
In Silico Study of Novel Cyclodextrin Inclusion Complexes of Polycaprolactone and Its Correlation with Skin Regeneration

 , , , and
, , , and
Abstract
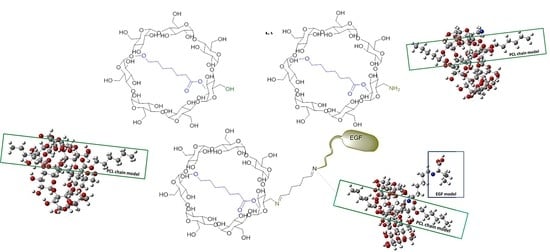
:
1. Introduction
2. Results and Discussion
2.1. Molecular Parameters
2.2. Vibrational Study
2.3. Molecular Reactivity
2.4. Molecular Electrostatic Potential Maps
3. Materials and Methods
3.1. Optimization
3.2. Interaction Energy Calculations
3.3. Vibrational Analysis
3.4. Thermochemical Parameters
3.5. Reactivity Parameters
3.6. Reactivity Descriptors
3.7. Biological Activity Predictions
3.8. Toxicological, Physicochemical and Metabolic Properties Prediction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saleh, T.S.; Badawi, A.K.; Salama, R.S.; Mostafa, M.M.M. Design and Development of Novel Composites Containing Nickel Ferrites Supported on Activated Carbon Derived from Agricultural Wastes and Its Application in Water Remediation. Materials 2023, 16, 2170. [Google Scholar] [CrossRef] [PubMed]
- Alasri, T.M.; Ali, S.L.; Salama, R.S.; Alshorifi, F.T. Band-Structure Engineering of TiO2 Photocatalyst by AuSe Quantum Dots for Efficient Degradation of Malachite Green and Phenol. J. Inorg. Organomet. Polym. Mater. 2023, 1–12. [Google Scholar] [CrossRef]
- Baaloudj, O.; Nasrallah, N.; Kenfoud, H.; Bourkeb, K.W.; Badawi, A.K. Polyaniline/Bi12TiO20 Hybrid System for Cefixime Removal by Combining Adsorption and Photocatalytic Degradation. Chemengineering 2023, 7, 4. [Google Scholar] [CrossRef]
- Kane, A.; Assadi, A.A.; El Jery, A.; Badawi, A.K.; Kenfoud, H.; Baaloudj, O.; Assadi, A.A. Advanced Photocatalytic Treatment of Wastewater Using Immobilized Titanium Dioxide as a Photocatalyst in a Pilot-Scale Reactor: Process Intensification. Materials 2022, 15, 4547. [Google Scholar] [CrossRef]
- Mostafa, M.M.M.; Alshehri, A.A.; Salama, R.S. High performance of supercapacitor based on alumina nanoparticles derived from Coca-Cola cans. J. Energy Storage 2023, 64, 107168. [Google Scholar] [CrossRef]
- Tu, Z.; Zhong, Y.; Hu, H.; Shao, D.; Haag, R.; Schirner, M.; Lee, J.; Sullenger, B.; Leong, K.W. Design of therapeutic biomaterials to control inflammation. Nat. Rev. Mater. 2022, 7, 557–574. [Google Scholar] [CrossRef]
- Joseph, X.; Akhil, V.; Arathi, A.; Mohanan, P. Nanobiomaterials in support of drug delivery related issues. Mater. Sci. Eng. B 2022, 279, 115680. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, H.; Yi, K.; Lv, S.; Hu, H.; Li, M.; Tao, Y. Applications of Nanobiomaterials in the Therapy and Imaging of Acute Liver Failure. Nano-Micro Lett. 2020, 13, 25. [Google Scholar] [CrossRef]
- Rodrigo-Navarro, A.; Sankaran, S.; Dalby, M.J.; del Campo, A.; Salmeron-Sanchez, M. Engineered living biomaterials. Nat. Rev. Mater. 2021, 6, 1175–1190. [Google Scholar] [CrossRef]
- Dulnik, J.; Sajkiewicz, P. Crosslinking of Gelatin in Bicomponent Electrospun Fibers. Materials 2021, 14, 3391. [Google Scholar] [CrossRef]
- Al-Kaabi, W.J.; Albukhaty, S.; Al-Fartosy, A.J.M.; Al-Karagoly, H.K.; Al-Musawi, S.; Sulaiman, G.M.; Dewir, Y.H.; Alwahibi, M.S.; Soliman, D.A. Development of Inula graveolens (L.) Plant Extract Electrospun/Polycaprolactone Nanofibers: A Novel Material for Biomedical Application. Appl. Sci. 2021, 11, 828. [Google Scholar] [CrossRef]
- Lim, W.L.; Chowdhury, S.R.; Ng, M.H.; Law, J.X. Physicochemical Properties and Biocompatibility of Electrospun Polycaprolactone/Gelatin Nanofibers. Int. J. Environ. Res. Public Health 2021, 18, 4764. [Google Scholar] [CrossRef] [PubMed]
- Malikmammadov, E.; Tanir, T.E.; Kiziltay, A.; Hasirci, V.; Hasirci, N. PCL and PCL-based materials in biomedical applications. J. Biomater. Sci. Polym. Ed. 2018, 29, 863–893. [Google Scholar] [CrossRef]
- Ghobeira, R.; Philips, C.; Liefooghe, L.; Verdonck, M.; Asadian, M.; Cools, P.; Declercq, H.; De Vos, W.H.; De Geyter, N.; Morent, R. Synergetic effect of electrospun PCL fiber size, orientation and plasma-modified surface chemistry on stem cell behavior. Appl. Surf. Sci. 2019, 485, 204–221. [Google Scholar] [CrossRef]
- Seddighian, A.; Ganji, F.; Baghaban-Eslaminejad, M.; Bagheri, F. Electrospun PCL scaffold modified with chitosan nanoparticles for enhanced bone regeneration. Prog. Biomater. 2021, 10, 65–76. [Google Scholar] [CrossRef]
- Choi, E.; Bae, S.; Kim, D.; Yang, G.H.; Lee, K.; You, H.-J.; Kang, H.J.; Gwak, S.-J.; An, S.; Jeon, H. Characterization and intracellular mechanism of electrospun poly (ε-caprolactone) (PCL) fibers incorporated with bone-dECM powder as a potential membrane for guided bone regeneration. J. Ind. Eng. Chem. 2021, 94, 282–291. [Google Scholar] [CrossRef]
- Jiang, C.; Wang, K.; Liu, Y.; Zhang, C.; Wang, B. Using Wet Electrospun PCL/Gelatin/CNT Yarns to Fabricate Textile-Based Scaffolds for Vascular Tissue Engineering. ACS Biomater. Sci. Eng. 2021, 7, 2627–2637. [Google Scholar] [CrossRef]
- Seema, K.; Mamba, B.; Njuguna, J.; Bakhtizin, R.; Mishra, A. Removal of lead (II) from aqeouos waste using (CD-PCL-TiO2) bio-nanocomposites. Int. J. Biol. Macromol. 2018, 109, 136–142. [Google Scholar] [CrossRef]
- Zhan, J.; Singh, A.; Zhang, Z.; Huang, L.; Elisseeff, J.H. Multifunctional aliphatic polyester nanofibers for tissue engineering. Biomatter 2012, 2, 202–212. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Nishiyama, T.; Okada, M.; Kamachi, M.; Harada, A. Complex Formation of Poly(ε-caprolactone) with Cyclodextrins. Macromolecules 2000, 33, 4472–4477. [Google Scholar] [CrossRef]
- Lindner, K.; Saenger, W. Crystal and molecular structure of cyclohepta-amylose dodecahydrate. Carbohydr. Res. 1982, 99, 103–115. [Google Scholar] [CrossRef]
- Ermer, O.; Dunitz, J.D.; Bernal, I. The structures of medium-ring compounds. XVIII. X-ray and neutron diffraction analysis of cyclodecane-1,6-trans-diol. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1973, 29, 2278–2285. [Google Scholar] [CrossRef]
- Steiner, T.; Koellner, G. Crystalline.beta.-Cyclodextrin Hydrate at Various Humidities: Fast, Continuous, and Reversible Dehydration Studied by X-ray Diffraction. J. Am. Chem. Soc. 1994, 116, 5122–5128. [Google Scholar] [CrossRef]
- Alvarez-Parrilla, E.; de la Rosa, L.A.; Rodrigo-García, J.; Escobedo-González, R.; Mercado-Mercado, G.; Moyers-Montoya, E.; Vázquez-Flores, A.; González-Aguilar, G.A. Dual effect of β-cyclodextrin (β-CD) on the inhibition of apple polyphenol oxidase by 4-hexylresorcinol (HR) and methyl jasmonate (MJ). Food Chem. 2007, 101, 1346–1356. [Google Scholar] [CrossRef]
- Silva, D.A.; Xavier, M.J.; Dutra, J.D.L.; Gimenez, I.F.; Freire, R.O.; da Costa, N.B. Prediction of correct intermolecular interactions in host-guest systems involving cyclodextrins. J. Mol. Struct. 2020, 1205, 127517. [Google Scholar] [CrossRef]
- Zhao, B.; Jiang, L.; Jia, Q. Advances in cyclodextrin polymers adsorbents for separation and enrichment: Classification, mechanism and applications. Chin. Chem. Lett. 2022, 33, 11–21. [Google Scholar] [CrossRef]
- Liu, Z.; Ye, L.; Xi, J.; Wang, J.; Feng, Z.-G. Cyclodextrin polymers: Structure, synthesis, and use as drug carriers. Prog. Polym. Sci. 2021, 118, 101408. [Google Scholar] [CrossRef]
- Li, X.-S.; Liu, L.; Mu, T.-W.; Guo, Q.-X. A Systematic Quantum Chemistry Study on Cyclodextrins. Monatsh. Chem. 2000, 131, 849–855. [Google Scholar] [CrossRef]
- Nicolás, I.; Vilchis, M.; Aragón, N.; Miranda, R.; Hojer, G.; Castro, M. Theoretical study of the structure and antimicrobial activity of horminone. Int. J. Quantum Chem. 2003, 93, 411–421. [Google Scholar] [CrossRef]
- Escobedo-González, R.G.; Bahena, L.; Arias Tellez, J.L.; Hinojosa Torres, J.; Ruvalcaba, R.M.; Aceves-Hernández, J.M. Characterization and comparison of perezone with some analogues. Experimental and theoretical study. J. Mol. Struct. 2015, 1097, 98–105. [Google Scholar] [CrossRef]
- Escobedo-González, R.; Méndez-Albores, A.; Villarreal-Barajas, T.; Aceves-Hernández, J.M.; Miranda-Ruvalcaba, R.; Nicolás-Vázquez, I. A Theoretical Study of 8-Chloro-9-Hydroxy-Aflatoxin B1, the Conversion Product of Aflatoxin B1 by Neutral Electrolyzed Water. Toxins 2016, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Escobedo-González, R.; Vargas-Requena, C.L.; Moyers-Montoya, E.; Aceves-Hernández, J.M.; Nicolás-Vázquez, M.I.; Miranda-Ruvalcaba, R. In silico Study of the Pharmacologic Properties and Cytotoxicity Pathways in Cancer Cells of Various Indolylquinone Analogues of Perezone. Molecules 2017, 22, 1060. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Albores, A.; Escobedo-González, R.; Aceves-Hernández, J.M.; García-Casillas, P.; Nicolás-Vázquez, M.I.; Miranda-Ruvalcaba, R. A Theoretical Study of the Adsorption Process of B-aflatoxins Using Pyracantha koidzumii (Hayata) Rehder Biomasses. Toxins 2020, 12, 283. [Google Scholar] [CrossRef] [PubMed]
- Escobedo-González, R.G.; Pérez Martínez, H.; Nicolás-Vázquez, M.I.; Martínez, J.; Gómez, G.; Serrano, J.N.; Carranza Téllez, V.; Vargas-Requena, C.L.; Miranda Ruvalcaba, R. Green Production of Indolylquinones, Derivatives of Perezone, and Related Molecules, Promising Antineoplastic Compounds. J. Chem. 2016, 2016, 3870529. [Google Scholar] [CrossRef]
- Martínez, J.; Rodríguez, M.H.; Escobedo-González, R.; Nicolás-Vázquez, M.I.; Saavedra-Leos, Z.; Ruvalcaba, R.M. Computational Characterization of Perezone, Isoperezone and their Sulfur-Derivatives: Anti-inflammatory Activity. Chemistryselect 2019, 4, 13333–13346. [Google Scholar] [CrossRef]
- Nicolás-Vázquez, I.; Méndez-Albores, A.; Moreno-Martínez, E.; Miranda, R.; Castro, M. Role of Lactone Ring in Structural, Electronic, and Reactivity Properties of Aflatoxin B1: A Theoretical Study. Arch. Environ. Contam. Toxicol. 2010, 59, 393–406. [Google Scholar] [CrossRef]
- Vazquez-Flores, A.A.; Martinez-Gonzalez, A.I.; Alvarez-Parrilla, E.; Díaz-Sánchez, G.; De La Rosa, L.A.; González-Aguilar, G.A.; Aguilar, C.N. Proanthocyanidins with a Low Degree of Polymerization are Good Inhibitors of Digestive Enzymes Because of their Ability to form Specific Interactions: A Hypothesis. J. Food Sci. 2018, 83, 2895–2902. [Google Scholar] [CrossRef]
- Makkar, P.; Ghosh, N.N. A review on the use of DFT for the prediction of the properties of nanomaterials. RSC Adv. 2021, 11, 27897–27924. [Google Scholar] [CrossRef]
- Schleder, G.R.; Padilha, A.C.M.; Acosta, C.M.; Costa, M.; Fazzio, A. From DFT to machine learning: Recent approaches to materials science–a review. J. Phys. Mater. 2019, 2, 032001. [Google Scholar] [CrossRef]
- Zhao, J.; Wu, L.; Zhan, C.; Shao, Q.; Guo, Z.; Zhang, L. Overview of polymer nanocomposites: Computer simulation understanding of physical properties. Polymer 2017, 133, 272–287. [Google Scholar] [CrossRef]
- Sathish, M.; Rajasekaran, L.; Shanthi, D.; Kanagathara, N.; Sarala, S.; Muthu, S. Spectroscopic (FT-IR, FT-Raman, UV-Vis) molecular structure, electronic, molecular docking, and thermodynamic investigations of indole-3-carboxylic acid by DFT method. J. Mol. Struct. 2021, 1234, 130182. [Google Scholar] [CrossRef]
- El Foujji, L.; El Bourakadi, K.; Mekhzoum, M.E.M.; Essassi, E.M.; Boeré, R.T.; Qaiss, A.E.K.; Bouhfid, R. Synthesis, crystal structure, spectroscopic, thermal properties and DFT calculation of a novel ethyl 2-(2-(thiazol-4-yl)-1H-benzimidazol-1-yl)acetate. J. Mol. Struct. 2020, 1209, 127939. [Google Scholar] [CrossRef]
- Garrain, P.A.; Costa, D.; Marcus, P. Biomaterial-Biomolecule Interaction: DFT-D Study of Glycine Adsorption on Cr2O3. J. Phys. Chem. C 2011, 115, 719–727. [Google Scholar] [CrossRef]
- López-Ramírez, E.; Chapa-González, C.; Perez, C.A.M.; Escobedo-González, R.; Vázquez, M.I.N.; Medellín-Rodríguez, F.; García-Casillas, P.E. Citrulline Malate transdermal delivery through integrating into polyvinyl alcohol (PVA) nanofibers. J. Drug Deliv. Sci. Technol. 2021, 64, 102630. [Google Scholar] [CrossRef]
- Alkan, F.; Aikens, C.M. TD-DFT and TD-DFTB Investigation of the Optical Properties and Electronic Structure of Silver Nanorods and Nanorod Dimers. J. Phys. Chem. C 2018, 122, 23639–23650. [Google Scholar] [CrossRef]
- Qin, W.; Li, X.; Bian, W.-W.; Fan, X.-J.; Qi, J.-Y. Density functional theory calculations and molecular dynamics simulations of the adsorption of biomolecules on graphene surfaces. Biomaterials 2010, 31, 1007–1016. [Google Scholar] [CrossRef]
- Moyers-Montoya, E.; García-Casillas, P.; Vargas-Requena, C.; Escobedo-González, R.; Martel-Estrada, S.-A.; Martínez-Pérez, C.A. Polycaprolactone/Amino-β-Cyclodextrin Inclusion Complex Prepared by an Electrospinning Technique. Polymers 2016, 8, 395. [Google Scholar] [CrossRef]
- Moyers-Montoya, E.D.; Escobedo-González, R.G.; Vargas-Requena, C.L.; Garcia-Casillas, P.E.; Martínez-Pérez, C.A. Epithelial Growth Factor-Anchored on Polycaprolactone/6-deoxy-6-amino-β-cyclodextrin Nanofibers: In Vitro and In Vivo Evaluation. Polymers 2021, 13, 1303. [Google Scholar] [CrossRef]
- Steffens, L.; Morás, A.M.; Arantes, P.R.; Masterson, K.; Cao, Z.; Nugent, M.; Moura, D.J. Electrospun PVA-Dacarbazine nanofibers as a novel nano brain-implant for treatment of glioblastoma: In silico and in vitro characterization. Eur. J. Pharm. Sci. 2020, 143, 105183. [Google Scholar] [CrossRef]
- Chatani, Y.; Shimane, Y.; Inagaki, T.; Ijitsu, T.; Yukinari, T.; Shikuma, H. Structural study on syndiotactic polystyrene: 2. Crystal structure of molecular compound with toluene. Polymer 1993, 34, 1620–1624. [Google Scholar] [CrossRef]
- Lindner, K.; Saenger, W. Topography of cyclodextrin inclusion complexes. XVI. Cyclic system of hydrogen bonds: Structure of α-cyclodextrin hexahydrate, form (II): Comparison with form (I). Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1982, 38, 203–210. [Google Scholar] [CrossRef]
- Ben Salah, A.M.; Naïli, H.; Mhiri, T.; Bataille, T. Synthesis and crystal structure of a chiral aromatic amine chloride salt (C8H12N)Cl. Crystallogr. Rep. 2015, 60, 1053–1057. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond: In Structural Chemistry and Biology; Oxford University, Ed.; International Union of Crystal: New York, NY, USA, 2001; Volume 9. [Google Scholar]
- Jayyinunnisya, H.; Solihat, L.S.; Sugimori, K.; Kawaguchi, K.; Nagao, H. Theoretical Study of Complex Aspirin and Hydroxypropyl-β-cyclodextrin in Solvent Phase. J. Phys. Conf. Ser. 2022, 2207, 012022. [Google Scholar] [CrossRef]
- Fragoso-Medina, A.J.; Escobedo-González, R.G.; Nicolás-Vázquez, M.I.; Arroyo-Razo, G.A.; Noguez-Córdova, M.O.; Miranda-Ruvalcaba, R. A DFT Study of the Geometrical, Spectroscopical and Reactivity Properties of Diindolylmethane-Phenylboronic Acid Hybrids. Molecules 2017, 22, 1744. [Google Scholar] [CrossRef] [PubMed]
- Kurt, M. An experimental and theoretical study of molecular structure and vibrational spectra of pentafluorophenylboronic acid molecule by density functional theory and ab initio Hartree Fock calculations. J. Mol. Struct. 2008, 874, 159–169. [Google Scholar] [CrossRef]
- Mohamed, A.; Finkenstadt, V.L.; Gordon, S.H.; Biresaw, G.; Palmquist, D.E.; Rayas-Duarte, P. Thermal properties of PCL/gluten bioblends characterized by TGA, DSC, SEM, and infrared-PAS. J. Appl. Polym. Sci. 2008, 110, 3256–3266. [Google Scholar] [CrossRef]
- Elzein, T.; Nasser-Eddine, M.; Delaite, C.; Bistac, S.; Dumas, P. FTIR study of polycaprolactone chain organization at interfaces. J. Colloid Interface Sci. 2004, 273, 381–387. [Google Scholar] [CrossRef]
- Stewart, S.; Fredericks, P. Surface-enhanced Raman spectroscopy of amino acids adsorbed on an electrochemically prepared silver surface. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1999, 55, 1641–1660. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J.; Bryce, D.L. Spectrometric Identification of Organic Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Egyed, O. Spectroscopic studies on β-cyclodextrin. Vib. Spectrosc. 1990, 1, 225–227. [Google Scholar] [CrossRef]
- Iwasaki, A.; Fujii, A.; Watanabe, T.; Ebata, T.; Mikami, N. Infrared Spectroscopy of Hydrogen-Bonded Phenol−Amine Clusters in Supersonic Jets. J. Phys. Chem. 1996, 100, 16053–16057. [Google Scholar] [CrossRef]
- Rygula, A.; Majzner, K.; Marzec, K.M.; Kaczor, A.; Pilarczyk, M.; Baranska, M. Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 2013, 44, 1061–1076. [Google Scholar] [CrossRef]
- Lima, J.; Freire, P.T.C.; Lima, R.J.C.; Moreno, A.J.D.; Filho, J.M.; Melo, F.E.A. Raman scattering ofL-valine crystals. J. Raman Spectrosc. 2005, 36, 1076–1081. [Google Scholar] [CrossRef]
- Lu, G.-H.; Wang, C.; Guo, X.-L. Prediction of Toxicity of Phenols and Anilines to Algae by Quantitative Structure-activity Relationship. Biomed. Environ. Sci. 2008, 21, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Hepokur, C.; Günsel, A.; Yarasir, M.N.; Bilgiçli, A.T.; Tüzün, B.; Tüzün, G.; Yaylim, I. Novel type ketone-substituted metallophthalocyanines: Synthesis, spectral, structural, computational and anticancer studies. RSC Adv. 2017, 7, 56296–56305. [Google Scholar] [CrossRef]
- Al-Sehemi, A.; Irfan, A.; Alrumman, S.A.; Hesham, A.E. Antibacterial activities, DFT and QSAR studies of quinazolinone compounds. Bull. Chem. Soc. Ethiop. 2016, 30, 307–316. [Google Scholar] [CrossRef]
- Gece, G.; Bilgiç, S. Quantum chemical study of some cyclic nitrogen compounds as corrosion inhibitors of steel in NaCl media. Corros. Sci. 2009, 51, 1876–1878. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 2008, 68, 3801–3807. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Sánchez-Bojorge, N.-A.; Flores-Holguín, N.; Glossman-Mitnik, D.; Rodríguez-Valdez, L.M. Computational note on the chemical reactivity of pyrrole derivatives. J. Mol. Struct. THEOCHEM 2009, 912, 119–120. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Giri, S.; Duley, S. Update 2 of: Electrophilicity Index. Chem. Rev. 2011, 111, PR43–PR75. [Google Scholar] [CrossRef]
- De Vleeschouwer, F.; Van Speybroeck, V.; Waroquier, M.; Geerlings, P.; De Proft, F. Electrophilicity and Nucleophilicity Index for Radicals. Org. Lett. 2007, 9, 2721–2724. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, U.; Padmanabhan, J.; Parthasarathi, R.; Subramanian, V.; Chattaraj, P. Toxicity analysis of polychlorinated dibenzofurans through global and local electrophilicities. J. Mol. Struct. THEOCHEM 2006, 758, 119–125. [Google Scholar] [CrossRef]
- Gersch, M.; Kreuzer, J.; Sieber, S.A. Electrophilic natural products and their biological targets. Nat. Prod. Rep. 2012, 29, 659–682. [Google Scholar] [CrossRef] [PubMed]
- Thanikaivelan, P.; Subramanian, V.; Rao, J.R.; Nair, B.U. Application of quantum chemical descriptor in quantitative structure activity and structure property relationship. Chem. Phys. Lett. 2000, 323, 59–70. [Google Scholar] [CrossRef]
- Kumar, A.; Mohan, C.; Mishra, P. Molecular electrostatic potential and field as descriptors of hydrogen bonding and molecular activity. Effects of hybridization displacement charge. J. Mol. Struct. THEOCHEM 1996, 361, 135–144. [Google Scholar] [CrossRef]
- Politzer, P.; Laurence, P.R.; Jayasuriya, K. Molecular electrostatic potentials: An effective tool for the elucidation of biochemical phenomena. Environ. Health Perspect. 1985, 61, 191–202. [Google Scholar] [CrossRef]
- Cisneros, G.I.V.; Vásquez-Pérez, J.M.; Cruz-Borbolla, J.; Gómez-Castro, C.Z.; Nicolás-Vázquez, M.I.; Ruvalcaba, R.M. Theoretical study: Electronic structure and receptor interaction of four type bis-1,4-dihydropyridine molecules. Comput. Theor. Chem. 2018, 1123, 102–110. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the Biological Activity Spectra of Organic Compounds Using the PASS Online Web Resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Yang, H.; Luo, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR. Available online: http://lmmd.ecust.edu.cn/admetsar2/about/ (accessed on 1 February 2023).
- Nguyen, V.-L.; Truong, C.-T.; Nguyen, B.C.Q.; Van Vo, T.-N.; Dao, T.-T.; Trinh, D.-T.T.; Huynh, H.K.; Bui, C.-B. Anti-inflammatory and wound healing activities of calophyllolide isolated from Calophyllum inophyllum Linn. PLoS ONE 2017, 12, e0185674. [Google Scholar] [CrossRef]
- Martín-Aragóna, S.; Marcos, E. Tratamiento de Las Cicatrices. Revisión. Farm. Prof. 2008, 22, 39–43. [Google Scholar]
- de Sousa, A.C.C.; Combrinck, J.M.; Maepa, K.; Egan, T.J. Virtual screening as a tool to discover new β-haematin inhibitors with activity against malaria parasites. Sci. Rep. 2020, 10, 3374. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Polkam, N.; Ramaswamy, V.R.; Rayam, P.; Allaka, T.R.; Anantaraju, H.S.; Dharmarajan, S.; Perumal, Y.; Gandamalla, D.; Yellu, N.R.; Balasubramanian, S.; et al. Synthesis, molecular properties prediction and anticancer, antioxidant evaluation of new edaravone derivatives. Bioorgan. Med. Chem. Lett. 2016, 26, 2562–2568. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Sander, T. OSIRIS Property Explorer. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 1 February 2016).
- Cheminformatics, M. Molinspiration. Available online: http://www.molinspiration.com/ (accessed on 2 February 2023).
- Tariq, M.; Sirajuddin, M.; Ali, S.; Khalid, N.; Tahir, M.N.; Khan, H.; Ansari, T.M. Pharmacological investigations and Petra/Osiris/Molinspiration (POM) analyses of newly synthesized potentially bioactive organotin(IV) carboxylates. J. Photochem. Photobiol. B Biol. 2016, 158, 174–183. [Google Scholar] [CrossRef]
- Pham-The, H.; González-Álvarez, I.; Bermejo, M.; Sanjuan, V.M.; Centelles, I.; Garrigues, T.M.; Cabrera-Pérez, M. In Silico Prediction of Caco-2 Cell Permeability by a Classification QSAR Approach. Mol. Inform. 2011, 30, 376–385. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Zhao, Q.; Peng, H.; Hou, T. ADME Evaluation in Drug Discovery. 10. Predictions of P-Glycoprotein Inhibitors Using Recursive Partitioning and Naive Bayesian Classification Techniques. Mol. Pharm. 2011, 8, 889–900. [Google Scholar] [CrossRef]
- Xu, M.; Liu, C.; Zhou, M.; Li, Q.; Wang, R.; Kang, J. Screening of Small-Molecule Inhibitors of Protein–Protein Interaction with Capillary Electrophoresis Frontal Analysis. Anal. Chem. 2016, 88, 8050–8057. [Google Scholar] [CrossRef]
- Broccatelli, F.; Carosati, E.; Neri, A.; Frosini, M.; Goracci, L.; Oprea, T.I.; Cruciani, G. A Novel Approach for Predicting P-Glycoprotein (ABCB1) Inhibition Using Molecular Interaction Fields. J. Med. Chem. 2011, 54, 1740–1751. [Google Scholar] [CrossRef]
- Deppmeier, B.J.; Driessen, A.J.; Hehre, T.S.; Hehre, W.J.; Johnson, J.A.; Klunzinger, P.E.; Leonard, J.M.; Pham, I.N.; Pietro, W.J.; Jianguo, Y.; et al. Spartan 06; Wavefunction Inc.: Irvine, CA, USA, 2006. [Google Scholar]
- Ramos, A.I.; Braga, T.M.; Silva, P.; Fernandes, J.A.; Ribeiro-Claro, P.; Lopes, M.d.F.S.; Paz, A.; Braga, S.S. CSD Entry: WEWTOJ. Available online: https://www.ccdc.cam.ac.uk/structures/search?id=doi:10.5517/cctln45&sid=DataCite (accessed on 13 April 2023).
- Ramos, A.I.; Braga, T.M.; Silva, P.; Fernandes, J.A.; Ribeiro-Claro, P.; Lopes, M.D.F.S.; Paz, F.A.A.; Braga, S.S. Chloramphenicol·cyclodextrin inclusion compounds: Co-dissolution and mechanochemical preparations and antibacterial action. Crystengcomm 2013, 15, 2822–2834. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Parr, R.G.; Yang, W. Density-Functional Theory of the Electronic Structure of Molecules. Annu. Rev. Phys. Chem. 1995, 46, 701–728. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chem. Accounts 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.T.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Nicolás-Vázquez, I.; Torres, J.H.; Borbolla, J.C.; Ruvalcaba, R.M.; Aceves-Hernández, J.M. Orlistat interaction with sibutramine and carnitine. A physicochemical and theoretical study. J. Mol. Struct. 2014, 1062, 1–12. [Google Scholar] [CrossRef]
- Handy, N.C.; Murray, C.W.; Amos, R.D. Study of methane, acetylene, ethene, and benzene using Kohn-Sham theory. J. Phys. Chem. 1993, 97, 4392–4396. [Google Scholar] [CrossRef]
- Devlin, F.J.; Finley, J.W.; Stephens, P.J.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields: A Comparison of Local, Nonlocal, and Hybrid Density Functionals. J. Phys. Chem. 1995, 99, 16883–16902. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Lee, S.Y.; Boo, B.H. Density Functional Theory Study of Vibrational Spectra of Anthracene Neutral and Radical Cation. Bull. Korean Chem. Soc. 1996, 17, 754–759. [Google Scholar] [CrossRef]
- Rauhut, G.; Pulay, P. Transferable Scaling Factors for Density Functional Derived Vibrational Force Fields. J. Phys. Chem. 1995, 99, 3093–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Erdogdu, Y.; Güllüoǧlu, M.T.; Kurt, M. DFT, FT-Raman, FT-IR and NMR studies of 2-fluorophenylboronic acid. J. Raman Spectrosc. 2009, 40, 1615–1623. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Glossman-Mitnik, D. G3-B3 calculation of the molecular structure and descriptors of isomeric thiadiazoles. J. Mol. Struct. THEOCHEM 2005, 725, 27–30. [Google Scholar] [CrossRef]
- Walch, S.P. Model calculations of the electron affinities and ionization potentials of DNA. Chem. Phys. Lett. 2003, 374, 496–500. [Google Scholar] [CrossRef]
- Petit, J.; Meurice, N.; Kaiser, C.; Maggiora, G. Softening the Rule of Five—Where to draw the line? Bioorgan. Med. Chem. 2012, 20, 5343–5351. [Google Scholar] [CrossRef]
- Anzali, S.; Barnickel, G.; Cezanne, B.; Krug, M.; Filimonov, D.; Poroikov, V. Discriminating between Drugs and Nondrugs by Prediction of Activity Spectra for Substances (PASS). J. Med. Chem. 2001, 44, 2432–2437. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef]
- Cheng, F.; Yu, Y.; Zhou, Y.; Shen, Z.; Xiao, W.; Liu, G.; Li, W.; Lee, P.W.; Tang, Y. Insights into Molecular Basis of Cytochrome P450 Inhibitory Promiscuity of Compounds. J. Chem. Inf. Model. 2011, 51, 2482–2495. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electronic Parameters | Electronic Energy (Hartree) | Interaction Energy (Einteracction) (Hartree) | Interaction Energy (Einteracction) (kcal/mol) | Dipole Moments (Debye) |
|---|---|---|---|---|
| PCL | −1007.39355 | N/A | N/A | 1.5227 |
| β–CD | −4275.32655 | N/A | N/A | 10.3339 |
| β–CDNH2 | −4255.47834 | N/A | N/A | 8.2063 |
| β–CD/PCL | −5282.81672 | −0.09662188 | −60.6 | 5.0998 |
| β–CDNH2/PCL | −5262.90531 | −0.03341895 | −20.9 | 5.9249 |
| EGF–β–CDNH2/PCL | −5897.57936 | −0.0272599 | −17.1 | 3.2688 |
| Molecules | ΔHormation (kcal/mol) | ΔGformation (kcal/mol) | ΔHprocess (kcal/mol) | ΔGprocess (kcal/mol) | Equilibrium Constant (Keq) |
|---|---|---|---|---|---|
| PCL model | −6.3139 × 105 | −6.3145 × 105 | N/A | N/A | N/A |
| β–CD | −2.6820 × 106 | −2.6821 × 106 | N/A | N/A | N/A |
| β–CDNH2 | −2.6678 × 106 | −2.6679 × 106 | N/A | N/A | N/A |
| β–CD/PCL | −3.3117 × 106 | −3.3118 × 106 | −50.6876 | −47.3919 | 5.5082 × 1034 |
| β–CDNH2/PCL | −3.2992 × 106 | −3.2992 × 106 | −19.4832 | −3.5309 | 3.8812 × 102 |
| EGF–β–CDNH2/PCL | −3.7203 × 106 | −3.7205 × 106 | −14.1420 | −5.6117 | 1.3074 × 104 |
| Energy (kcal/mol) | |||
|---|---|---|---|
| Material | EHOMO | ELUMO | GAP (kcal/mol) |
| PCL | −165.2734 | 3.7712 | 169.0446 |
| β–CD/PCL | −155.3905 | −2.0017 | 153.3889 |
| β–CDNH2/PCL | −133.0207 | −12.2924 | 120.7282 |
| EGF–β–CDNH2/PCL | −150.8978 | −1.7758 | 149.1220 |
| Parameters | PCL | β–CD | β–CDNH2 | β–CD/PCL | β–CDNH2/PCL | EGF–β–CDNH2/PCL | |
|---|---|---|---|---|---|---|---|
| Energy (Hartrees) | Neutral | −1007.39355 | −4275.32655 | −4255.47834 | −5282.81672 | −5262.90531 | −5897.57936 |
| Positive | −1007.06894 | −4275.04279 | −4255.20427 | −5282.53199 | −5262.63330 | −5897.302952 | |
| Negative | −1007.33909 | −4275.2557 | −4255.40896 | −5282.77010 | −5262.87433 | −5897.541407 | |
| Reactivity parameters | EA | 0.0545 | 0.0708 | 0.06934 | 0.047 | 0.031 | 0.0380 |
| I | 0.325 | 0.284 | 0.274 | 0.285 | 0.272 | 0.276 | |
| η | 0.135 | 0.106 | 0.102 | 0.119 | 0.121 | 0.119 | |
| μ | −0.190 | −0.177 | −0.172 | −0.166 | −0.151 | −0.157 | |
| Ω | 0.133 | 0.148 | 0.144 | 0.115 | 0.095 | 0.104 | |
| (a) | (b) | (c) | |||
|---|---|---|---|---|---|
| Pa | Biological Activity | Pa | Biological Activity | Pa | Biological Activity |
| 0.953 | All-trans-retinyl-palmitate hydrolase inhibitor | 0.962 | Anti-inflammatory | 0.972 | Pullulanase inhibitor |
| 0.940 | Sugar-phosphatase inhibitor | 0.934 | Sugar-phosphatase inhibitor | 0.968 | Glucan 1,4-alpha-maltotriohydrolase inhibitor |
| 0.939 | Alkenylglycerophosphocholine hydrolase inhibitor | 0.905 | UDP-N-acetylglucosamine 4-epimerase inhibitor | 0.918 | Anti-inflammatory |
| 0.935 | Alkylacetylglycerophosphatase inhibitor | 0.901 | Alkenylglycerophosphocholine hydrolase inhibitor | 0.91 | 4-Alpha-glucanotransferase inhibitor |
| 0.934 | Acylcarnitine hydrolase inhibitor | 0.898 | Apoptosis agonist | 0.906 | Beta-amylase inhibitor |
| 0.931 | Cutinase inhibitor | 0.866 | Exoribonuclease II inhibitor | 0.877 | Amylo-alpha-1,6-glucosidase inhibitor |
| β–CD | β–CD-NH2 | PCL Model | ||
|---|---|---|---|---|
| Physicochemical properties | cLog P | −12.86 | −13.257 | 6.4546 |
| cLog S | 1.57 | 1.494 | −4.57 | |
| Drug-likeness | −10.037 | −9.5768 | −27.332 | |
| Drug score | 0.2499585 | 0.24995251 | 0.13383782 | |
| Toxicity risks | Mutagenic | N | N | N |
| Tumorigenic | N | N | N | |
| Reproductive effects | N | N | N | |
| Irritant | N | N | H |
| Absorption Model | (a) | (b) | (c) | |||
|---|---|---|---|---|---|---|
| Results | P | Results | P | Results | P | |
| Blood–brain barrier | BBB+ | 0.954 | BBB+ | 0.584 | BBB− | 0.903 |
| Human intestinal absorption | HIA+ | 0.949 | HIA− | 0.814 | HIA− | 0.863 |
| Caco-2 permeability | Caco2+ | 0.659 | Caco2− | 0.775 | Caco2− | 0.733 |
| P-glycoprotein substrate | Non-substrate | 0.604 | Non-substrate | 0.567 | Non-substrate | 0.571 |
| P-glycoprotein inhibitor | Non-inhibitor | 0.849 | Non-inhibitor | 0.886 | Non-inhibitor | 0.873 |
| Non-inhibitor | 0.879 | Non-inhibitor | 0.981 | Non-inhibitor | 0.961 | |
| Renal organic cation transporter | Non-inhibitor | 0.879 | Non-inhibitor | 0.818 | Non-inhibitor | 0.820 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escobedo-González, R.G.; Moyers-Montoya, E.D.; Martínez-Pérez, C.A.; García-Casillas, P.E.; Miranda-Ruvalcaba, R.; Nicolás-Vázquez, M.I.N. In Silico Study of Novel Cyclodextrin Inclusion Complexes of Polycaprolactone and Its Correlation with Skin Regeneration. Int. J. Mol. Sci. 2023, 24, 8932. https://doi.org/10.3390/ijms24108932
Escobedo-González RG, Moyers-Montoya ED, Martínez-Pérez CA, García-Casillas PE, Miranda-Ruvalcaba R, Nicolás-Vázquez MIN. In Silico Study of Novel Cyclodextrin Inclusion Complexes of Polycaprolactone and Its Correlation with Skin Regeneration. International Journal of Molecular Sciences. 2023; 24(10):8932. https://doi.org/10.3390/ijms24108932
Chicago/Turabian StyleEscobedo-González, René Gerardo, Edgar Daniel Moyers-Montoya, Carlos Alberto Martínez-Pérez, Perla Elvia García-Casillas, René Miranda-Ruvalcaba, and María Inés Nicolás Nicolás-Vázquez. 2023. "In Silico Study of Novel Cyclodextrin Inclusion Complexes of Polycaprolactone and Its Correlation with Skin Regeneration" International Journal of Molecular Sciences 24, no. 10: 8932. https://doi.org/10.3390/ijms24108932