
Adhesion Molecule Profile and the Effect of Anti-VLA-4 mAb Treatment in Experimental Autoimmune Encephalomyelitis, a Mouse Model of Multiple Sclerosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
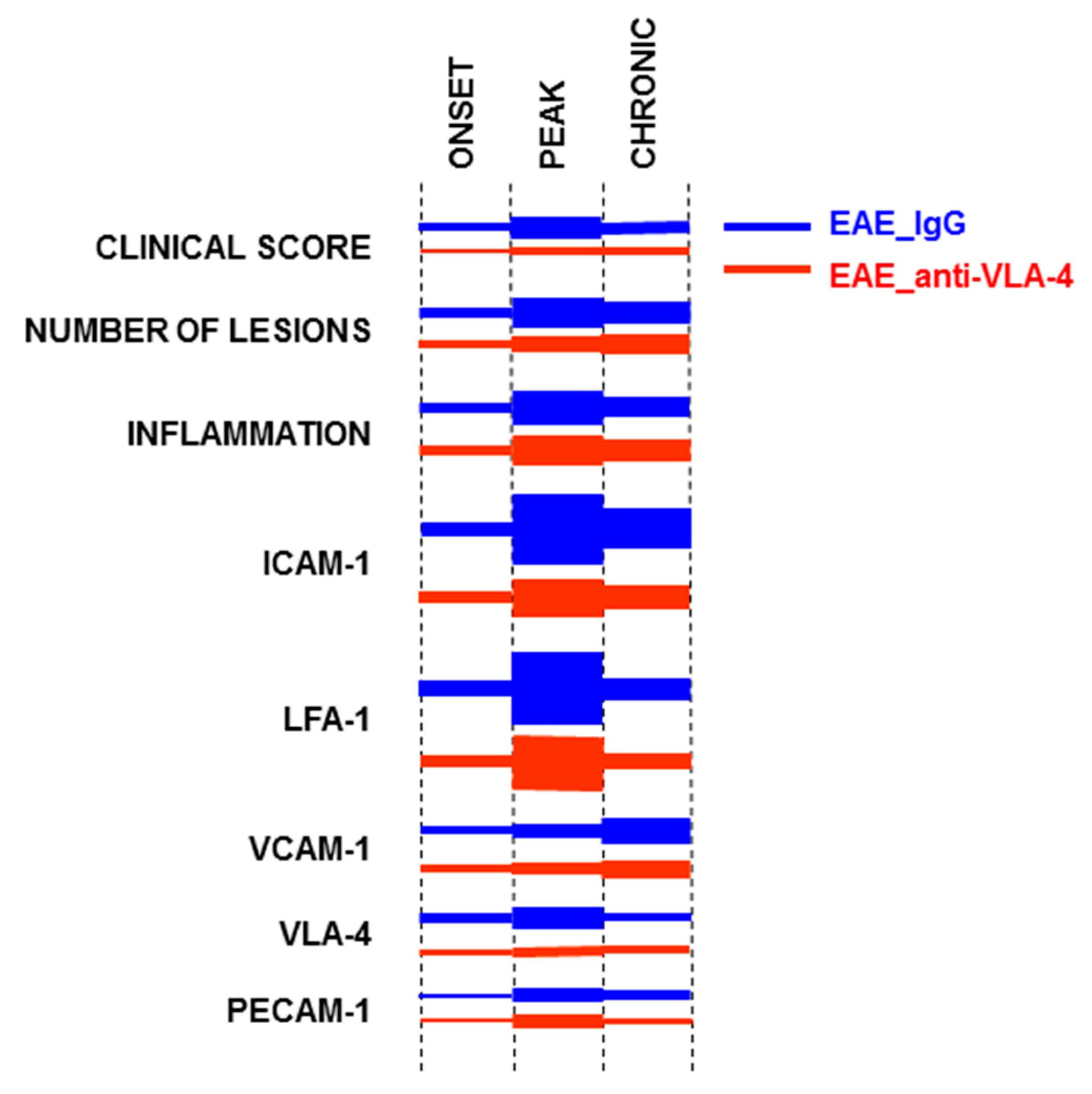
2.1. Effect of Anti-VLA-4 mAb and IgG Treatment on the Development of Progressive EAE
2.2. CD45 Expression
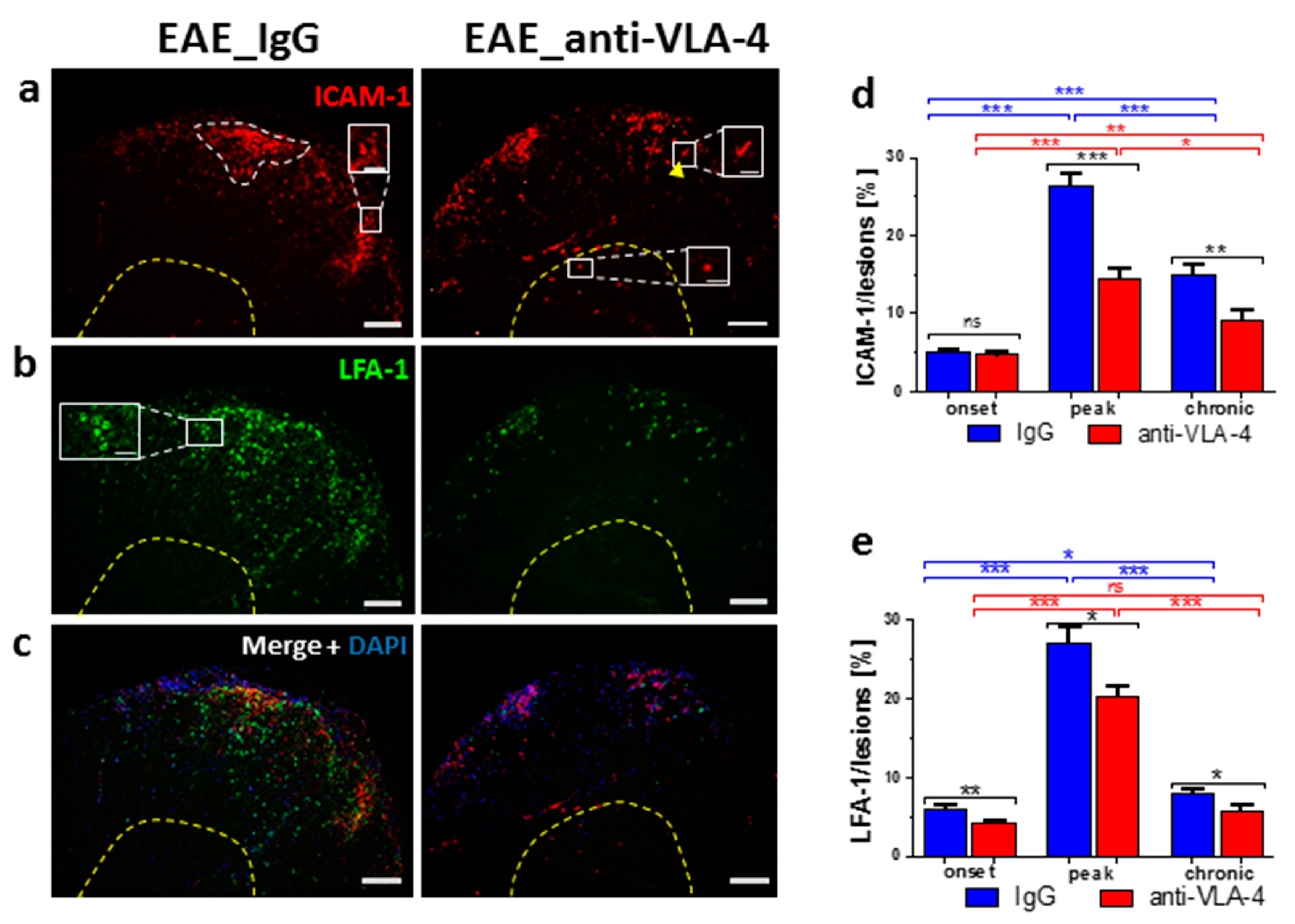
2.3. ICAM-1/LFA-1 Expression
2.4. VCAM-1/VLA-4 Expression
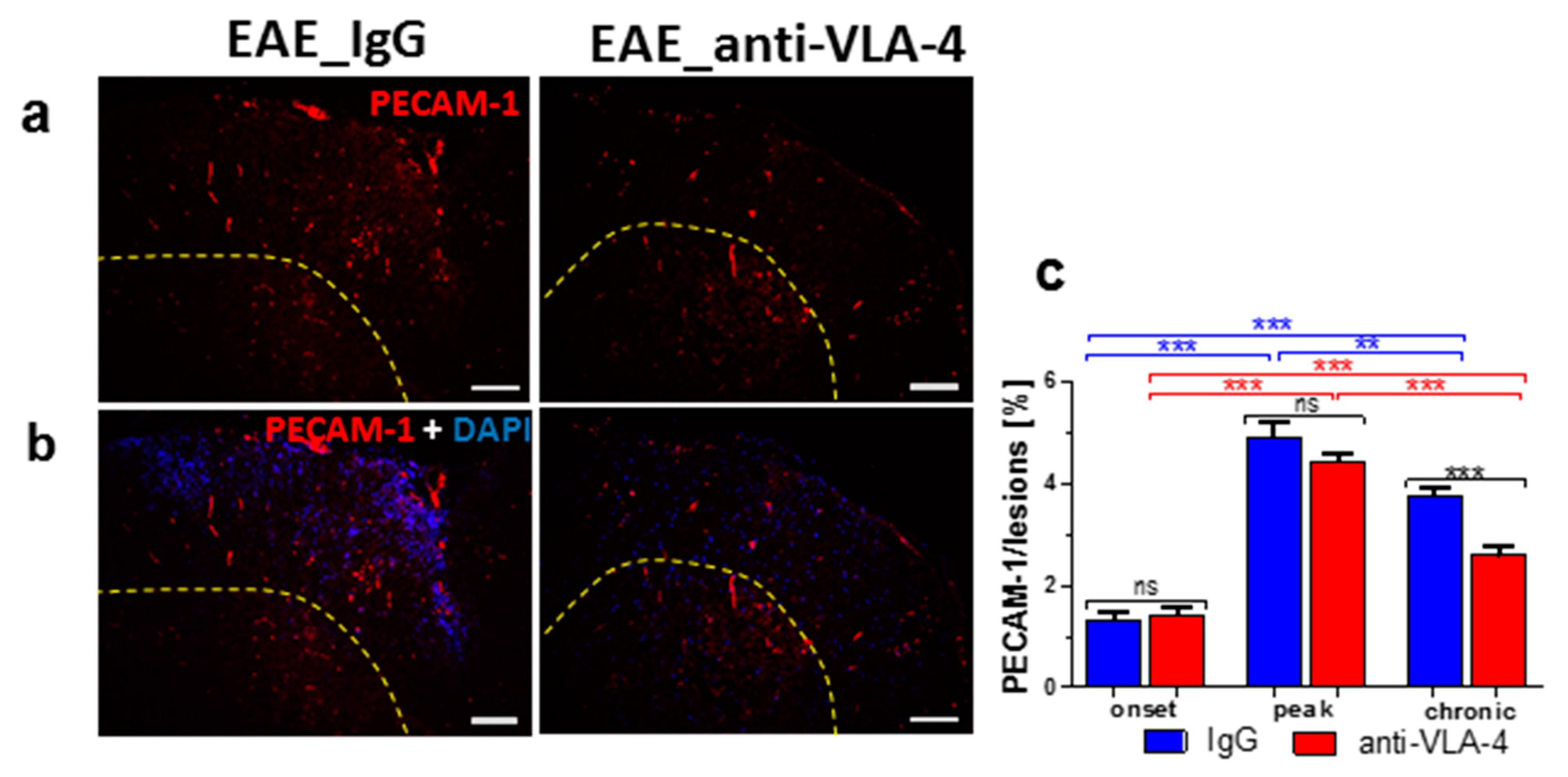
2.5. PECAM-1 Expression
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of EAE
4.3. Evaluation of EAE
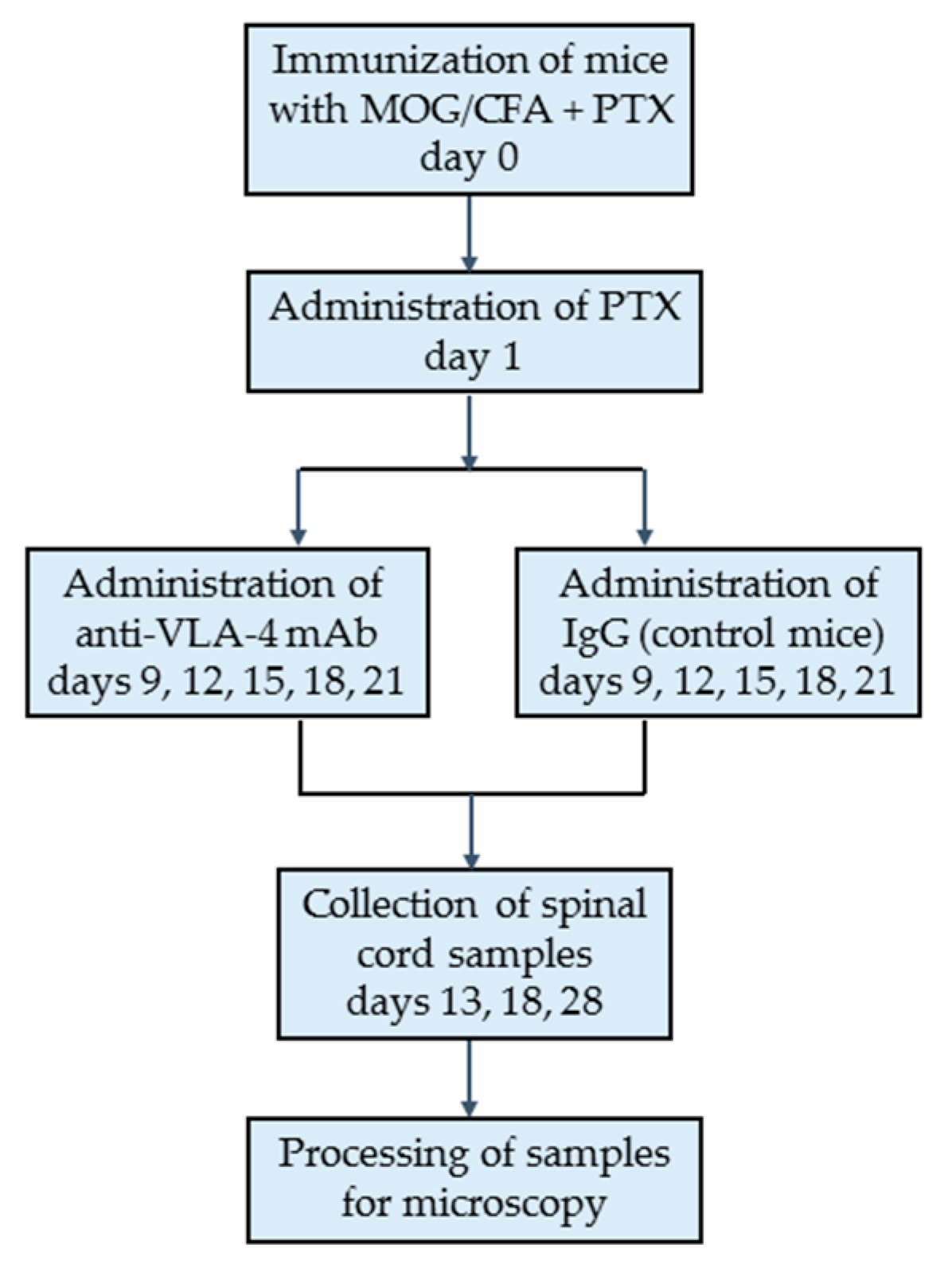
4.4. Experimental Groups
4.5. Tissue Sampling and Processing
4.6. Immunohistochemistry
4.7. Microscopy, Morphometry and Image Collection
4.8. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Cannella, B.; Raine, C.S. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann. Neurol. 1995, 37, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Conley, F.K.; Butcher, E.C. Cell adhesion molecules on vessels during inflammation in the mouse central nervous system. J. Neuroimmunol. 1994, 51, 199–208. [Google Scholar] [CrossRef]
- Archelos, J.J.; Hartung, H.P. The role of adhesion molecules in multiple sclerosis: Biology, pathogenesis and therapeutic implications. Mol. Med. Today 1997, 3, 310–321. [Google Scholar] [CrossRef]
- Rice, G.P.; Hartung, H.P.; Calabresi, P.A. Anti-alpha4 integrin therapy for multiple sclerosis: Mechanisms and rationale. Neurology 2005, 64, 1336–1342. [Google Scholar] [CrossRef]
- Yusuf-Makagiansar, H.; Anderson, M.E.; Yakovleva, T.V.; Murray, J.S.; Siahaan, T.J. Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as a therapeutic approach to inflammation and autoimmune diseases. Med. Res. Rev. 2002, 22, 146–167. [Google Scholar] [CrossRef]
- Wimmer, I.; Tietz, S.; Nishihara, H.; Deutsch, U.; Sallusto, F.; Gosselet, F.; Lyck, R.; Muller, W.A.; Lassmann, H.; Engelhardt, B. PECAM-1 Stabilizes Blood-Brain Barrier Integrity and Favors Paracellular T-Cell Diapedesis Across the Blood-Brain Barrier during Neuroinflammation. Front. Immunol. 2019, 10, 711. [Google Scholar] [CrossRef] [Green Version]
- Paddock, C.; Zhou, D.; Lertkiatmongkol, P.; Newman, P.J.; Zhu, J. Structural basis for PECAM-1 homophilic binding. Blood 2016, 127, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Steinman, L. Blocking adhesion molecules as therapy for multiple sclerosis: Natalizumab. Nat. Rev. Drug Discov. 2005, 4, 510–518. [Google Scholar] [CrossRef]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against a4b1 integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef]
- Clerico, M.; Artusi, C.A.; Di Liberto, A.; Rolla, S.; Bardina, V.; Barbero, P.; De Mercanti, S.F.; Durelli, L. Long-term safety evaluation of natalizumab for the treatment of multiple sclerosis. Expert. Opin. Drug Saf. 2017, 16, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Pyka-Fosciak, G.; Lis, G.J.; Litwin, J.A. Effect of natalizumab treatment on metalloproteinases and their inhibitors in a mouse model of multiple sclerosis. J. Physiol. Pharmacol. 2020, 71, 265–273. [Google Scholar]
- Theien, B.E.; Vanderlugt, C.L.; Eagar, T.N.; Nickerson-Nutter, C.; Nazareno, R.; Kuchroo, V.; Miller, S. Discordant effects of anti-VLA-4 treatment before and after onset of relapsing experimental autoimmune encephalomyelitis. J. Clin. Investig. 2001, 107, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannella, B.; Gaupp, S.; Tilton, R.G.; Raine, C.S. Differential efficacy of a synthetic antagonist of VLA-4 during the course of chronic relapsing experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2003, 71, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef] [Green Version]
- Linker, R.A.; Lee, D.H. Models of autoimmune demyelination in the central nervous system: On the way to translational medicine. Exp. Transl. Stroke Med. 2009, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Archelos, J.J.; Jung, S.; Mäurer, M.; Schmied, M.; Lassmann, H.; Tamatani, T.; Miyasaka, M.; Toyka, K.V.; Hartung, H.P. Inhibition of experimental autoimmune encephalomyelitis by an antibody to the intercellular adhesion molecule ICAM-1. Ann. Neurol. 1993, 34, 145–154. [Google Scholar] [CrossRef]
- Ran, Z.; Yue-Bei, L.; Qiu-Ming, Z.; Huan, Y. Regulatory B cells and its role in central nervous system inflammatory demyelinating diseases. Front. Immunol. 2020, 11, 1884. [Google Scholar] [CrossRef]
- Pyka-Fosciak, G.; Stasiolek, M.; Litwin, J.A. Immunohistochemical analysis of spinal cord components in mouse model of experimental autoimmune encephalomyelitis. Folia Histochem. Cytobiol. 2018, 56, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, J.; Wang, Y.; Calder, V.L. Lymphocyte adhesion and transendothelial migration in the central nervous system—The role of LFA-1, ICAM-1, VLA-4 and VCAM-1. Immunology 1995, 86, 408–415. [Google Scholar]
- Wong, D.; Prameya, R.; Dorovini-Zis, K. In Vitro adhesion and migration of T lymphocytes across monolayers of human brain microvessel endothelial cells: Regulation by ICAM-1, VCAM-1, E-selectin and PECAM-1. J. Neuropathol. Exp. Neurol. 1999, 58, 138–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dopp, J.M.; Brenemann, S.M.; Olschowka, J.A. Expression of ICAM-1, VCAM-1, L-selectin, and leukosialin in the mouse central nervous system during the induction and remission stages of experimental allergic encephalomyelitis. J. Neuroimmunol. 1994, 54, 129–144. [Google Scholar] [CrossRef]
- Graesser, D.; Solowiej, A.; Bruckner, M.; Osterweil, E.; Juedes, A.; Davis, S.; Ruddle, N.H.; Engelhardt, B.; Madri, J.A. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1–deficient mice. J. Clin. Investig. 2002, 109, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Cannella, B.; Cross, A.H.; Raine, C.S. Adhesion-related molecules in the central nervous system. Upregulation correlates with inflammatory cell influx during relapsing experimental autoimmune encephalomyelitis. Lab. Investig. 1991, 65, 23–31. [Google Scholar] [PubMed]
- Ledeboer, A.; Wierinckx, A.; Bol, J.G.; Floris, S.; Renardel de Lavalette, C.; De Vries, H.E.; van den Berg, T.K.; Dijkstra, C.D.; Tilders, F.J.; van Dam, A.M. Regional and temporal expression patterns of interleukin-10, interleukin-10 receptor and adhesion molecules in the rat spinal cord during chronic relapsing EAE. J. Neuroimmunol. 2003, 136, 94–103. [Google Scholar] [CrossRef]
- Williams, K.C.; Zhao, R.W.; Ueno, K.; Hickey, W.F. PECAM-1 (CD31) expression in the central nervous system and its role in experimental allergic encephalomyelitis in the rat. J. Neurosci. Res. 1996, 45, 747–757. [Google Scholar] [CrossRef]
- Bullard, D.C.; Hu, X.; Schoeb, T.R.; Collins, R.G.; Beaudet, A.L.; Barnum, S.R. Intercellular adhesion molecule-1 expression is required on multiple cell types for the development of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 178, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Baron, J.L.; Madri, J.A.; Ruddle, N.H.; Hashim, G.; Janeway, C.A., Jr. Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J. Exp. Med. 1993, 177, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Hikita, S.T.; Cann, G.M.; Wingerd, K.L.; Mullick, L.H.; Wayne, W.C.; Webb, S.W.; Clegg, D.O. Integrin alpha4beta1 (VLA-4) expression and activity in retinal and peripheral neurons. Mol. Cell Neurosci. 2003, 23, 427–439. [Google Scholar] [CrossRef]
- Khademi, M.; Bornsen, L.; Rafatnia, F.; Andersson, M.; Brundin, L.; Piehl, F.; Sellebjerg, F.; Olsson, T. The effects of natalizumab on inflammatory mediators in multiple sclerosis: Prospects for treatment-sensitive biomarkers. Eur. J. Neurol. 2009, 16, 528–536. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyka-Fościak, G.; Lis, G.J.; Litwin, J.A. Adhesion Molecule Profile and the Effect of Anti-VLA-4 mAb Treatment in Experimental Autoimmune Encephalomyelitis, a Mouse Model of Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 4637. https://doi.org/10.3390/ijms23094637
Pyka-Fościak G, Lis GJ, Litwin JA. Adhesion Molecule Profile and the Effect of Anti-VLA-4 mAb Treatment in Experimental Autoimmune Encephalomyelitis, a Mouse Model of Multiple Sclerosis. International Journal of Molecular Sciences. 2022; 23(9):4637. https://doi.org/10.3390/ijms23094637
Chicago/Turabian StylePyka-Fościak, Grażyna, Grzegorz J. Lis, and Jan A. Litwin. 2022. "Adhesion Molecule Profile and the Effect of Anti-VLA-4 mAb Treatment in Experimental Autoimmune Encephalomyelitis, a Mouse Model of Multiple Sclerosis" International Journal of Molecular Sciences 23, no. 9: 4637. https://doi.org/10.3390/ijms23094637