/stromal cells: A new therapeutic paradigm. Biomark. Res. 2019, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.T.; Hosseini-Beheshti, E.; Afrose, D.; Ding, X.; Xia, B.; Grau, G.E.; Little, C.B.; McClements, L.; Li, J.J. Extracellular Vesicles from Mesenchymal Stromal Cells for the Treatment of Inflammation-Related Conditions. Int. J. Mol. Sci. 2021, 22, 3023. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Liu, Y.; Ji, W.; Zhuang, J.; Chen, X.; Gong, B.; Chu, J.; Liang, W.; Gao, J.; Yin, Y. Engineered mesenchymal stem cell-derived extracellular vesicles: A state-of-the-art multifunctional weapon against Alzheimer’s disease. Theranostics 2023, 13, 1264–1285. [Google Scholar] [CrossRef]
- Li, X.; Ji, M.; Zhang, H.; Liu, Z.; Chai, Y.; Cheng, Q.; Yang, Y.; Cordato, D.; Gao, J. Non-drug Therapies for Alzheimer’s Disease: A Review. Neurol. Ther. 2023, 12, 39–72. [Google Scholar] [CrossRef]
- Bahar-Fuchs, A.; Clare, L.; Woods, B. Cognitive training and cognitive rehabilitation for mild to moderate Alzheimer’s disease and vascular dementia. Cochrane Database Syst. Rev. 2013, 2013, CD003260. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve. Neuropsychologia 2009, 47, 2015–2028. [Google Scholar] [CrossRef]
- Spector, A.; Thorgrimsen, L.; Woods, B.; Royan, L.; Davies, S.; Butterworth, M.; Orrell, M. Efficacy of an evidence-based cognitive stimulation therapy programme for people with dementia: Randomised controlled trial. Br. J. Psychiatry 2003, 183, 248–254. [Google Scholar] [CrossRef]
- Baird, A.; Samson, S. Music and dementia. Prog. Brain Res. 2015, 217, 207–235. [Google Scholar] [CrossRef]
- Rebok, G.W.; Ball, K.; Guey, L.T.; Jones, R.N.; Kim, H.Y.; King, J.W.; Marsiske, M.; Morris, J.N.; Tennstedt, S.L.; Unverzagt, F.W.; et al. Ten-year effects of the advanced cognitive training for independent and vital elderly cognitive training trial on cognition and everyday functioning in older adults. J. Am. Geriatr. Soc. 2014, 62, 16–24. [Google Scholar] [CrossRef]
- Kishita, N.; Backhouse, T.; Mioshi, E. Nonpharmacological Interventions to Improve Depression, Anxiety, and Quality of Life (QoL) in People with Dementia: An Overview of Systematic Reviews. J. Geriatr. Psychiatry Neurol. 2020, 33, 28–41. [Google Scholar] [CrossRef]
- Meng, Q.; Lin, M.S.; Tzeng, I.S. Relationship between Exercise and Alzheimer’s Disease: A Narrative Literature Review. Front. Neurosci. 2020, 14, 131. [Google Scholar] [CrossRef]
- Padala, K.P.; Padala, P.R.; Lensing, S.Y.; Dennis, R.A.; Bopp, M.M.; Roberson, P.K.; Sullivan, D.H. Home-Based Exercise Program Improves Balance and Fear of Falling in Community-Dwelling Older Adults with Mild Alzheimer’s Disease: A Pilot Study. J. Alzheimer’s Dis. 2017, 59, 565–574. [Google Scholar] [CrossRef]
- Öhman, H.; Savikko, N.; Strandberg, T.; Kautiainen, H.; Raivio, M.; Laakkonen, M.L.; Tilvis, R.; Pitkälä, K.H. Effects of Exercise on Functional Performance and Fall Rate in Subjects with Mild or Advanced Alzheimer’s Disease: Secondary Analyses of a Randomized Controlled Study. Dement. Geriatr. Cogn. Disord. 2016, 41, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.H.; Pai, M.C.; Lin, H.S.; Sung, P.S.; Wang, J.J. Effects of walking on sundown syndrome in community-dwelling people with Alzheimer’s disease. Int. J. Older People Nurs. 2020, 15, e12292. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef]
- Chang, C.H.; Lane, H.Y.; Lin, C.H. Brain stimulation in Alzheimer’s disease. Front. Psychiatry 2018, 9, 201. [Google Scholar] [CrossRef]
- Perestelo-Pérez, L.; Rivero-Santana, A.; Pérez-Ramos, J.; Serrano-Pérez, P.; Panetta, J.; Hilarion, P. Deep brain stimulation in Parkinson’s disease: Meta-analysis of randomized controlled trials. J. Neurol. 2014, 261, 2051–2060. [Google Scholar] [CrossRef]
- Leoutsakos, J.M.S.; Yan, H.; Anderson, W.S.; Asaad, W.F.; Baltuch, G.; Burke, A.; Chakravarty, M.M.; Drake, K.E.; Foote, K.D.; Fosdick, L.; et al. Deep Brain Stimulation Targeting the Fornix for Mild Alzheimer Dementia (the ADvance Trial): A Two Year Follow-up Including Results of Delayed Activation. J. Alzheimer’s Dis. 2018, 64, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Stagg, C.J.; Nitsche, M.A. Physiological basis of transcranial direct current stimulation. Neuroscientist 2011, 17, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Matt, E.; Dörl, G.; Beisteiner, R. Transcranial pulse stimulation (TPS) improves depression in AD patients on state-of-the-art treatment. Alzheimer’s Dement. 2022, 8, e12245. [Google Scholar] [CrossRef]
- Smith, A.D.; Smith, S.M.; de Jager, C.A.; Whitbread, P.; Johnston, C.; Agacinski, G.; Oulhaj, A.; Bradley, K.M.; Jacoby, R.; Refsum, H. Homocysteine-lowering by b vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS ONE 2010, 5, e12244. [Google Scholar] [CrossRef] [PubMed]
- Scarmeas, N.; Stern, Y.; Mayeux, R.; Manly, J.J.; Schupf, N.; Luchsinger, J.A. Mediterranean diet and mild cognitive impairment. Arch. Neurol. 2009, 66, 216–225. [Google Scholar] [CrossRef]
- Yoon, G.; Song, J. Intermittent Fasting: A Promising Approach for Preventing Vascular Dementia. J. Lipid Atheroscler. 2019, 8, 1–7. [Google Scholar] [CrossRef]
- Toups, K.; Hathaway, A.; Gordon, D.; Chung, H.; Raji, C.; Boyd, A.; Hill, B.D.; Hausman-Cohen, S.; Attarha, M.; Chwa, W.J.; et al. Precision Medicine Approach to Alzheimer’s Disease: Successful Pilot Project. J. Alzheimer’s Dis. 2022, 88, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Treasure, J. Review: Exploration of psychological and physical health differences between caregivers and non-caregivers: Commentary. Evid. Based. Ment. Health 2004, 7, 28. [Google Scholar] [CrossRef]
- Hampel, H.; Au, R.; Mattke, S.; van der Flier, W.M.; Aisen, P.; Apostolova, L.; Chen, C.; Cho, M.; De Santi, S.; Gao, P.; et al. Designing the next-generation clinical care pathway for Alzheimer’s disease. Nat. Aging 2022, 2, 692–703. [Google Scholar] [CrossRef]
- Goldoni, R.; Dolci, C.; Boccalari, E.; Inchingolo, F.; Paghi, A.; Strambini, L.; Galimberti, D.; Tartaglia, G.M. Salivary biomarkers of neurodegenerative and demyelinating diseases and biosensors for their detection. Ageing Res. Rev. 2022, 76, 101587. [Google Scholar] [CrossRef] [PubMed]
- Buccellato, F.R.; Galimberti, D.; Tartaglia, G.M. Beyond dentistry: Could prevention and screening for neurodegenerative diseases start in the dental office? Neural Regen. Res. 2024, 19, 156–157. [Google Scholar] [CrossRef]


{kind=link}
| Medication | Mechanism of Action | Clinical Trial Number | Estimated Completion Date/Current Status |
|---|---|---|---|
| Aducanumab | Anti-amyloid mAb directed at plaques and oligomers | NCT04241068 NCT05310071 | October 2023 December 2025 |
| Donanemab | Anti-amyloid mAb specific for pyroglutamate modified Aβ plaque proteins | NCT04437511 NCT05026866 NCT05508789 | Completed October 2027 April 2027 |
| Gantenerumab | Anti-amyloid mAb directed at plaques and oligomers | NCT04339413 NCT04374253 NCT05256134 NCT05552157 | Terminated Terminated Terminated Suspended |
| Lecanemab | Anti-amyloid mAb directed at protofibrils and plaques | NCT03887455 NCT04468659 NCT05269394 NCT01760005 | September 2027 October 2027 October 2027 October 2027 |
| Remternetug | Anti-amyloid mAb recognizing pyroglutamated form of Aβ plaques | NCT05463731 | October 2026 |
| Solanezumab | Anti-amyloid mAb targeting soluble Aβ | NCT02008357 NCT01760005 | Terminated October 2027 |
| E2814 | Anti-tau mAb | NCT05269394 | October 2027 |
| Other biologic agents | |||
| Semaglutide | glucagon-like peptide-1 receptor agonist | NCT04777396 NCT04777409 | |
| Tertomotide | Vaccine, 16aa peptide from human telomerase reverse transcriptase | NCT05303701 | April 2026 |
| Small Molecules | |||
|---|---|---|---|
| Hydralazine hydrocloride | Free radical scavenger | NCT04842552 | December 2023 |
| Icosapent ethyl | Purified form of the omega-3 fatty acid eicosapentaenoic acid | NCT02719327 | September 2023 |
| Metformin | Insulin sensitizer | NTC04098666 | April 2026 |
| AGB101 | Anti-seizure medicament | NCT03486938 | November 2022, Ended |
| Blarcamesine (ANAVEX 2-73) | Mixed ligand of the sigma-1 receptor (σ1R) and the M2 muscarinic receptor | NCT04314934 | July 2024 |
| Fosfogonimenton (ATH-1017, NDX-1017) | HGF/MET system | NCT04488419 | February 2024 |
| Masitinib | Tyrosine kinase inhibitor | NCT05564169 | December 2025 |
| NE3107 | Derivative of β-androstenetriol | NCT04669028 | October 2023 |
| Nilotinib | c-Abl tyrosine kinase inhibitor | NCT05143528 | June 2026 |
| Piromelatine | Agonist of melatonin and serotonin receptors | NCT05267535 | June 2025 |
| Simufilam | Inhibitor of scaffold protein Filamin A | NCT05575076 | July 2027 |
| Valiltramiprosate (ALZ-801) | Prevents neurotoxic Aβ formation with no plaque interaction | NCT04770220 | June 2024 |
| Hydromethylthionine mesylate | Tau aggregation inhibitor | NCT03446001 | Completed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buccellato, F.R.; D’Anca, M.; Tartaglia, G.M.; Del Fabbro, M.; Scarpini, E.; Galimberti, D. Treatment of Alzheimer’s Disease: Beyond Symptomatic Therapies. Int. J. Mol. Sci. 2023, 24, 13900. https://doi.org/10.3390/ijms241813900
Buccellato FR, D’Anca M, Tartaglia GM, Del Fabbro M, Scarpini E, Galimberti D. Treatment of Alzheimer’s Disease: Beyond Symptomatic Therapies. International Journal of Molecular Sciences. 2023; 24(18):13900. https://doi.org/10.3390/ijms241813900
Chicago/Turabian StyleBuccellato, Francesca R., Marianna D’Anca, Gianluca Martino Tartaglia, Massimo Del Fabbro, Elio Scarpini, and Daniela Galimberti. 2023. "Treatment of Alzheimer’s Disease: Beyond Symptomatic Therapies" International Journal of Molecular Sciences 24, no. 18: 13900. https://doi.org/10.3390/ijms241813900