

Structural Factors That Determine the Activity of the Xenobiotic Reductase B Enzyme from Pseudomonas putida on Nitroaromatic Compounds

 , ,
, ,  ,
,  , , and
, , and
Abstract
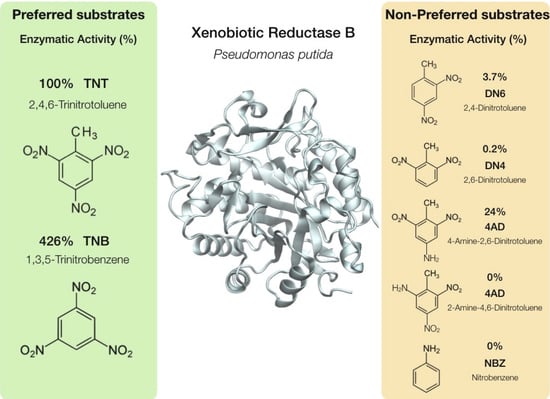
:
1. Introduction
2. Results and Discussion
2.1. Molecular Dynamics Simulations
Trajectory and Cluster Analysis
2.2. Quantum Chemical Reactivity Indexes
2.2.1. Global Reactivity Molecular Descriptors
2.2.2. Molecular Electrostatic Potential Maps
2.2.3. Ring Current Densities Calculations
3. Materials and Methods
3.1. Preparation of the Systems
3.2. Molecular Dynamics (MD) Simulation Protocol
3.3. Cluster Analysis
3.4. Density Functional Theory (DFT) Calculations
3.5. Ring Current Densities Calculations
3.6. Non-Covalent Interactions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wittich, R.M.; Haidour, A.; Van Dillewijn, P.; Ramos, J.L. OYE flavoprotein reductases initiate the condensation of TNT-derived intermediates to secondary diarylamines and nitrite. Environ. Sci. Technol. 2008, 42, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Avellaneda, H.; Arbeli, Z.; Teran, W.; Roldan, F. Transformation of TNT, 2,4-DNT, and PETN by Raoultella planticola M30b and Rhizobium radiobacter M109 and exploration of the associated enzymes. World J. Microbiol. Biotechnol. 2020, 36, 190. [Google Scholar] [CrossRef] [PubMed]
- Gungor, T.; Yetis, G.; Onder, F.C.; Tokay, E.; Tok, T.T.; Celik, A.; Ay, M.; Kockar, F. Prodrugs for Nitroreductase Based Cancer Therapy- 1: Metabolite Profile, Cell Cytotoxicity and Molecular Modeling Interactions of Nitro Benzamides with Ssap-NtrB. Med. Chem. 2018, 14, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Um, Y.; Han, S.; Hilberath, T.; Kim, Y.H.; Hollmann, F.; Park, C.B. Unbiased Photoelectrode Interfaces for Solar Coupling of Lignin Oxidation with Biocatalytic C horizontal lineC Bond Hydrogenation. ACS Appl. Mater. Interfaces 2022, 14, 11465–11473. [Google Scholar] [CrossRef]
- Ball, P.; Halliwell, J.; Anderson, S.; Gwenin, V.; Gwenin, C. Evaluation of two xenobiotic reductases from Pseudomonas putida for their suitability for magnetic nanoparticle-directed enzyme prodrug therapy as a novel approach to cancer treatment. Microbiologyopen 2020, 9, e1110. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [CrossRef]
- Pratap, S.; Katiki, M.; Gill, P.; Kumar, P.; Golemi-Kotra, D. Active-Site Plasticity Is Essential to Carbapenem Hydrolysis by OXA-58 Class D beta-Lactamase of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2016, 60, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Alcolombri, U.; Elias, M.; Tawfik, D.S. Directed evolution of sulfotransferases and paraoxonases by ancestral libraries. J. Mol. Biol. 2011, 411, 837–853. [Google Scholar] [CrossRef]
- Yasutake, Y.; Yao, M.; Sakai, N.; Kirita, T.; Tanaka, I. Crystal structure of the Pyrococcus horikoshii isopropylmalate isomerase small subunit provides insight into the dual substrate specificity of the enzyme. J. Mol. Biol. 2004, 344, 325–333. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Understanding the mechanism of cytochrome P450 3A4: Recent advances and remaining problems. Dalton Trans. 2013, 42, 3116–3126. [Google Scholar] [CrossRef]
- Yao, J.; Guo, H.; Chaiprasongsuk, M.; Zhao, N.; Chen, F.; Yang, X.; Guo, H. Substrate-Assisted Catalysis in the Reaction Catalyzed by Salicylic Acid Binding Protein 2 (SABP2), a Potential Mechanism of Substrate Discrimination for Some Promiscuous Enzymes. Biochemistry 2015, 54, 5366–5375. [Google Scholar] [CrossRef] [PubMed]
- van Dillewijn, P.; Wittich, R.M.; Caballero, A.; Ramos, J.L. Subfunctionality of hydride transferases of the old yellow enzyme family of flavoproteins of Pseudomonas putida. Appl. Environ. Microbiol. 2008, 74, 6703–6708. [Google Scholar] [CrossRef] [Green Version]
- Fuller, M.E.; McClay, K.; Hawari, J.; Paquet, L.; Malone, T.E.; Fox, B.G.; Steffan, R.J. Transformation of RDX and other energetic compounds by xenobiotic reductases XenA and XenB. Appl. Microbiol. Biot. 2009, 84, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, J.W.; Knoke, K.L.; Noguera, D.R.; Fox, B.G.; Chambliss, G.H. Transformation of 2,4,6-trinitrotoluene by purified xenobiotic reductase B from Pseudomonas fluorescens I-C. Appl. Environ. Microbiol. 2000, 66, 4742–4750. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhu, Z.H.; Chen, M.W.; Chen, Q.A.; Zhou, Y.G. Catalytic Biomimetic Asymmetric Reduction of Alkenes and Imines Enabled by Chiral and Regenerable NAD(P)H Models. Angew. Chem. Int. Ed. Engl. 2019, 58, 1813–1817. [Google Scholar] [CrossRef] [PubMed]
- Knaus, T.; Paul, C.E.; Levy, C.W.; de Vries, S.; Mutti, F.G.; Hollmann, F.; Scrutton, N.S. Better than Nature: Nicotinamide Biomimetics That Outperform Natural Coenzymes. J. Am. Chem. Soc. 2016, 138, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Osorio, M.I.; Cabrera, M.A.; Gonzalez-Nilo, F.; Perez-Donoso, J.M. Odd Loop Regions of XenA and XenB Enzymes Modulate Their Interaction with Nitro-explosives Compounds and Provide Structural Support for Their Regioselectivity. J. Chem. Inf. Model. 2019, 59, 3860–3870. [Google Scholar] [CrossRef]
- Wei, T.; Yang, Z.; Zhou, M.; Zhou, Y. Comparison of biotransformation mechanisms of 2, 4, 6-trinitrotoluene and its hydride-Meisenheimer metabolite by the old yellow enzyme family of flavoproteins. Energ. Mater. Front. 2020, 1, 216–226. [Google Scholar] [CrossRef]
- Zhou, L.J.; Han, P.; Zhao, M.; Yu, Y.; Sun, D.; Hou, L.; Liu, M.; Zhao, Q.; Tang, X.; Klumper, U.; et al. Biotransformation of lincomycin and fluoroquinolone antibiotics by the ammonia oxidizers AOA, AOB and comammox: A comparison of removal, pathways, and mechanisms. Water Res. 2021, 196, 117003. [Google Scholar] [CrossRef]
- Schubert, E.; Sander, J.; Ester, M.; Kriegel, H.P.; Xu, X.W. DBSCAN Revisited, Revisited: Why and How You Should (Still) Use DBSCAN. ACM T. Database Syst. 2017, 42, 1–21. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Parthasarathi, R.; Elango, M.; Subramanian, V.; Krishnamoorthy, B.S.; Gutierrez-Oliva, S.; Toro-Labbé, A.; Roy, D.R.; Chattaraj, P.K. Multiphilic Descriptor for Chemical Reactivity and Selectivity. J. Phys. Chem. A 2007, 111, 9130–9138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, R.; Domingo, L.R.; Andrés, J.; Pérez, P.; Tapia, O. Nonlocal (Pair Site) Reactivity from Second-Order Static Density Response Function: Gas- and Solution-Phase Reactivity of the Acetaldehyde Enolate as a Test Case. J. Phys. Chem. A 1999, 103, 1367–1375. [Google Scholar] [CrossRef]
- Teles Fujishima, M.A.; Silva, N.; Ramos, R.D.S.; Batista Ferreira, E.F.; Santos, K.; Silva, C.; Silva, J.O.D.; Campos Rosa, J.M.; Santos, C. An Antioxidant Potential, Quantum-Chemical and Molecular Docking Study of the Major Chemical Constituents Present in the Leaves of Curatella americana Linn. Pharmaceuticals 2018, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- Baez-Grez, R.; Inostroza, D.; Garcia, V.; Vasquez-Espinal, A.; Donald, K.J.; Tiznado, W. Aromatic ouroboroi: Heterocycles involving a sigma-donor-acceptor bond and 4n + 2 pi-electrons. Phys. Chem. Chem. Phys. 2020, 22, 1826–1832. [Google Scholar] [CrossRef]
- Torres-Vega, J.J.; Alcoba, D.R.; Oña, O.B.; Vásquez-Espinal, A.; Báez-Grez, R.; Lain, L.; Torre, A.; García, V.; Tiznado, W. Analysis of Local and Global Aromaticity in Si3C5 and Si4C8 Clusters. Aromatic Species Containing Planar Tetracoordinate Carbon. Chemistry 2021, 3, 1101–1112. [Google Scholar] [CrossRef]
- Leyva-Parra, L.; Inostroza, D.; Yañez, O.; Cruz, J.C.; Garza, J.; García, V.; Tiznado, W. Persistent Planar Tetracoordinate Carbon in Global Minima Structures of Silicon-Carbon Clusters. Atoms 2022, 10, 27. [Google Scholar] [CrossRef]
- Inostroza, D.; García, V.; Yañez, O.; Torres-Vega, J.J.; Vásquez-Espinal, A.; Pino-Rios, R.; Báez-Grez, R.; Tiznado, W. On the NICS limitations to predict local and global current pathways in polycyclic systems. New J. Chem. 2021, 45, 8345–8351. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Osorio, M.I.; Yanez, O.; Gallardo, M.; Zuniga-Bustos, M.; Mulia-Rodriguez, J.; Lopez-Rendon, R.; Garcia-Beltran, O.; Gonzalez-Nilo, F.; Perez-Donoso, J.M. Search for Novel Potent Inhibitors of the SARS-CoV-2 Papain-like Enzyme: A Computational Biochemistry Approach. Pharmaceuticals 2022, 15, 986. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, D.; Liu, X. A simple and accurate algorithm for path integral molecular dynamics with the Langevin thermostat. J. Chem. Phys. 2016, 145, 024103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wires Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical hardness and density functional theory. J. Chem. Sci. 2005, 117, 369–377. [Google Scholar] [CrossRef]
- Fliegl, H.; Taubert, S.; Lehtonen, O.; Sundholm, D. The gauge including magnetically induced current method. Phys. Chem. Chem. Phys. 2011, 13, 20500–20518. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware, Inc.: Clifton Park, NY, USA, 2015. [Google Scholar]
- Ahrens, J.; Geveci, B.; Law, C. 36—ParaView: An End-User Tool for Large-Data Visualization. In Visualization Handbook; Hansen, C.D., Johnson, C.R., Eds.; Butterworth-Heinemann: Oxford, UK; Burlington Township: Burlington, NJ, USA, 2005; pp. 717–731. [Google Scholar]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Estructure | % Activity | |
|---|---|---|
| 2,4,6-Trinitrotoluene (TNT) |  | 100 |
| 2,4-Dinitrotoluene (DN6) |  | 3.7 |
| 2,6-Dinitrotoluene (DN4) |  | 0.2 |
| 4-Amino-2,6-dinitrotoluene (4AD) |  | 24 |
| 2-Amino-4,6-dinitrotoluene (2AD) |  | 0 |
| 1,3,5-Trinitrobenzene (TNB) |  | 426 |
| Nitrobenzene (NBZ) |  | 0 |
| [13] |
| Compound | χ | η | ω | ω− | ω+ | Δω;± | |||
|---|---|---|---|---|---|---|---|---|---|
| 2,4,6 Trinitrotolueno (TNT) | −9.0 | −3.5 | 5.5 | 6.2 | 2.7 | 7.0 | 10.5 | 4.3 | 14.7 |
| 2,4 Dinitrotolueno (DN6) | −8.5 | −3.0 | 5.6 | 5.8 | 2.8 | 5.9 | 9.1 | 3.4 | 12.5 |
| 2,6 Dinitrotolueno (DN4) | −8.3 | −2.8 | 5.6 | 5.6 | 2.8 | 5.6 | 8.7 | 3.1 | 11.8 |
| 4-amino-2,6-Dinitrotolueno (4AD) | −7.0 | −2.6 | 4.4 | 4.8 | 2.2 | 5.3 | 7.9 | 3.1 | 11.1 |
| 2-amino-4,6-Dinitrotolueno (2AD) | −7.1 | −2.8 | 4.4 | 4.9 | 2.2 | 5.6 | 8.3 | 3.4 | 11.7 |
| 1,3,5-trinitrobenzeno (TNB) | −9.6 | −3.7 | 5.9 | 6.6 | 3.0 | 7.4 | 11.1 | 4.5 | 15.6 |
| Nitrobenzeno (NBZ) | −8.1 | −2.5 | 5.7 | 5.3 | 2.8 | 5.0 | 8.0 | 2.7 | 10.6 |
| Koopmans’ Theorem | Reference | |
|---|---|---|
| Global Hardness (η) | [7,8,9,10,11,12] | |
| Electronegativity (χ) | [8,13,14] | |
| Electrophilicity (ω) | [15] | |
| Electron Acceptor (ω+) | [15] | |
| Electron Donator (ω−) | [15] | |
| Net Electrophilicity (Δω±) | [16] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osorio, M.I.; Bruna, N.; García, V.; González-Rodríguez, L.; Leal, M.S.; Salgado, F.; Vargas-Reyes, M.; González-Nilo, F.; Pérez-Donoso, J.M.; Yáñez, O. Structural Factors That Determine the Activity of the Xenobiotic Reductase B Enzyme from Pseudomonas putida on Nitroaromatic Compounds. Int. J. Mol. Sci. 2023, 24, 400. https://doi.org/10.3390/ijms24010400
Osorio MI, Bruna N, García V, González-Rodríguez L, Leal MS, Salgado F, Vargas-Reyes M, González-Nilo F, Pérez-Donoso JM, Yáñez O. Structural Factors That Determine the Activity of the Xenobiotic Reductase B Enzyme from Pseudomonas putida on Nitroaromatic Compounds. International Journal of Molecular Sciences. 2023; 24(1):400. https://doi.org/10.3390/ijms24010400
Chicago/Turabian StyleOsorio, Manuel I., Nicolás Bruna, Víctor García, Lisdelys González-Rodríguez, Matías S. Leal, Francisco Salgado, Matías Vargas-Reyes, Fernando González-Nilo, José M. Pérez-Donoso, and Osvaldo Yáñez. 2023. "Structural Factors That Determine the Activity of the Xenobiotic Reductase B Enzyme from Pseudomonas putida on Nitroaromatic Compounds" International Journal of Molecular Sciences 24, no. 1: 400. https://doi.org/10.3390/ijms24010400