
Endogenous TOM20 Proximity Labeling: A Swiss-Knife for the Study of Mitochondrial Proteins in Human Cells
 , , , , and
, , , , and 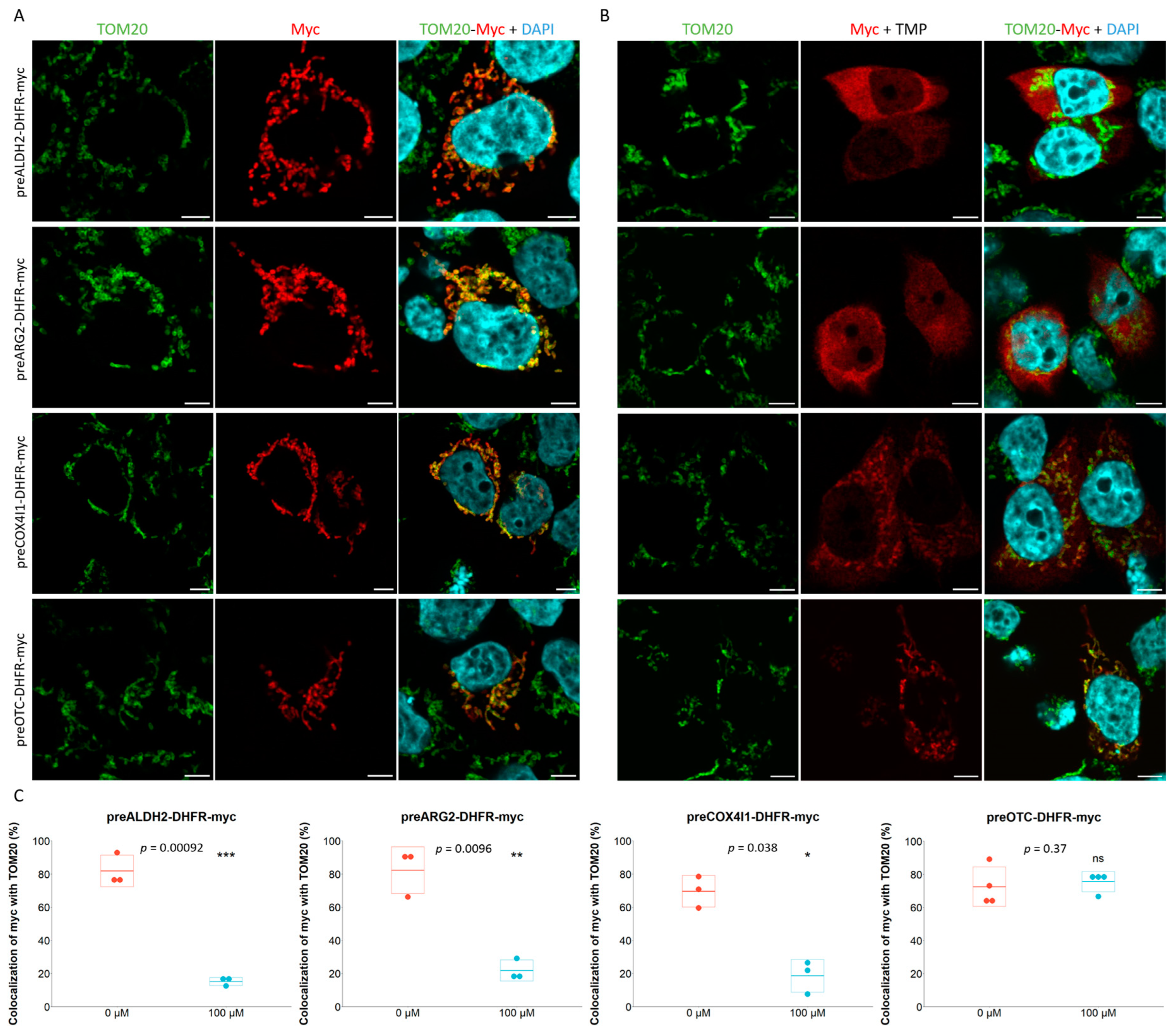{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
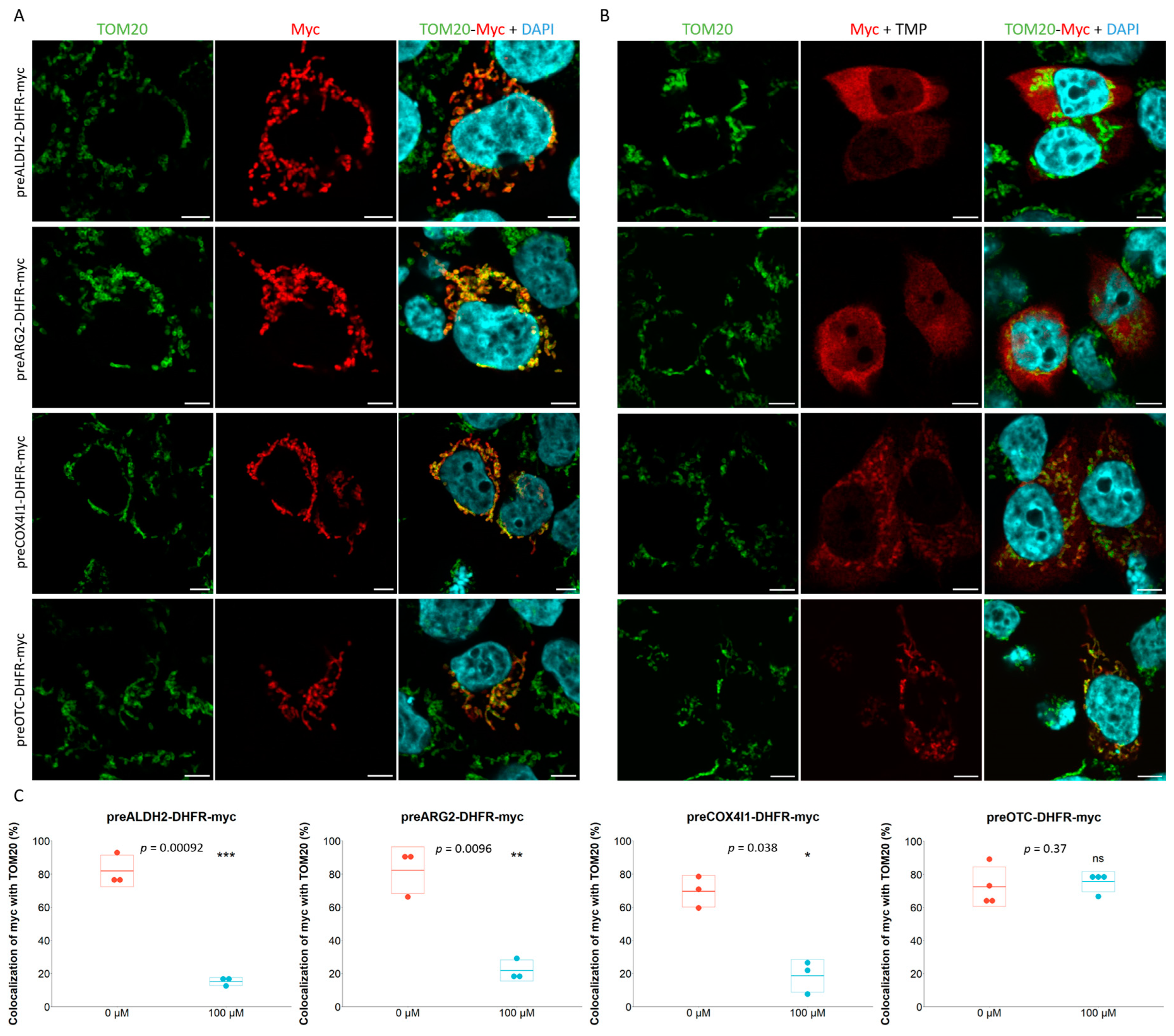
2.1. Characterization of Mitochondrial Post- and Co-Translational Import Reporters
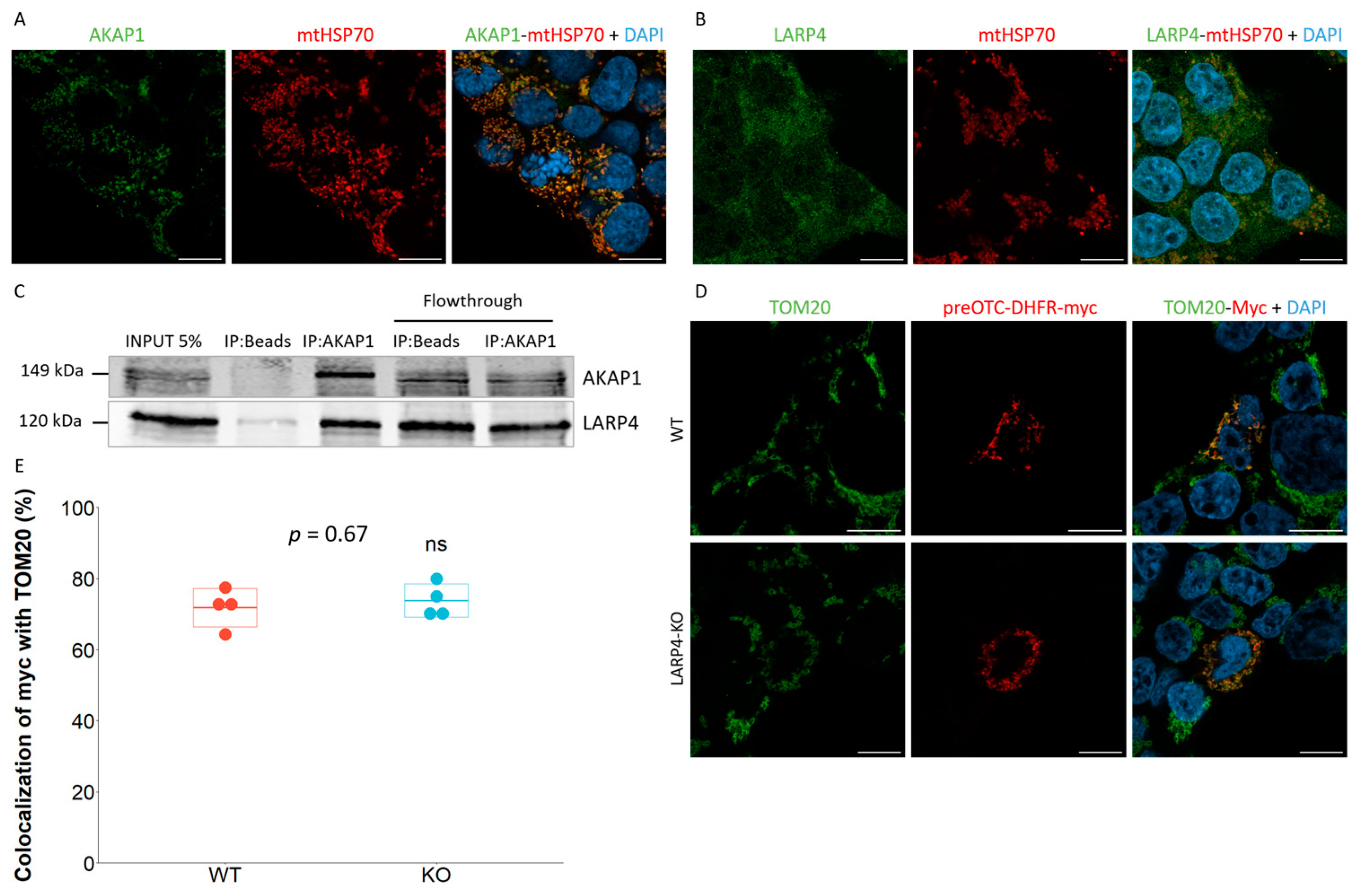
2.2. LARP4 Does Not Participate in the Co-Translational Import of preOTC-DHFR Reporter
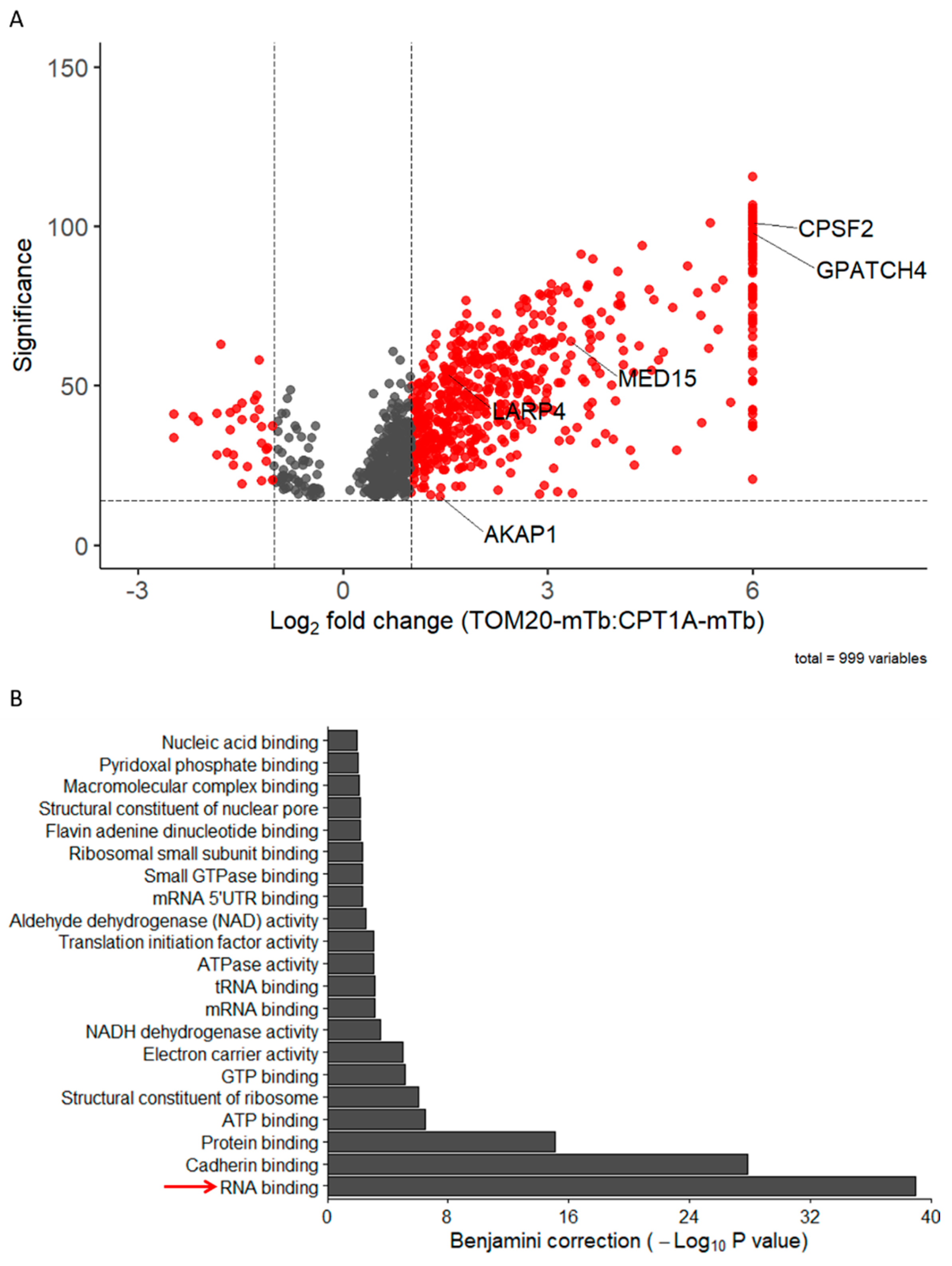
2.3. TOM20 Proxisome Characterization as a Strategy to Identify Effectors of the Mitochondrial Co-Translational Import
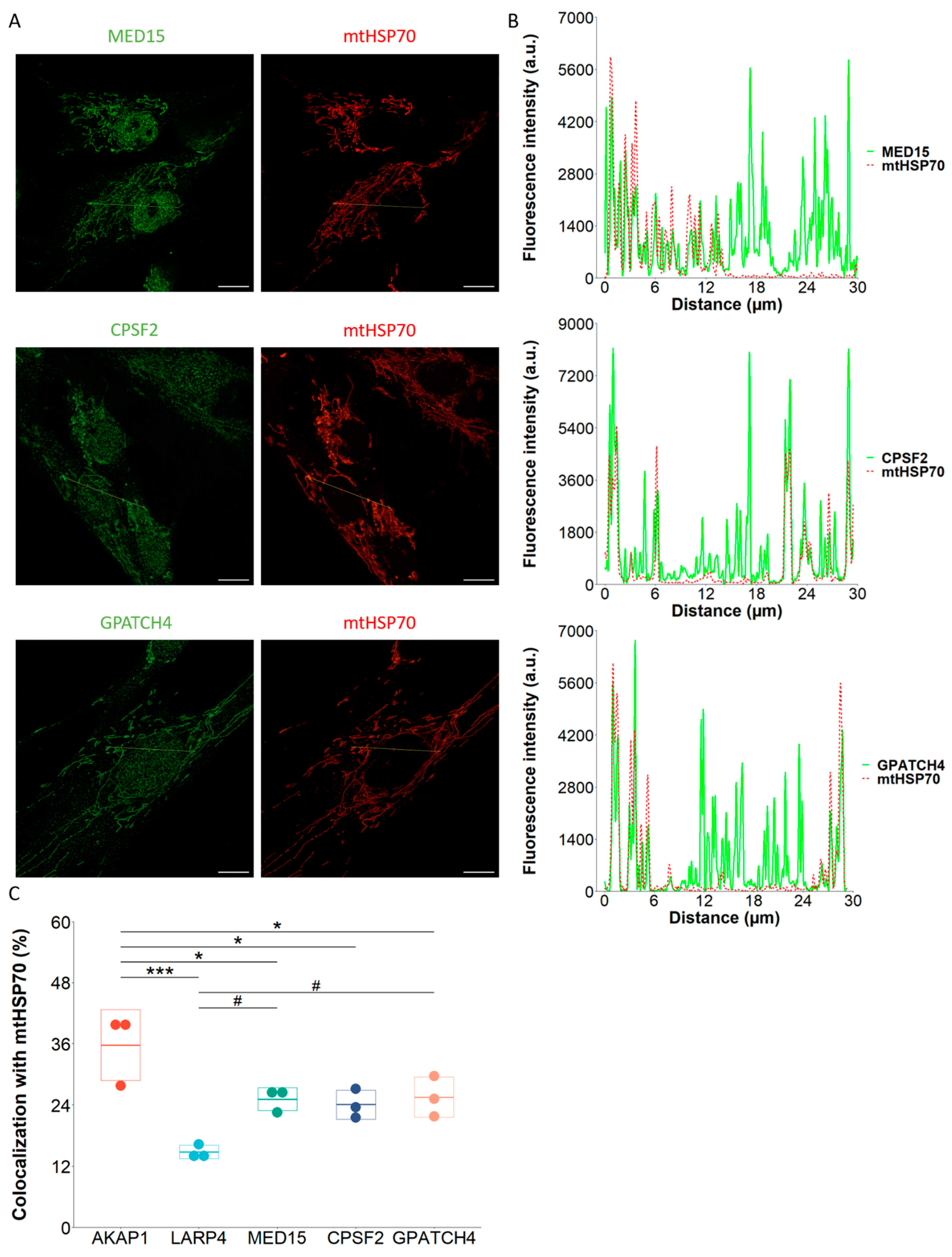
2.4. TOM20-mTb-Based BioID Unravels New Proteins Localized in the Mitochondria
2.5. TOM20-mTb as a Tool to Detect Protein Entry Inside Mitochondria
3. Discussion
4. Materials and Methods
4.1. Isolation of Primary Fibroblasts
4.2. Cell Culture
4.3. Generation of KO Cell Lines
4.4. Generation of Endogenous BioID Cell Lines
4.5. Repair Template Plasmids and Reporters Cloning
4.6. Proximity Labeling
4.7. Mass Spectrometry Analyses
4.8. Cell Transfection with Reporter Constructs
4.9. Immunofluorescence Analysis and Confocal Microscopy Observation
4.10. Image Analysis
4.11. Western Blotting
4.12. Immunoprecipitation
4.13. Data Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vandemoortele, G.; De Sutter, D.; Moliere, A.; Pauwels, J.; Gevaert, K.; Eyckerman, S. A Well-Controlled BioID Design for Endogenous Bait Proteins. J. Proteome Res. 2018, 18, 95–106. [Google Scholar] [CrossRef]
- Sen, A.; Kallabis, S.; Gaedke, F.; Jüngst, C.; Boix, J.; Nüchel, J.; Maliphol, K.; Hofmann, J.; Schauss, A.C.; Krüger, M.; et al. Mitochondrial Membrane Proteins and VPS35 Orchestrate Selective Removal of MtDNA. Nat. Commun. 2022, 13, 6704. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaka, T.; Kosako, H.; Yoshizumi, T.; Furukawa, R.; Hirano, Y.; Kuge, O.; Tamada, T.; Koshiba, T. Structural Basis of Mitochondrial Scaffolds by Prohibitin Complexes: Insight into a Role of the Coiled-Coil Region. iScience 2019, 19, 1065–1078. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Koolmeister, C.; Misic, J.; Siira, S.; Kühl, I.; Ramos, E.S.; Miranda, M.; Jiang, M.; Posse, V.; Lytovchenko, O.; et al. TEFM Regulates Both Transcription Elongation and RNA Processing in Mitochondria. EMBO Rep. 2019, 20, e48101. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef]
- Kim, D.I.; Birendra, K.C.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing Nuclear Pore Complex Architecture with Proximity-Dependent Biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef]
- Kaewsapsak, P.; Shechner, D.M.; Mallard, W.; Rinn, J.L.; Ting, A.Y. Live-Cell Mapping of Organelle-Associated RNAs via Proximity Biotinylation Combined with Protein-RNA Crosslinking. eLife 2017, 6, 29224. [Google Scholar] [CrossRef]
- Kwak, C.; Shin, S.; Park, J.S.; Jung, M.; My Nhung, T.T.; Kang, M.G.; Lee, C.; Kwon, T.H.; Park, S.K.; Mun, J.Y.; et al. Contact-ID, a Tool for Profiling Organelle Contact Sites, Reveals Regulatory Proteins of Mitochondrial-Associated Membrane Formation. Proc. Natl. Acad. Sci. USA 2020, 117, 12109–12120. [Google Scholar] [CrossRef]
- Cho, K.F.; Branon, T.C.; Rajeev, S.; Svinkina, T.; Udeshi, N.D.; Thoudam, T.; Kwak, C.; Rhee, H.W.; Lee, I.K.; Carr, S.A.; et al. Split-TurboID Enables Contact-Dependent Proximity Labeling in Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 12143–12154. [Google Scholar] [CrossRef]
- Kim, J.; Cantor, A.B.; Orkin, S.H.; Wang, J. Use of in Vivo Biotinylation to Study Protein–Protein and Protein–DNA Interactions in Mouse Embryonic Stem Cells. Nat. Protoc. 2009, 4, 506–517. [Google Scholar] [CrossRef]
- Titeca, K.; Lemmens, I.; Tavernier, J.; Eyckerman, S. Discovering Cellular Protein-protein Interactions: Technological Strategies and Opportunities. Mass Spectrom. Rev. 2019, 38, 79–111. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T.; Kosako, H. Mass Spectrometry-Based Methods for Analysing the Mitochondrial Interactome in Mammalian Cells. J. Biochem. 2020, 167, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Lönn, P.; Landegren, U. Close Encounters—Probing Proximal Proteins in Live or Fixed Cells. Trends Biochem. Sci. 2017, 42, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Hung, V.; Lam, S.S.; Udeshi, N.D.; Svinkina, T.; Guzman, G.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic Mapping of Cytosol-Facing Outer Mitochondrial and ER Membranes in Living Human Cells by Proximity Biotinylation. eLife 2017, 6, e24463. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Udeshi, N.D.; Deerinck, T.J.; Svinkina, T.; Ellisman, M.H.; Carr, S.A.; Ting, A.Y. Proximity Biotinylation as a Method for Mapping Proteins Associated with MtDNA in Living Cells. Cell Chem. Biol. 2017, 24, 404–414. [Google Scholar] [CrossRef]
- Fazal, F.M.; Han, S.; Parker, K.R.; Kaewsapsak, P.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 2019, 178, 473–490.e26. [Google Scholar] [CrossRef]
- Williams, C.C.; Jan, C.H.; Weissman, J.S. Targeting and Plasticity of Mitochondrial Proteins Revealed by Proximity-Specific Ribosome Profiling. Science 2014, 346, 748–751. [Google Scholar] [CrossRef]
- Vardi-Oknin, D.; Arava, Y. Characterization of Factors Involved in Localized Translation Near Mitochondria by Ribosome-Proximity Labeling. Front. Cell Dev. Biol. 2019, 7, 305. [Google Scholar] [CrossRef]
- Yoo, C.M.; Rhee, H.W. APEX, a Master Key to Resolve Membrane Topology in Live Cells. Biochemistry 2020, 59, 250–259. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kang, M.G.; Park, J.S.; Lee, G.; Ting, A.Y.; Rhee, H.W. APEX Fingerprinting Reveals the Subcellular Localization of Proteins of Interest. Cell Rep. 2016, 15, 1837–1847. [Google Scholar] [CrossRef]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient Proximity Labeling in Living Cells and Organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–898. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bitsch, S.; Kubitz, L.; Schmitt, K.; Deweid, L.; Roehrig, A.; Barazzone, E.C.; Valerius, O.; Kolmar, H.; Béthune, J. UltraID: A Compact and Efficient Enzyme for Proximity-Dependent Biotinylation in Living Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Avendaño-Monsalve, M.C.; Ponce-Rojas, J.C.; Funes, S. From Cytosol to Mitochondria: The Beginning of a Protein Journey. Biol. Chem. 2020, 401, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Proteins: From Biogenesis to Functional Networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Becker, T.; Song, J.; Pfanner, N. Versatility of Preprotein Transfer from the Cytosol to Mitochondria. Trends Cell Biol. 2019, 29, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Geissler, A. Versatility of the Mitochondrial Protein Import Machinery. Nat. Rev. Mol. Cell Biol. 2001, 2, 339–349. [Google Scholar] [CrossRef]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial Protein Import: Common Principles and Physiological Networks. Biochim. Et Biophys. Acta—Mol. Cell Res. 2013, 1833, 274–285. [Google Scholar] [CrossRef]
- Tucker, K.; Park, E. Cryo-EM Structure of the Mitochondrial Protein-Import Channel TOM Complex at near-Atomic Resolution. Nat. Struct. Mol. Biol. 2019, 26, 1158–1166. [Google Scholar] [CrossRef]
- Araiso, Y.; Tsutsumi, A.; Qiu, J.; Imai, K.; Shiota, T.; Song, J.; Lindau, C.; Wenz, L.-S.; Sakaue, H.; Yunoki, K.; et al. Structure of the Mitochondrial Import Gate Reveals Distinct Preprotein Paths. Nature 2019, 575, 395–401. [Google Scholar] [CrossRef]
- Opaliński, Ł.; Song, J.; Priesnitz, C.; Wenz, L.-S.; Oeljeklaus, S.; Warscheid, B.; Pfanner, N.; Becker, T. Recruitment of Cytosolic J-Proteins by TOM Receptors Promotes Mitochondrial Protein Biogenesis. Cell Rep. 2018, 25, 2036–2043.e5. [Google Scholar] [CrossRef]
- Hansen, K.G.; Herrmann, J.M. Transport of Proteins into Mitochondria. Protein J. 2019, 38, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, H. Translational Regulation of Mitochondrial Biogenesis. Biochem. Soc. Trans. 2016, 44, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Lesnik, C.; Golani-Armon, A.; Arava, Y. Localized Translation near the Mitochondrial Outer Membrane: An Update. RNA Biol. 2015, 12, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Lesnik, C.; Cohen, Y.; Atir-Lande, A.; Schuldiner, M.; Arava, Y. OM14 Is a Mitochondrial Receptor for Cytosolic Ribosomes That Supports Co-Translational Import into Mitochondria. Nat. Commun. 2014, 5, 5711. [Google Scholar] [CrossRef] [PubMed]
- Quenault, T.; Lithgow, T.; Traven, A. PUF Proteins: Repression, Activation and MRNA Localization. Trends Cell Biol. 2011, 21, 104–112. [Google Scholar] [CrossRef]
- Saint-Georges, Y.; Garcia, M.; Delaveau, T.; Jourdren, L.; Le Crom, S.; Lemoine, S.; Tanty, V.; Devaux, F.; Jacq, C. Yeast Mitochondrial Biogenesis: A Role for the PUF RNA-Binding Protein Puf3p in MRNA Localization. PLoS ONE 2008, 3, e2293. [Google Scholar] [CrossRef]
- Devaux, F.; Lelandais, G.; Garcia, M.; Goussard, S.; Jacq, C. Posttranscriptional Control of Mitochondrial Biogenesis: Spatio-Temporal Regulation of the Protein Import Process. FEBS Lett. 2010, 584, 4273–4279. [Google Scholar] [CrossRef]
- Lapointe, C.P.; Stefely, J.A.; Jochem, A.; Hutchins, P.D.; Wilson, G.M.; Kwiecien, N.W.; Coon, J.J.; Wickens, M.; Pagliarini, D.J. Multi-Omics Reveal Specific Targets of the RNA-Binding Protein Puf3p and Its Orchestration of Mitochondrial Biogenesis. Cell Syst. 2018, 6, 125–135.e6. [Google Scholar] [CrossRef]
- Eliyahu, E.; Pnueli, L.; Melamed, D.; Scherrer, T.; Gerber, A.P.; Pines, O.; Rapaport, D.; Arava, Y. Tom20 Mediates Localization of MRNAs to Mitochondria in a Translation-Dependent Manner. Mol. Cell. Biol. 2010, 30, 284–294. [Google Scholar] [CrossRef]
- Gehrke, S.; Wu, Z.; Klinkenberg, M.; Sun, Y.; Auburger, G.; Guo, S.; Lu, B. PINK1 and Parkin Control Localized Translation of Respiratory Chain Component MRNAs on Mitochondria Outer Membrane. Cell Metab. 2015, 21, 95–108. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.H.; Liu, Y.; Chen, Y.; Sun, N.; Gucek, M.; Zhang, F.; Xu, H. PINK1 Inhibits Local Protein Synthesis to Limit Transmission of Deleterious Mitochondrial DNA Mutations. Mol. Cell 2019, 73, 1127–1137.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Gucek, M.; Xu, H. The Mitochondrial Outer Membrane Protein MDI Promotes Local Protein Synthesis and MtDNA Replication. EMBO J. 2016, 35, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Kalvakuri, S.; Bodmer, R.; Cox, R.T. Clueless, a Protein Required for Mitochondrial Function, Interacts with the PINK1-Parkin Complex in Drosophila. Dis. Model. Mech. 2015, 8, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Cox, R.T. Clueless Is a Conserved Ribonucleoprotein That Binds the Ribosome at the Mitochondrial Outer Membrane. Biol. Open 2016, 5, 195–203. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Ni, L.; Weiner, H. A Co-Translational Model to Explain the in Vivo Import of Proteins into HeLa Cell Mitochondria. Biochem. J. 2004, 382, 385–392. [Google Scholar] [CrossRef]
- Sylvestre, J.; Margeot, A.; Jacq, C.; Dujardin, G.; Corral-Debrinski, M. The Role of the 3′ Untranslated Region in MRNA Sorting to the Vicinity of Mitochondria Is Conserved from Yeast to Human Cells. Mol. Biol. Cell 2003, 14, 3848–3856. [Google Scholar] [CrossRef]
- Matsumoto, S.; Uchiumi, T.; Saito, T.; Yagi, M.; Takazaki, S.; Kanki, T.; Kang, D. Localization of MRNAs Encoding Human Mitochondrial Oxidative Phosphorylation Proteins. Mitochondrion 2012, 12, 391–398. [Google Scholar] [CrossRef]
- Jan, C.H.; Williams, C.C.; Weissman, J.S. Principles of ER Cotranslational Translocation Revealed by Proximity-Specific Ribosome Profiling. Science 2014, 346, 1257521. [Google Scholar] [CrossRef]
- Grevel, A.; Pfanner, N.; Becker, T. Coupling of Import and Assembly Pathways in Mitochondrial Protein Biogenesis. Biol. Chem. 2019, 401, 117–129. [Google Scholar] [CrossRef]
- Priesnitz, C.; Becker, T. Pathways to Balance Mitochondrial Translation and Protein Import. Genes Dev. 2018, 401, 1285–1296. [Google Scholar] [CrossRef]
- Fujiki, M.; Verner, K. Coupling of Cytosolic Protein Synthesis and Mitochondrial Protein Import in Yeast. Evidence for Cotranslational Import in Vivo. J. Biol. Chem. 1993, 268, 1914–1920. [Google Scholar] [CrossRef]
- Baker, D.J.; Beddell, C.R.; Champness, J.N.; Goodford, P.J.; Norrington, F.E.A.; Smith, D.R.; Stammers, D.K. The Binding of Trimethoprim to Bacterial Dihydrofolate Reductase. FEBS Lett. 1981, 126, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Gabrovsek, L.; Collins, K.B.; Aggarwal, S.; Saunders, L.M.; Lau, H.T.; Suh, D.; Sancak, Y.; Trapnell, C.; Ong, S.E.; Smith, F.D.; et al. A-Kinase-Anchoring Protein 1 (DAKAP1)-Based Signaling Complexes Coordinate Local Protein Synthesis at the Mitochondrial Surface. J. Biol. Chem. 2020, 295, 10749–10765. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, M.L.L.; Hughes, L.E.; Luke, G.; Mendoza, H.; Ten Dam, E.; Gani, D.; Ryan, M.D. The “cleavage” Activities of Foot-and-Mouth Disease Virus 2A Site-Directed Mutants and Naturally Occurring “2A-like” Sequences. J. Gen. Virol. 2001, 82, 1027–1041. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An Updated Inventory of Mammalian Mitochondrial Proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Chan, X.C.Y.; Black, C.M.; Lin, A.J.; Ping, P.; Lau, E. Mitochondrial Protein Turnover: Methods to Measure Turnover Rates on a Large Scale. J. Mol. Cell. Cardiol. 2015, 78, 54–61. [Google Scholar] [CrossRef]
- Li, J.; Cai, Z.; Vaites, L.P.; Shen, N.; Mitchell, D.C.; Huttlin, E.L.; Paulo, J.A.; Harry, B.L.; Gygi, S.P. Proteome-Wide Mapping of Short-Lived Proteins in Human Cells. Mol. Cell 2021, 81, 4722–4735.e5. [Google Scholar] [CrossRef]
- Mathieson, T.; Franken, H.; Kosinski, J.; Kurzawa, N.; Zinn, N.; Sweetman, G.; Poeckel, D.; Ratnu, V.S.; Schramm, M.; Becher, I.; et al. Systematic Analysis of Protein Turnover in Primary Cells. Nat. Commun. 2018, 9, 689. [Google Scholar] [CrossRef]
- Richter-Dennerlein, R.; Oeljeklaus, S.; Lorenzi, I.; Ronsör, C.; Bareth, B.; Schendzielorz, A.B.; Wang, C.; Warscheid, B.; Rehling, P.; Dennerlein, S. Mitochondrial Protein Synthesis Adapts to Influx of Nuclear-Encoded Protein. Cell 2016, 167, 471–483.e10. [Google Scholar] [CrossRef]
- Couvillion, M.T.; Soto, I.C.; Shipkovenska, G.; Churchman, L.S. Synchronized Mitochondrial and Cytosolic Translation Programs. Nature 2016, 533, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.; Haupt, A.; Tucker, A.; Grancharova, T.; Arakaki, J.; Fuqua, M.A.; Nelson, A.; Hookway, C.; Ludmann, S.A.; Mueller, I.A.; et al. Systematic Gene Tagging Using CRISPR/Cas9 in Human Stem Cells to Illuminate Cell Organization. Mol. Biol. Cell 2017, 28, 2854–2874. [Google Scholar] [CrossRef] [PubMed]
- Tytgat, H.L.P.; Schoofs, G.; Driesen, M.; Proost, P.; Van Damme, E.J.M.; Vanderleyden, J.; Lebeer, S. Endogenous Biotin-Binding Proteins: An Overlooked Factor Causing False Positives in Streptavidin-Based Protein Detection. Microb. Biotechnol. 2015, 8, 164–168. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, O.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Ruth Vaseghi, H.; Qian, L.; Liu, J. Systematic Comparison of 2A Peptides for Cloning Multi-Genes in a Polycistronic Vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef]
- Gold, V.A.; Chroscicki, P.; Bragoszewski, P.; Chacinska, A. Visualization of Cytosolic Ribosomes on the Surface of Mitochondria by Electron Cryo-Tomography. EMBO Rep. 2017, 18, 1786–1800. [Google Scholar] [CrossRef]
- Pla-Martín, D.; Schatton, D.; Wiederstein, J.L.; Marx, M.; Khiati, S.; Krüger, M.; Rugarli, E.I. CLUH Granules Coordinate Translation of Mitochondrial Proteins with MTORC1 Signaling and Mitophagy. EMBO J. 2020, 39, e102731. [Google Scholar] [CrossRef]
- Youn, D.Y.; Xiaoli, A.M.; Pessin, J.E.; Yang, F. Regulation of Metabolism by the Mediator Complex. Biophys. Rep. 2016, 2, 69–77. [Google Scholar] [CrossRef]
- Yang, F.; Vought, B.W.; Satterlee, J.S.; Walker, A.K.; Jim Sun, Z.Y.; Watts, J.L.; DeBeaumont, R.; Mako Saito, R.; Hyberts, S.G.; Yang, S.; et al. An ARC/Mediator Subunit Required for SREBP Control of Cholesterol and Lipid Homeostasis. Nature 2006, 442, 700–704. [Google Scholar] [CrossRef]
- Tuttle, L.M.; Pacheco, D.; Warfield, L.; Wilburn, D.B.; Hahn, S.; Klevit, R.E. Mediator Subunit Med15 Dictates the Conserved “Fuzzy” Binding Mechanism of Yeast Transcription Activators Gal4 and Gcn4. Nat. Commun. 2021, 12, 2220. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Shi, Y.; Walz, T.; Tong, L. Structural Insights into the Human Pre-MRNA 3′-End Processing Machinery. Mol. Cell 2020, 77, 800–809.e6. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Delaleau, M.; Borden, K.L.B. Nuclear EIF4E Stimulates 3′-End Cleavage of Target RNAs. Cell Rep. 2019, 27, 1397–1408.e4. [Google Scholar] [CrossRef] [PubMed]
- Hirawake-Mogi, H.; Thanh Nhan, N.T.; Okuwaki, M. G-Patch Domain-Containing Protein 4 Localizes to Both the Nucleoli and Cajal Bodies and Regulates Cell Growth and Nucleolar Structure. Biochem. Biophys. Res. Commun. 2021, 559, 99–105. [Google Scholar] [CrossRef]
- Morgenstern, M.; Peikert, C.D.; Lübbert, P.; Suppanz, I.; Klemm, C.; Alka, O.; Steiert, C.; Naumenko, N.; Schendzielorz, A.; Melchionda, L.; et al. Quantitative High-Confidence Human Mitochondrial Proteome and Its Dynamics in Cellular Context. Cell Metab. 2021, 33, 2464–2483.e18. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Haley, J.D. Pulse-Chase SILAC-Based Analyses Reveal Selective Oversynthesis and Rapid Turnover of Mitochondrial Protein Components of Respiratory Complexes. J. Biol. Chem. 2020, 295, 2544–2554. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Ostermeyer-Fay, A.G.; Haley, J.D.; Garcia-Diaz, M. Kinetics and Mechanism of Mammalian Mitochondrial Ribosome Assembly. Cell Rep. 2018, 22, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Tigges, J.; Weighardt, H.; Wolff, S.; Götz, C.; Förster, I.; Kohne, Z.; Huebenthal, U.; Merk, H.F.; Abel, J.; Haarmann-Stemmann, T.; et al. Aryl Hydrocarbon Receptor Repressor (AhRR) Function Revisited: Repression of CYP1 Activity in Human Skin Fibroblasts Is Not Related to AhRR Expression. J. Invest. Dermatol. 2013, 133, 87–96. [Google Scholar] [CrossRef]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive Guide Selection for CRISPR/Cas9 Genome Editing Experiments and Screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef]
- Vandemoortele, G.; De Sutter, D.; Eyckerman, S. Robust Generation of Knock-in Cell Lines Using CRISPR-Cas9 and RAAV-Assisted Repair Template Delivery. Bio-Protocol 2017, 7, e2211. [Google Scholar] [CrossRef]
- Czlapinski, J.L.; Schelle, M.W.; Miller, L.W.; Laughlin, S.T.; Kohler, J.J.; Cornish, V.W.; Bertozzi, C.R. Conditional Glycosylate in Eukaryotic Cells Using a Biocompatible Chemical Inducer of Dimerization. J. Am. Chem. Soc. 2008, 130, 13186–13187. [Google Scholar] [CrossRef]
- Le Sage, V.; Cinti, A.; Mouland, A.J. Proximity-Dependent Biotinylation for Identification of Interacting Proteins. Curr. Protoc. Cell Biol. 2016, 73, 17.19.1–17.19.12. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteomics 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; He, L.; Ma, B. A Combinatorial Approach to the Peptide Feature Matching Problem for Label-Free Quantification. Bioinformatics 2013, 29, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Rana, S.; Turkes, E.; Ostenforf, B.; Grioni, A.; Lewis, M. Publication-Ready Volcano Plots with Enhanced Colouring and Labeling; Bioconductor: Boston, MA, USA, 2023. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Katrukha, E. Ekatrukha/ComDet: ComDet 0.5.3; Zenodo: Geneva, Switzerland, 2020. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meurant, S.; Mauclet, L.; Dieu, M.; Arnould, T.; Eyckerman, S.; Renard, P. Endogenous TOM20 Proximity Labeling: A Swiss-Knife for the Study of Mitochondrial Proteins in Human Cells. Int. J. Mol. Sci. 2023, 24, 9604. https://doi.org/10.3390/ijms24119604
Meurant S, Mauclet L, Dieu M, Arnould T, Eyckerman S, Renard P. Endogenous TOM20 Proximity Labeling: A Swiss-Knife for the Study of Mitochondrial Proteins in Human Cells. International Journal of Molecular Sciences. 2023; 24(11):9604. https://doi.org/10.3390/ijms24119604
Chicago/Turabian StyleMeurant, Sébastien, Lorris Mauclet, Marc Dieu, Thierry Arnould, Sven Eyckerman, and Patricia Renard. 2023. "Endogenous TOM20 Proximity Labeling: A Swiss-Knife for the Study of Mitochondrial Proteins in Human Cells" International Journal of Molecular Sciences 24, no. 11: 9604. https://doi.org/10.3390/ijms24119604