
PLA2G12A as a Novel Biomarker for Colorectal Cancer with Prognostic Relevance
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Screening of Candidate Genes Involved in Tumor Cell Dissemination Using an In Vivo CRC Model
2.2. Identification of PLA2G12A as a Potential Tumor Suppressor Gene Associated with Prognosis in Human CRC Cohorts
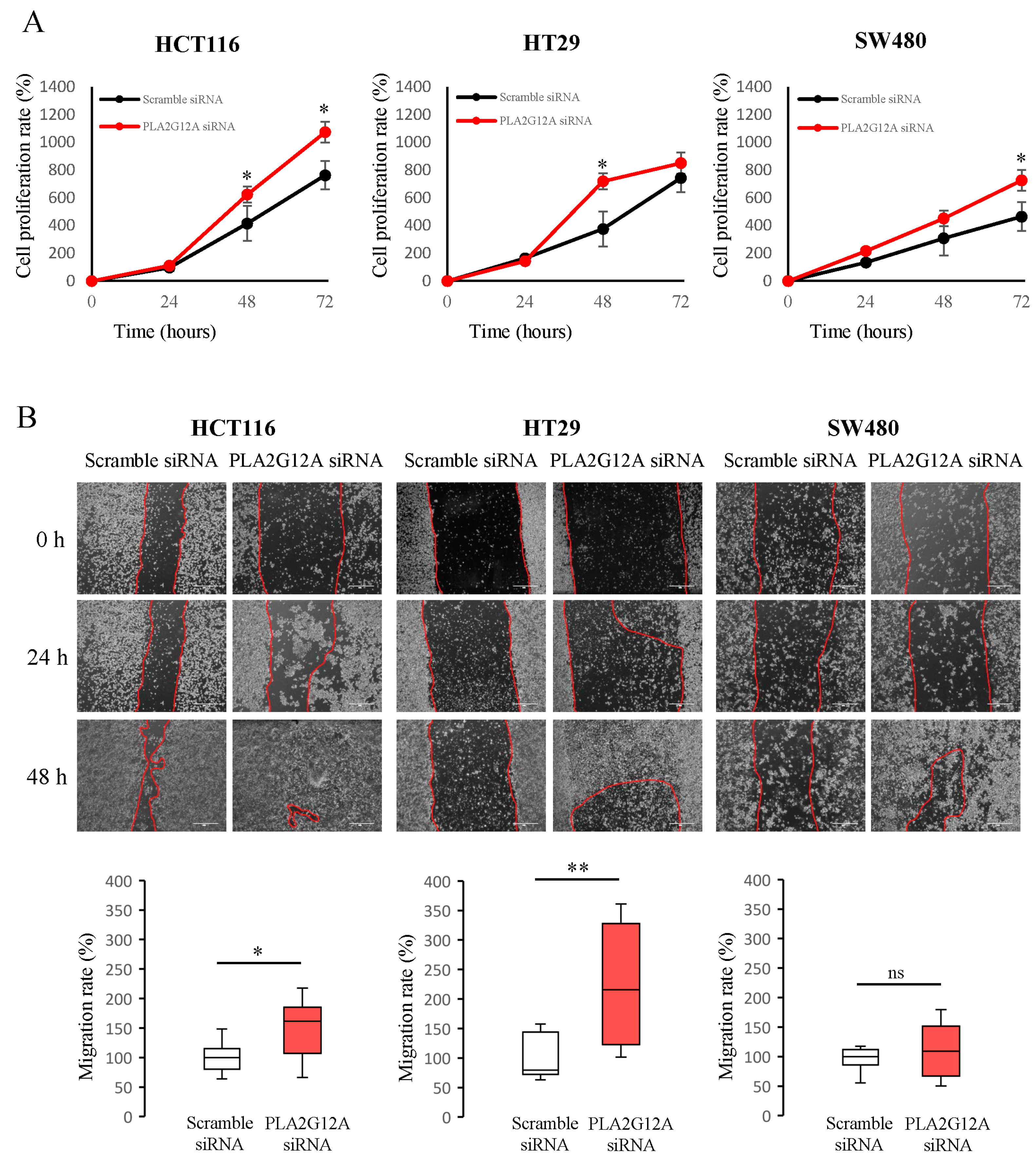
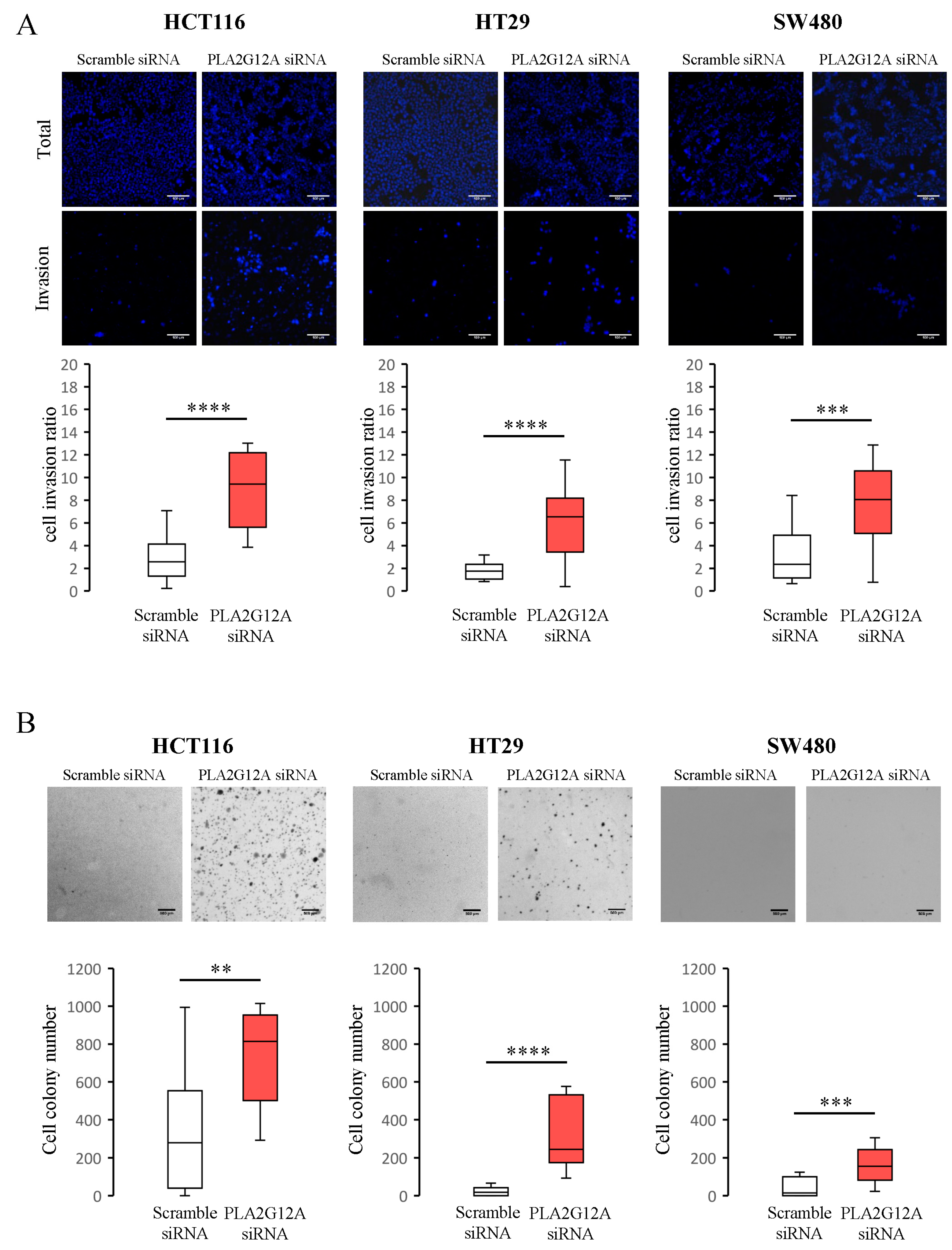
2.3. Functional Characterization of PLA2G12A in CRC Preclinical Models
3. Discussion
4. Materials and Methods
4.1. Fly Strains
4.2. Luciferase Assay
4.3. Human Samples
4.4. Real-Time PCR (RT-PCR)
4.5. TCGA Data Analysis
4.6. Cell Culture Assays
4.7. Proliferative Assays
4.8. Cell Migration Assays
4.9. Invasion Assays
4.10. Anchorage-Independent Cell Growth Assays
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Eng, C.; Nieman, L.Z. Trends in Colorectal Cancer Incidence by Anatomic Site and Disease Stage in the United States. Am. J. Clin. Oncol. 2011, 34, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Miller, K.; Sandeep Wagle, N.; Jemal DVM, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.; Hu, C.; You, N.; Bednarski, B.; Rodriguez-Bigas, M.; Skibber, J.; Cantor, S.; Chang, G. Increasing Disparities in Age-Related Incidence of Colon and Rectal Cancer in the United States, 1975-2010. JAMA Surg 2015, 150, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Cunningham, D.; Atkin, W.; Lenz, H.J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef]
- Benson, A.B.; Schrag, D.; Somerfield, M.R.; Cohen, A.M.; Figueredo, A.T.; Flynn, P.J.; Krzyzanowska, M.K.; Maroun, J.; McAllister, P.; Van Cutsem, E.; et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J. Clin. Oncol. 2004, 22, 3408–3419. [Google Scholar] [CrossRef]
- Argilés, G.; Tabernero, J.; Labianca, R.; Hochhauser, D.; Salazar, R.; Iveson, T.; Laurent-Puig, P.; Quirke, P.; Yoshino, T.; Taieb, J.; et al. Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2020, 31, 1291–1305. [Google Scholar] [CrossRef]
- André, T.; De Gramont, A.; Vernerey, D.; Chibaudel, B.; Bonnetain, F.; Tijeras-raballand, A.; Scriva, A.; Hickish, T.; Tabernero, J.; Van Laethem, J.L.; et al. Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF Mutation and Mismatch Repair Status of the MOSAIC Study. J. Clin. Oncol. 2023, 33, 4176–4187. [Google Scholar] [CrossRef]
- Baxter, N.N.; Kennedy, E.B.; Bergsland, E.; Berlin, J.; George, T.J. Adjuvant Therapy for Stage II Colon Cancer: ASCO Guideline Update. J. Clin. Oncol. 2023, 40, 892–910. [Google Scholar] [CrossRef]
- Grothey, A.; Sobrero, A.F.; Shields, A.F.; Yoshino, T.; Paul, J.; Taieb, J.; Souglakos, J.; Shi, Q.; Labianca, R.; Meyerhardt, J.A.; et al. Duration of Adjuvant Chemotherapy for Stage III Colon Cancer. N. Engl. J. Med. 2019, 378, 1177–1188. [Google Scholar] [CrossRef]
- Wang, Y.S.; Kou, Y.; Zhu, R.T.; Han, B.W.; Li, C.H.; Wang, H.J.; Wu, H.B.; Xia, T.M.; Che, X.M. CDX2 as a Predictive Biomarker Involved in Immunotherapy Response Suppresses Metastasis through EMT in Colorectal Cancer. Dis. Markers 2022, 2022. [Google Scholar] [CrossRef]
- Dalerba, P.; Sahoo, D.; Soonmyung, P.; Guo, X.; Yothers, G.; Song, N.; Wilcox-Fogel, N.; Forgó, E.; Rajendran, P.S.; Miranda, S.P.; et al. CDX2 as a Prognostic Biomarker in Stage II and Stage III Colon Cancer. N. Engl. J. Med. 2016, 374, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Silliman, N.; Tacey, M.; Wong, H.; Christie, M.; Skinner, I.; et al. Circulating Tumor DNA Analysis. Sci. Transl. Med. 2017, 8. [Google Scholar]
- Cavalieri, D.; Dolara, P.; Mini, E.; Luceri, C.; Castagnini, C.; Toti, S.; Maciag, K.; De Filippo, C.; Nobili, S.; Morganti, M.; et al. Analysis of gene expression profiles reveals novel correlations with the clinical course of colorectal cancer. Oncol. Res. 2007, 16, 535–548. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.J.; Lavery, I.; Yothers, G.; Paik, S.; Clark-Langone, K.M.; Lopatin, M.; Watson, D.; Baehner, F.L.; Shak, S.; Baker, J.; et al. Relationship between tumor gene expression and recurrence in four independent studies of patients with stage II/III colon cancer treated with surgery alone or surgery plus adjuvant fluorouracil plus leucovorin. J. Clin. Oncol. 2010, 28, 3937–3944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, R.; Roepman, P.; Capella, G.; Moreno, V.; Simon, I.; Dreezen, C.; Lopez-Doriga, A.; Santos, C.; Marijnen, C.; Westerga, J.; et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J. Clin. Oncol. 2011, 29, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, R.D.; Bylesjo, M.; Kerr, P.; Davison, T.; Black, J.M.; Kay, E.W.; Holt, R.J.; Proutski, V.; Ahdesmaki, M.; Farztdinov, V.; et al. Development and independent validation of a prognostic assay for stage ii colon cancer using formalin-fixed paraffin-embedded tissue. J. Clin. Oncol. 2011, 29, 4620–4626. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Xu, E.; Liu, H.; Wan, L.; Lai, M. Epithelial-mesenchymal transition in colorectal cancer metastasis: A system review. Pathol. Res. Pract. 2015, 211, 557–569. [Google Scholar] [CrossRef]
- Khanna, C.; Hunter, K. Modeling metastasis in vivo. Carcinogenesis 2005, 26, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Astell, K.R.; Sieger, D. Zebrafish In Vivo Models of Cancer and Metastasis. Cold Spring Harb Perspect Med 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, N.; Mani, K.; Fay, D. Cancer models in C. elegans. Dev. Dyn. 2010, 239, 1413–1448. [Google Scholar] [PubMed] [Green Version]
- Chakraborty, U.; Dinh, T.; Alani, E. Genomic Instability Promoted by Overexpression of Mismatch Repair Factors in Yeast: A Model for Understanding Cancer Progression. Genetics 2018, 209, 439–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brumby, A.M.; Richardson, H.E. Using Drosophila melanogaster to map human cancer pathways. Nat. Rev. Cancer 2005, 5, 626–639. [Google Scholar] [CrossRef]
- Gonzalez, C. Drosophila melanogaster: A model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. Cancer 2013, 13, 172–183. [Google Scholar] [CrossRef]
- Martorell, Ò.; Merlos-Suárez, A.; Campbell, K.; Barriga, F.M.; Christov, C.P.; Miguel-Aliaga, I.; Batlle, E.; Casanova, J.; Casali, A. Conserved mechanisms of tumorigenesis in the Drosophila adult midgut. PLoS ONE 2014, 9, e88413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K.; Rossi, F.; Adams, J.; Pitsidianaki, I.; Barriga, F.M.; Garcia-Gerique, L.; Batlle, E.; Casanova, J.; Casali, A. Collective cell migration and metastases induced by an epithelial-to-mesenchymal transition in Drosophila intestinal tumors. Nat. Commun. 2019, 10, 2311. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Chang, Y.; Fan, J.; Ji, W.; Su, C. Phospholipase A2 superfamily in cancer. Cancer Lett. 2021, 497, 165–177. [Google Scholar] [CrossRef]
- Brglez, V.; Lambeau, G.; Petan, T. Secreted phospholipases A2 in cancer: Diverse mechanisms of action. Biochimie 2014, 107, 114–123. [Google Scholar] [CrossRef]
- Adams, J.; Casali, A.; Campbell, K. Sensitive high-throughput assays for tumour burden reveal the response of a drosophila melanogaster model of colorectal cancer to standard chemotherapies. Int. J. Mol. Sci. 2021, 22, 5101. [Google Scholar] [CrossRef]
- Miles, W.O.; Dyson, N.J.; Walker, J.A. Modeling tumor invasion and metastasis in Drosophila. DMM Dis. Model. Mech. 2011, 4, 753–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brumby, A.M.; Richardson, H.E. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003, 22, 5769–5779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, M.; Cagan, R.L. Drosophila models for cancer research. Curr. Opin. Genet. Dev. 2006, 16, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Januschke, J.; Gonzalez, C. Drosophila asymmetric division, polarity and cancer. Oncogene 2008, 27, 6994–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevalainen, T.J.; Cardoso, J.C.R.; Riikonen, P.T. Conserved domains and evolution of secreted phospholipases A 2. FEBS J. 2012, 279, 636–649. [Google Scholar] [CrossRef] [Green Version]
- Park, J.B.; Lee, C.S.; Jang, J.H.; Ghim, J.; Kim, Y.J.; You, S.; Hwang, D.; Suh, P.G.; Ryu, S.H. Phospholipase signalling networks in cancer. Nat. Rev. Cancer 2012, 12, 782–792. [Google Scholar] [CrossRef]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Hirabayashi, T.; Yamamoto, K. Recent progress in phospholipase A2 research: From cells to animals to humans. Prog. Lipid Res. 2011, 50, 152–192. [Google Scholar] [CrossRef]
- Suh, P.G.; Park, J.I.; Manzoli, L.; Cocco, L.; Peak, J.C.; Katan, M.; Fukami, K.; Kataoka, T.; Yun, S.; Sung, H.R. Multiple roles of phosphoinositide-specific phospholipase C isozymes. J. Biochem. Mol. Biol. 2008, 41, 415–434. [Google Scholar] [CrossRef] [Green Version]
- Aoki, J.; Inoue, A.; Makide, K.; Saiki, N.; Arai, H. Structure and function of extracellular phospholipase A1 belonging to the pancreatic lipase gene family. Biochimie 2007, 89, 197–204. [Google Scholar] [CrossRef]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Yamamoto, K.; Lambeau, G. Emerging roles of secreted phospholipase A2 enzymes: The 3rd edition. Biochimie 2014, 107, 105–113. [Google Scholar] [CrossRef]
- Gelb, M.H.; Valentin, E.; Ghomashchi, F.; Lazdunski, M.; Pharmacologie, I. De Cloning and Recombinant Expression of a Structurally Novel Human Secreted Phospholipase A2. J Biol Chem 2000, 275, 39823–39826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, M.; Bollinger, J.G.; Lazdunski, M.; Gelb, M.H.; Lambeau, G. Novel mammalian group XII secreted phospholipase A2 lacking enzymatic activity. Biochemistry 2003, 42, 11494–11503. [Google Scholar] [CrossRef] [PubMed]
- Peuravuori, H.; Kollanus, S.; Nevalainen, T.J. Expression of group XIIA phospholipase A2 in human digestive organs. Apmis 2014, 122, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.; Arm, J.P.; Iii, C.O.B.; Choi, A.; Austen, K.F.; Glimcher, L.H. A Novel Group of Phospholipase A 2 s Preferentially Expressed in Type 2 Helper T Cells *. J. Biol. Chem. 2001, 276, 18321–18326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schewe, M.; Franken, P.F.; Sacchetti, A.; Schmitt, M.; Joosten, R.; Böttcher, R.; van Royen, M.E.; Jeammet, L.; Payré, C.; Scott, P.M.; et al. Secreted Phospholipases A2 Are Intestinal Stem Cell Niche Factors with Distinct Roles in Homeostasis, Inflammation, and Cancer. Cell Stem Cell 2016, 19, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, K.; Ivanova, T.; Wu, Y.; Rajasegaran, V.; Wu, J.; Ming, H.L.; Yu, K.; Sun, Y.R.; Hyun, C.C.; Ylstra, B.; et al. Inhibition of gastric cancer invasion and metastasis by PLA2G2A, a novel β-catenin/TCF target gene. Cancer Res. 2008, 68, 4277–4286. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Gandhi, C.R.; Gao, Z.H. Involvement of cytosolic phospholipase A 2 alpha signalling pathway in spontaneous and transforming growth factor-beta-induced activation of rat hepatic stellate cells. Liver Int. 2011, 31, 1565–1573. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Orozco, R.; Navarro-Tito, N.; Soto-Guzman, A.; Castro-Sanchez, L.; Perez Salazar, E. Arachidonic acid promotes epithelial-to-mesenchymal-like transition in mammary epithelial cells MCF10A. Eur. J. Cell Biol. 2010, 89, 476–488. [Google Scholar] [CrossRef]
- Villegas-Comonfort, S.; Castillo-Sanchez, R.; Serna-Marquez, N.; Cortes-Reynosa, P.; Salazar, E.P. Arachidonic acid promotes migration and invasion through a PI3K/Akt-dependent pathway in MDA-MB-231 breast cancer cells. Prostaglandins Leukot. Essent. Fat. Acids 2014, 90, 169–177. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Luo, L. Mosaic analysis with a repressible neurotechnique cell marker for studies of gene function in neuronal morphogenesis. Neuron 1999, 22, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics. Cancer Discov. 2017, 32, 736–740. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal Complementary Data Sources and Analysis Options. Sci. Signal. 2014, 6, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjeed, S.; Ebert, B.L.; Gillettea, M.A.; Paulovichg, A.; Pomeroyh, S.L.; Golub, T.R.; Landera, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drosophila Orthologue | Human Gene | TTR | OS | ||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | p | HR | 95% CI | p | ||
| Cont | CNTN4 | 0.677 | 0.432–1.060 | 0.088 | 0.602 | 0.342–1.060 | 0.079 |
| Dl | DLL1 | 1.316 | 0.840–2.060 | 0.230 | 1.597 | 0.910–2.805 | 0.103 |
| Src42 | FRK | 0.847 | 0.542–1.322 | 0.464 | 0.961 | 0.551–1.675 | 0.887 |
| Ipk2 | IPMK | 1.078 | 0.690–1.685 | 0.740 | 1.044 | 0.599–1.820 | 0.879 |
| Dp110 | PIK3CD | 1.088 | 0.697–1.698 | 0.711 | 0.855 | 0.490–1.493 | 0.582 |
| GXIVsPLA2 | PLA2G12A | 0.590 | 0.375–0.928 | 0.022 | 0.546 | 0.308–0.967 | 0.038 |
| Gap1 | RASA3 | 0.984 | 0.631–1.534 | 0.943 | 1.287 | 0.738–2.243 | 0.375 |
| CG9098 | SH2D3C | 0.833 | 0.533–1.300 | 0.421 | 1.122 | 0.644–1.957 | 0.684 |
| Mal-A1 | SLC3A1 | 1.037 | 0.665–1.617 | 0.872 | 0.929 | 0.532–1.620 | 0.794 |
| Bai | TMED10 | 1.225 | 0.783–1.915 | 0.375 | 1.064 | 0.609–1.859 | 0.828 |
| Characteristic | High | Low | p Value | |
|---|---|---|---|---|
| Sex | Male (%) | 45 (58.4) | 51 (67.1) | 0.317 |
| Female (%) | 32 (41.6) | 25 (32.9) | ||
| Age | Median (years) | 74.8 | 75.4 | 0.755 |
| Anatomic location | Cecum (%) | 7 (9.1) | 3 (3.9) | 0.507 |
| Ascending colon (%) | 17 (22.1) | 24 (31.6) | ||
| Hepatic flexure (%) | 1 (1.3) | 2 (2.6) | ||
| Transverse colon (%) | 2 (2.6) | 2 (2.6) | ||
| Splenic flexure (%) | 1 (1.3) | 3 (3.9) | ||
| Descending colon (%) | 6 (7.8) | 3 (3.9) | ||
| Sigmoid colon (%) | 25 (31.5) | 28 (36.8) | ||
| Rectosigmoid junction (%) | 1 (1.3) | 5 (6.6) | ||
| Rectum (%) | 17 (22.1) | 6 (7.9) | ||
| Stage | I (%) | 4 (5.2) | 2 (2.6) | 0.063 |
| II (%) | 29 (37.7) | 19 (25) | ||
| III (%) | 44 (57.1) | 55 (72.4) | ||
| Histology | Adenocarcinoma (%) | 63 (81.8) | 65 (85.5) | 0.663 |
| Mucinous adenocarcinoma (%) | 13 (16.9) | 11 (14.5) | ||
| Signet ring cell carcinoma (%) | 1 (1.3) | 0 (0) | ||
| Differentiation degree | Low-grade (%) | 65 (84.4) | 68 (89.5) | 0.473 |
| High-grade (%) | 12 (15.6) | 8 (10.5) | ||
| Venous invasion [missing 1] | Yes (%) | 21 (27.6) | 33 (43.4) | 0.062 |
| No (%) | 55 (72.4) | 43 (56.6) | ||
| Lymphatic invasion [missing 3] | Yes (%) | 27 (35.5) | 34 (45.9) | 0.245 |
| No (%) | 49 (64.5) | 40 (54.1) | ||
| Perineural invasion [missing 3] | Yes (%) | 21 (28) | 28 (37.3) | 0.296 |
| No (%) | 54 (72) | 47 (62.7) | ||
| Microsatellite status [missing 80] | MSS (%) | 39 (95.1) | 30 (93.8) | 1 |
| MSI (%) | 2 (4.9) | 2 (6.3) | ||
| RAS [missing 122] | RAS mut (%) | 6 (60) | 12 (57.1) | 1 |
| RAS wt (%) | 4 (40) | 9 (42.9) | ||
| Adjuvant chemotherapy | Yes (%) | 38 (49.4) | 47 (61.8) | 0.144 |
| No (%) | 39 (50.6) | 29 (38.2) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parisi, E.; Hidalgo, I.; Montal, R.; Pallisé, O.; Tarragona, J.; Sorolla, A.; Novell, A.; Campbell, K.; Sorolla, M.A.; Casali, A.; et al. PLA2G12A as a Novel Biomarker for Colorectal Cancer with Prognostic Relevance. Int. J. Mol. Sci. 2023, 24, 10889. https://doi.org/10.3390/ijms241310889
Parisi E, Hidalgo I, Montal R, Pallisé O, Tarragona J, Sorolla A, Novell A, Campbell K, Sorolla MA, Casali A, et al. PLA2G12A as a Novel Biomarker for Colorectal Cancer with Prognostic Relevance. International Journal of Molecular Sciences. 2023; 24(13):10889. https://doi.org/10.3390/ijms241310889
Chicago/Turabian StyleParisi, Eva, Ivan Hidalgo, Robert Montal, Ona Pallisé, Jordi Tarragona, Anabel Sorolla, Anna Novell, Kyra Campbell, Maria Alba Sorolla, Andreu Casali, and et al. 2023. "PLA2G12A as a Novel Biomarker for Colorectal Cancer with Prognostic Relevance" International Journal of Molecular Sciences 24, no. 13: 10889. https://doi.org/10.3390/ijms241310889