
Novel Inhibitor for Downstream Targeting of Transforming Growth Factor-β Signaling to Suppress Epithelial to Mesenchymal Transition and Cell Migration

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
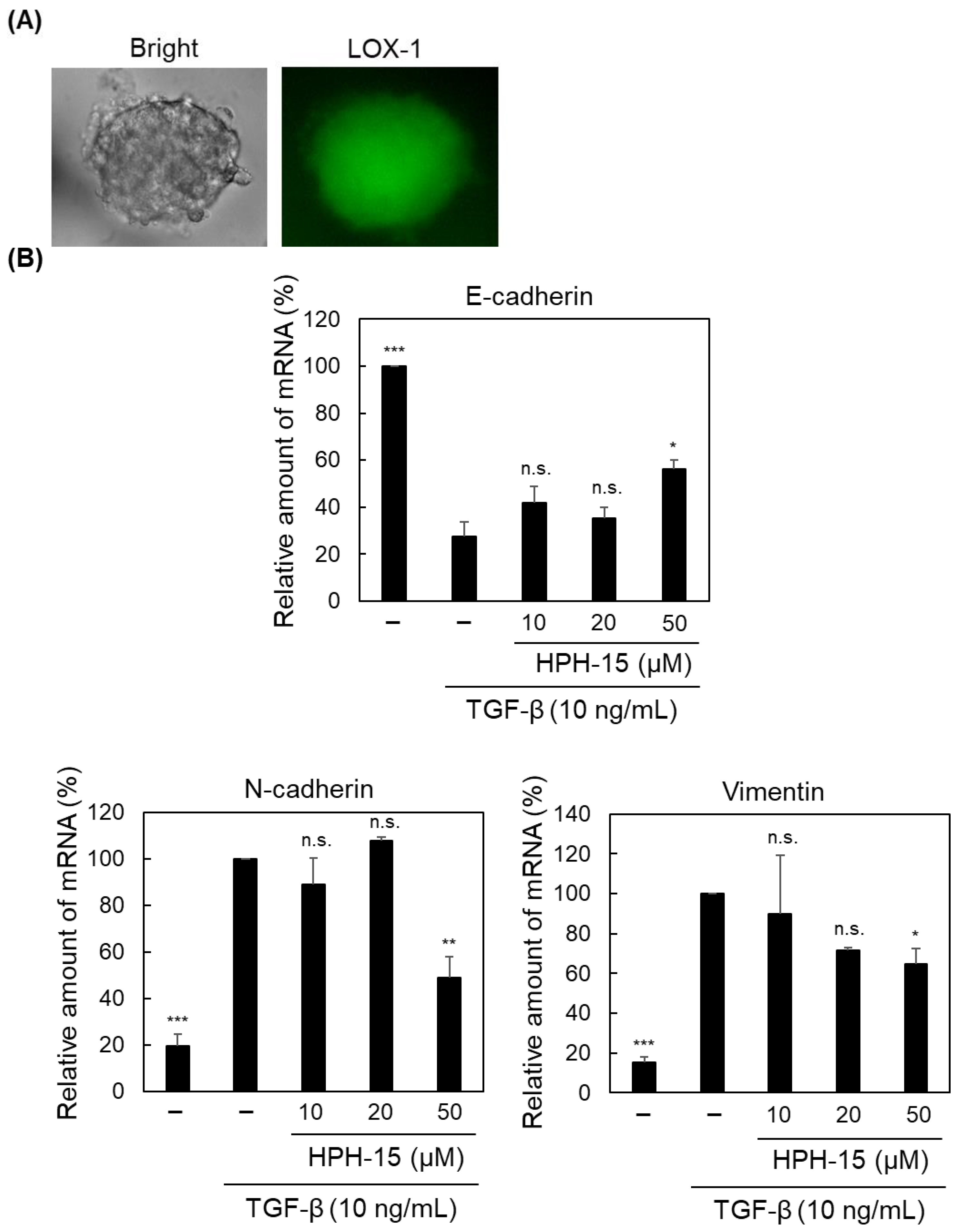
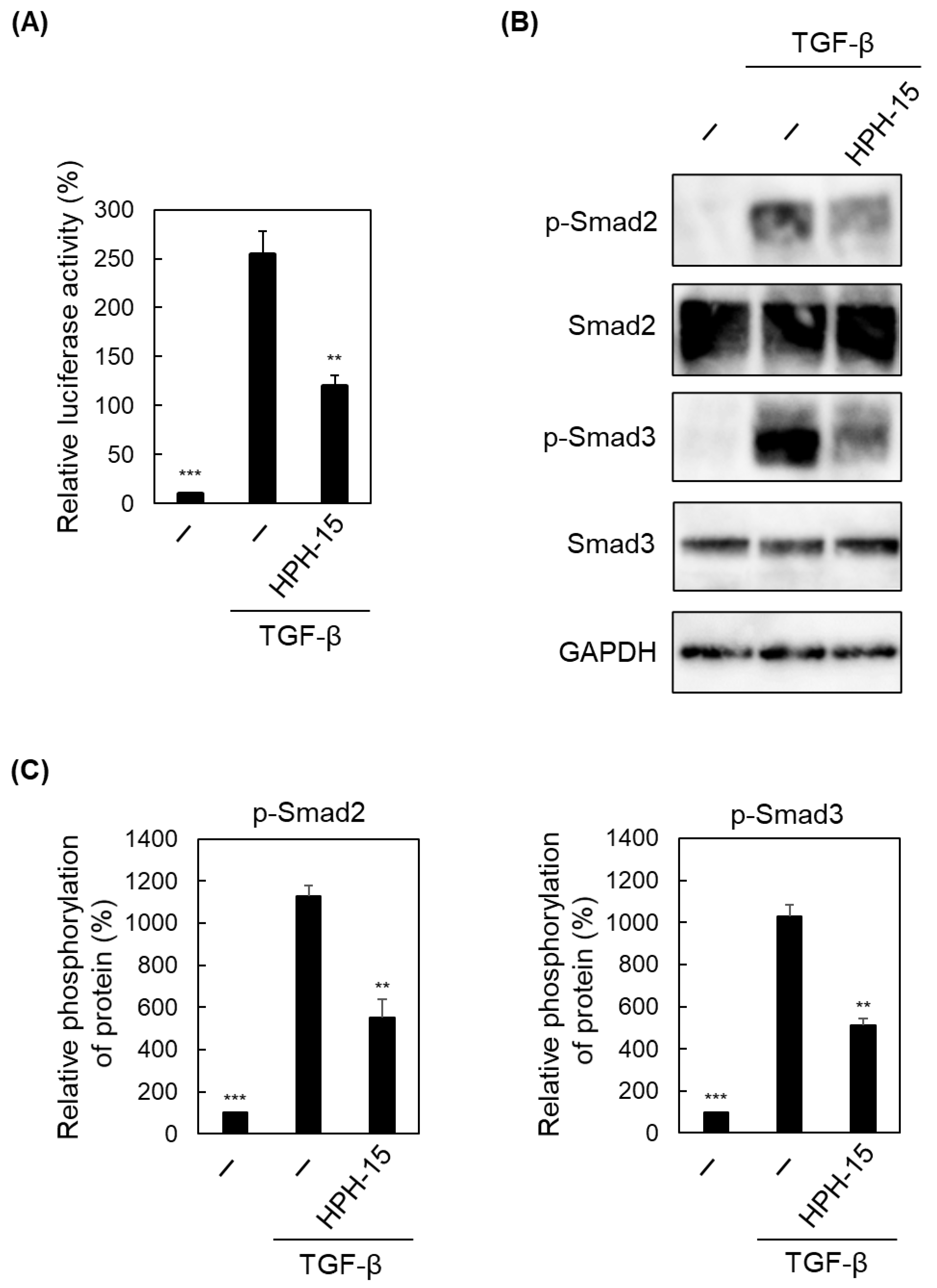
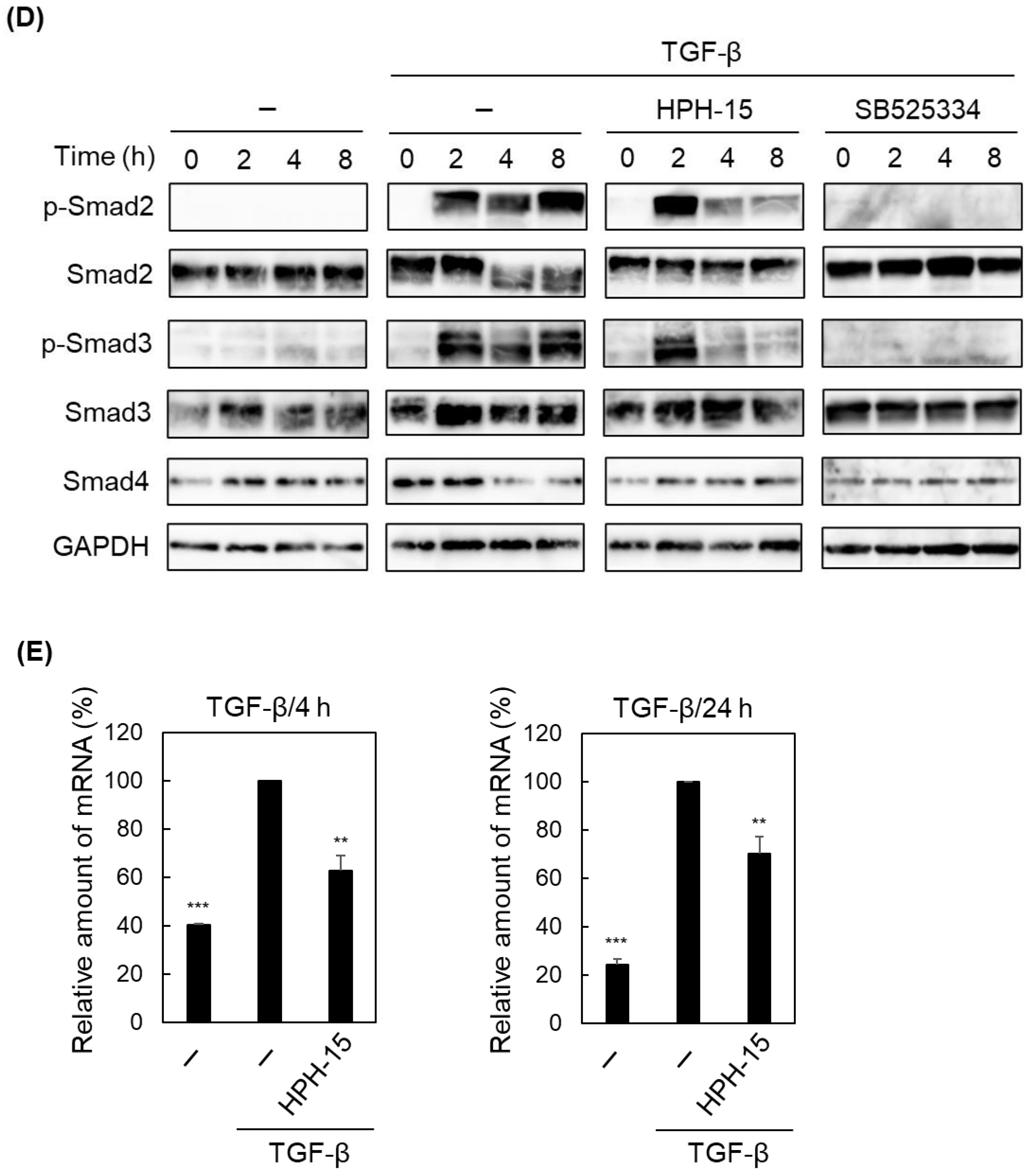
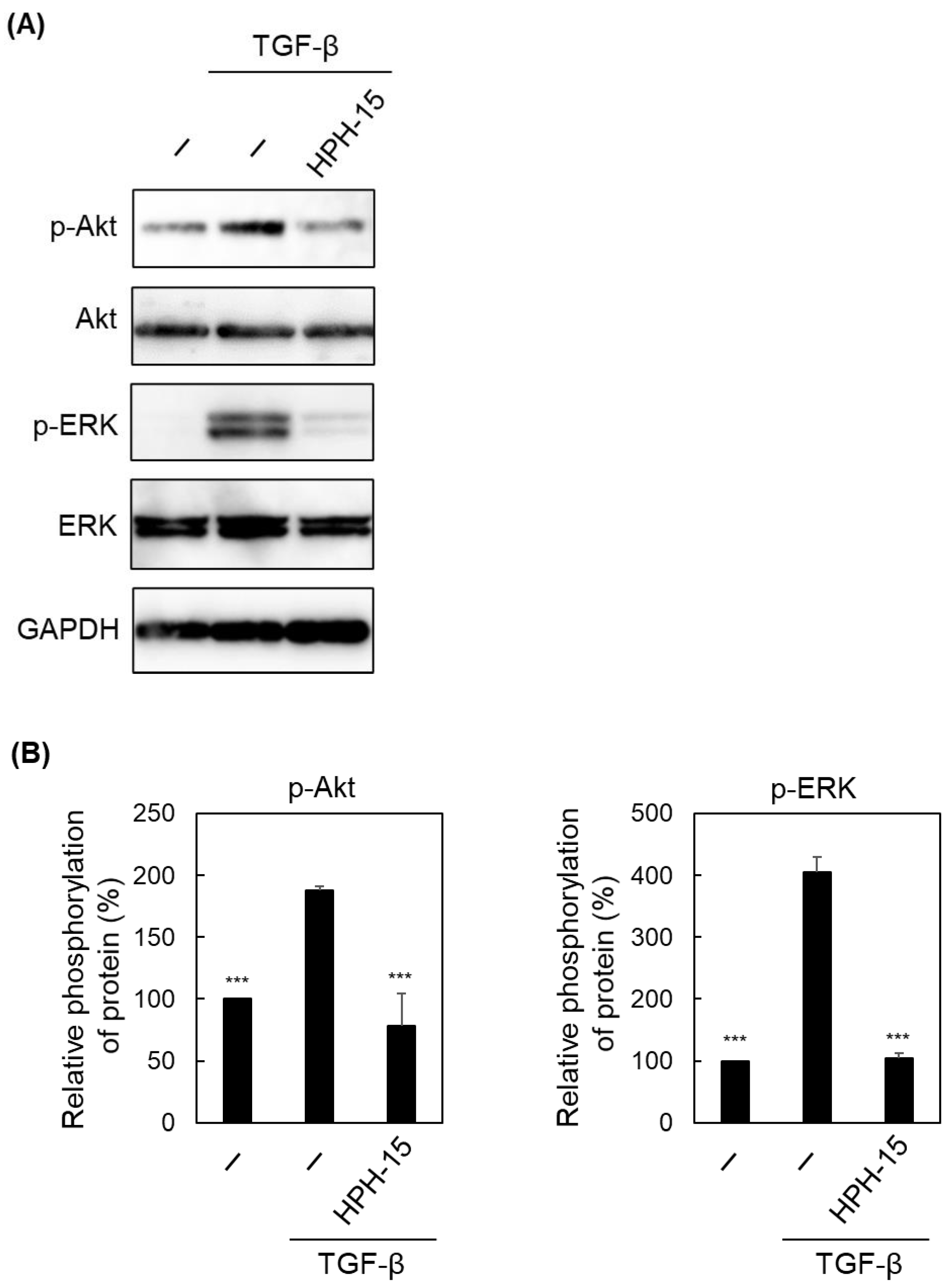
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals and a Cytokine
4.2. Cell Culture and Viability
4.3. Generation of Cancer Spheroids and Hypoxia Assay
4.4. Protein Knockdown
4.5. In Vitro Scratch Assay
4.6. Immunostaining of Cells
4.7. Immunoblot Analysis
4.8. RT-PCR Analysis
4.9. Luciferase Activity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics-Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.L.; Balasas, T.; Callaghan, J.; Coombes, R.C.; Evans, J.; Hall, J.A.; Kinrade, S.; Jones, D.; Jones, P.S.; Jones, R.; et al. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 2019, 16, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Karlsson, M.C.; Gonzalez, S.F.; Welin, J.; Fuxe, J. Epithelial-mesenchymal transition in cancer metastasis through the lymphatic system. Mol. Oncol. 2017, 11, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adh. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Gheldof, A.; Berx, G. Cadherins and epithelial-to-mesenchymal transition. Prog. Mol. Biol. Transl. Sci. 2013, 116, 317–336. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhan, L.; Yang, T.; Wang, L.; Li, C.; Zhao, J.; Lei, Z.; Li, X.; Zhang, H.T. Ski prevents TGF-β-induced EMT and cell invasion by repressing SMAD-dependent signaling in non-small cell lung cancer. Oncol. Rep. 2015, 34, 87–94. [Google Scholar] [CrossRef]
- Lee, J.H.; Chinnathambi, A.; Alharbi, S.A.; Shair, O.H.M.; Sethi, G.; Ahn, K.S. Farnesol abrogates epithelial to mesenchymal transition process through regulating Akt/mTOR pathway. Pharmacol. Res. 2019, 150, 104504. [Google Scholar] [CrossRef]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFβ: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, Y.; Wang, H.; Guo, Z.; Wang, X.; Li, X.; Chang, S.; Sun, T.; Yu, Z.; Xu, T.; et al. Synthesis and biological evaluation of 4-(pyridin-4-oxy)-3-(3,3-difluorocyclobutyl)-pyrazole derivatives as novel potent transforming growth factor-β type 1 receptor inhibitors. Eur. J. Med. Chem. 2020, 198, 112354. [Google Scholar] [CrossRef]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]
- Luong, V.H.; Chino, T.; Oyama, N.; Matsushita, T.; Sasaki, Y.; Ogura, D.; Niwa, S.I.; Biswas, T.; Hamasaki, A.; Fujita, M.; et al. Blockade of TGF-β/Smad signaling by the small compound HPH-15 ameliorates experimental skin fibrosis. Arthritis Res. Ther. 2018, 20, 46. [Google Scholar] [CrossRef]
- Hosono, T.; Yokomizo, K.; Hamasaki, A.; Okamoto, Y.; Okawara, T.; Otsuka, M.; Mukai, R.; Suzuki, K. Antiviral activities against herpes simplex virus type 1 by HPH derivatives and their structure-activity relationships. Bioorg. Med. Chem. Lett. 2008, 18, 371–374. [Google Scholar] [CrossRef]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Karacosta, L.G.; Anchang, B.; Ignatiadis, N.; Kimmey, S.C.; Benson, J.A.; Shrager, J.B.; Tibshirani, R.; Bendall, S.C.; Plevritis, S.K. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat. Commun. 2019, 10, 5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Hosaka, M.; Yoshihara, T.; Negishi, K.; Iida, Y.; Tobita, S.; Takeuchi, T. Phosphorescent light-emitting iridium complexes serve as a hypoxia-sensing probe for tumor imaging in living animals. Cancer Res. 2010, 70, 4490–4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwagi, I.; Morita, R.; Schichita, T.; Komai, K.; Saeki, K.; Matsumoto, M.; Takeda, K.; Nomura, M.; Hayashi, A.; Kanai, T.; et al. Smad2 and Smad3 Inversely Regulate TGF-β Autoinduction in Clostridium butyricum-Activated Dendritic Cells. Immunity 2015, 43, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Grygielko, E.T.; Martin, W.M.; Tweed, C.; Thornton, P.; Harling, J.; Brooks, D.P.; Laping, N.J. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-beta type I receptor kinase in puromycin-induced nephritis. J. Pharmacol. Exp. Ther. 2005, 313, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Li, N.; He, H.; Ying, Y.; Sunkara, S.; Luo, L.; Lv, N.; Huang, D.; Luo, Z. AMPK Inhibits the Stimulatory Effects of TGF-β on Smad2/3 Activity, Cell Migration, and Epithelial-to-Mesenchymal Transition. Mol. Pharmacol. 2015, 88, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Otsuka, M.; Sugiura, Y. Metal-chelating inhibitors of a zinc finger protein HIV-EP1. Remarkable potentiation of inhibitory activity by introduction of SH groups. J. Med. Chem. 1996, 39, 503–507. [Google Scholar] [CrossRef]
- Ejima, T.; Hirota, M.; Mizukami, T.; Otsuka, M.; Fujita, M. An anti-HIV-1 compound that increases steady-state expression of apoplipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G. Int. J. Mol. Med. 2011, 28, 613–616. [Google Scholar] [CrossRef]
- Koga, R.; Radwan, M.O.; Ejima, T.; Kanemaru, Y.; Tateishi, H.; Ali, T.F.S.; Ciftci, H.I.; Shibata, Y.; Taguchi, Y.; Inoue, J.I.; et al. A Dithiol Compound Binds to the Zinc Finger Protein TRAF6 and Suppresses Its Ubiquitination. ChemMedChem 2017, 12, 1935–1941. [Google Scholar] [CrossRef]
- Hamasaki, A.; Naka, H.; Tamanoi, F.; Umezawa, K.; Otsuka, M. A novel metal-chelating inhibitor of protein farnesyltransferase. Bioorg. Med. Chem. Lett. 2003, 13, 1523–1526. [Google Scholar] [CrossRef]
- Tanaka, A.; Radwan, M.O.; Hamasaki, A.; Ejima, A.; Obata, E.; Koga, R.; Tateishi, H.; Okamoto, Y.; Fujita, M.; Nakao, M.; et al. A novel inhibitor of farnesyltransferase with a zinc site recognition moiety and a farnesyl group. Bioorg. Med. Chem. Lett. 2017, 27, 3862–3866. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.O.; Koga, R.; Hida, T.; Ejima, T.; Kanemaru, Y.; Tateishi, H.; Okamoto, Y.; Inoue, J.I.; Fujita, M.; Otsuka, M. Minimum structural requirements for inhibitors of the zinc finger protein TRAF6. Bioorg. Med. Chem. Lett. 2019, 29, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, H.; Tateishi, M.; Radwan, M.O.; Masunaga, T.; Kawatashiro, K.; Oba, Y.; Oyama, M.; Inoue-Kitahashi, N.; Fujita, M.; Okamoto, Y.; et al. A New Inhibitor of ADAM17 Composed of a Zinc-Binding Dithiol Moiety and a Specificity Pocket-Binding Appendage. Chem. Pharm. Bull. (Tokyo) 2021, 69, 1123–1130. [Google Scholar] [CrossRef]
- Alexander, M.; Kim, S.Y.; Cheng, H. Update 2020: Management of Non-Small Cell Lung Cancer. Lung 2020, 198, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.Y.; Zhou, Q.; Yang, J.J.; Zhong, W.Z.; Chen, Z.H.; Deng, W.; He, Y.Y.; Chen, H.J.; Zeng, Z.; Ke, E.E.; et al. Distribution and prognosis of uncommon metastases from non-small cell lung cancer. BMC Cancer 2016, 16, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mujoomdar, A.; Austin, J.H.; Malhotra, R.; Powell, C.A.; Pearson, G.D.; Shiau, M.C.; Raftopoulos, H. Clinical predictors of metastatic disease to the brain from non-small cell lung carcinoma: Primary tumor size, cell type, and lymph node metastases. Radiology 2007, 242, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Fontoura, J.C.; Viezzer, C.; Dos Santos, F.G.; Ligabue, R.A.; Weinlich, R.; Puga, R.D.; Antonow, D.; Severino, P.; Bonorino, C. Comparison of 2D and 3D cell culture models for cell growth, gene expression and drug resistance. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 107, 110264. [Google Scholar] [CrossRef] [PubMed]
- Oshikata, A.; Matsushita, T.; Ueoka, R. Enhancement of drug efflux activity via MDR1 protein by spheroid culture of human hepatic cancer cells. J. Biosci. Bioeng. 2011, 111, 590–593. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Ozturk, S.E.; Ali, T.F.S.; Radwan, M.O.; Tateishi, H.; Koga, R.; Ocak, Z.; Can, M.; Otsuka, M.; Fujita, M. The First Pentacyclic Triterpenoid Gypsogenin Derivative Exhibiting Anti-ABL1 Kinase and Anti-chronic Myelogenous Leukemia Activities. Biol. Pharm. Bull. 2018, 41, 570–574. [Google Scholar] [CrossRef] [Green Version]
- Kamo, M.; Ito, M.; Toma, T.; Gotoh, H.; Shimozono, R.; Nakagawa, R.; Koga, R.; Monde, K.; Tateishi, H.; Misumi, S.; et al. Discovery of anti-cell migration activity of an anti-HIV heterocyclic compound by identification of its binding protein hnRNP M. Bioorg. Chem. 2021, 107, 104627. [Google Scholar] [CrossRef]
- Yamamoto, M.; Koga, R.; Fujino, H.; Shimagaki, K.; Ciftci, H.I.; Kamo, M.; Tateishi, H.; Otsuka, M.; Fujita, M. Zinc-binding site of human immunodeficiency virus 2 Vpx prevents instability and dysfunction of the protein. J. Gen. Virol. 2017, 98, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Bhattacharya, R.; Ray, U.; Mukhopadhyay, S.; Chatterjee, U.; Roy, S.S. Invasion of ovarian cancer cells is induced byPITX2-mediated activation of TGF-β and Activin-A. Mol. Cancer 2015, 14, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, D.S.; Handa, R.J.; Yang, R.S.; Campain, J.A. Gene expression patterns as potential molecular biomarkers for malignant transformation in human keratinocytes treated with MNNG, arsenic, or a metal mixture. Toxicol. Sci. 2003, 74, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.R.; Terra, L.F.; Wailemann, R.A.; Labriola, L.; Sogayar, M.C. TGF-β1 modulates the homeostasis between MMPs and MMP inhibitors through p38 MAPK and ERK1/2 in highly invasive breast cancer cells. BMC Cancer 2012, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Valente, V.; Teixeira, S.A.; Neder, L.; Okamoto, O.K.; Oba-Shinjo, S.M.; Marie, S.K.; Scrideli, C.A.; Paçó-Larson, M.L.; Carlotti, C.G., Jr. Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT-PCR. BMC Mol. Biol. 2009, 10, 17. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toma, T.; Tateishi, H.; Kawakami, K.; Ali, T.F.S.; Kamo, M.; Monde, K.; Nakashima, Y.; Fujita, M.; Otsuka, M. Novel Inhibitor for Downstream Targeting of Transforming Growth Factor-β Signaling to Suppress Epithelial to Mesenchymal Transition and Cell Migration. Int. J. Mol. Sci. 2022, 23, 5047. https://doi.org/10.3390/ijms23095047
Toma T, Tateishi H, Kawakami K, Ali TFS, Kamo M, Monde K, Nakashima Y, Fujita M, Otsuka M. Novel Inhibitor for Downstream Targeting of Transforming Growth Factor-β Signaling to Suppress Epithelial to Mesenchymal Transition and Cell Migration. International Journal of Molecular Sciences. 2022; 23(9):5047. https://doi.org/10.3390/ijms23095047
Chicago/Turabian StyleToma, Tsugumasa, Hiroshi Tateishi, Kensaku Kawakami, Taha F. S. Ali, Masahiro Kamo, Kazuaki Monde, Yuta Nakashima, Mikako Fujita, and Masami Otsuka. 2022. "Novel Inhibitor for Downstream Targeting of Transforming Growth Factor-β Signaling to Suppress Epithelial to Mesenchymal Transition and Cell Migration" International Journal of Molecular Sciences 23, no. 9: 5047. https://doi.org/10.3390/ijms23095047