
Cyto-Genotoxic and Transcriptomic Alterations in Human Liver Cells by Tris (2-Ethylhexyl) Phosphate (TEHP): A Putative Hepatocarcinogen

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Cytotoxicity Assays
2.2. Quantification of ROS, NO, ΔΨm, Ca++ Influx, and Esterase Level
2.3. DNA Damage Analysis
2.4. Cell Cycle Dysfunction and Apoptosis Analysis
2.5. Apoptotic Protein Analysis
2.6. Transcriptomic Analysis by RT-qPCR
3. Results
3.1. Cytotoxicity Induced by TEHP
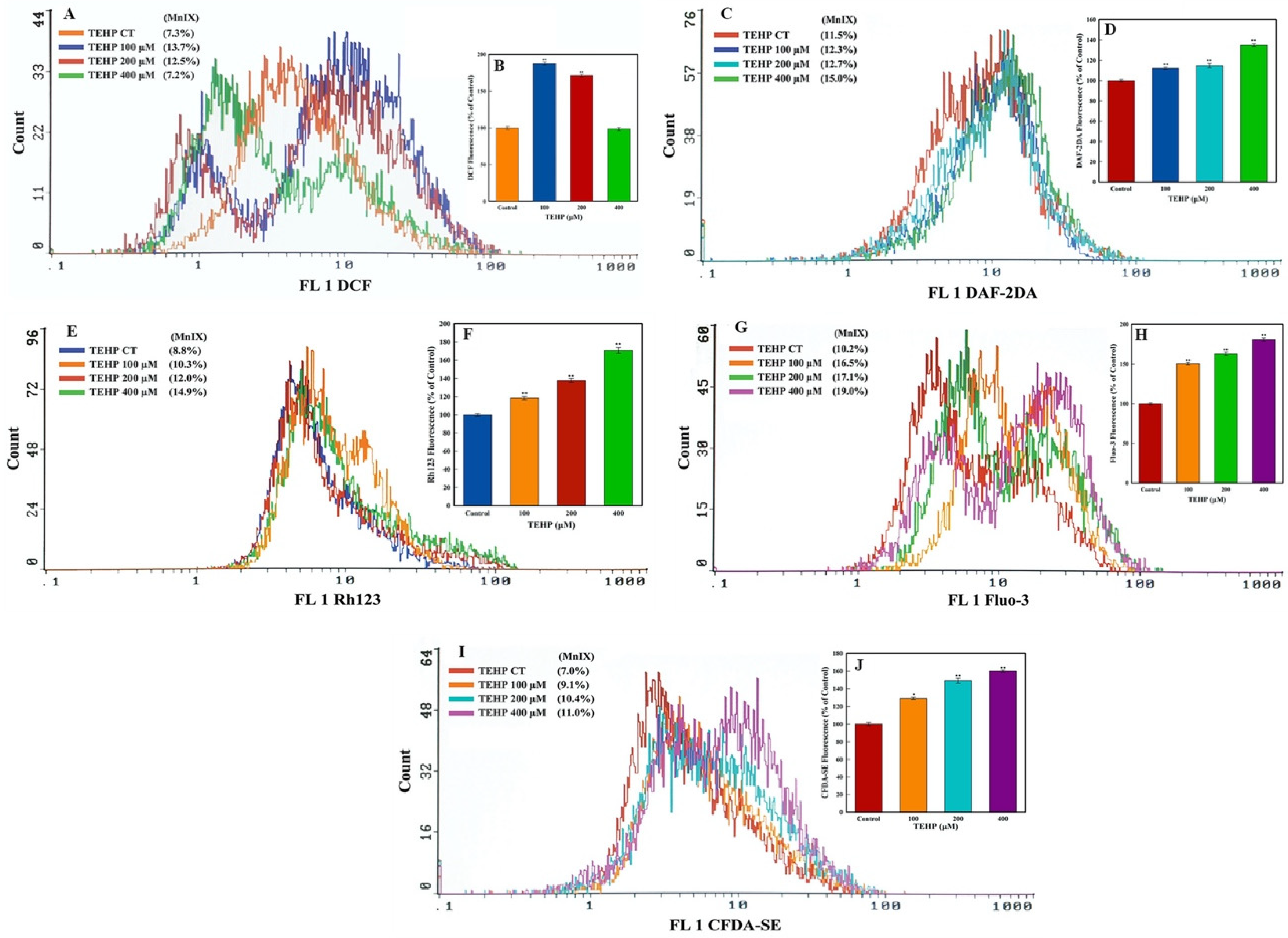
3.2. Intracellular Quantification of ROS, NO, ΔΨm, Ca++ Influx, and Esterase
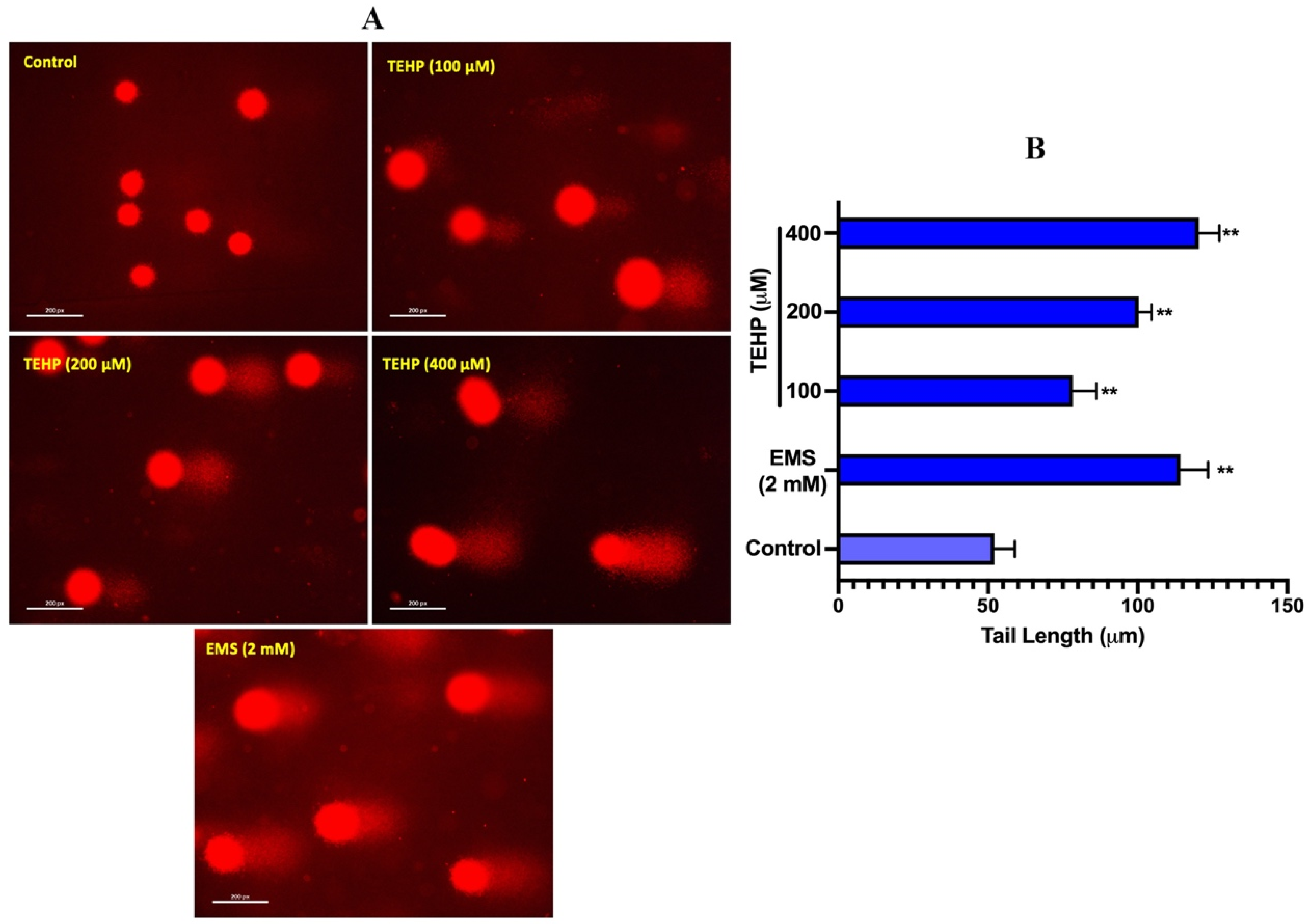
3.3. DNA Damage Measurement
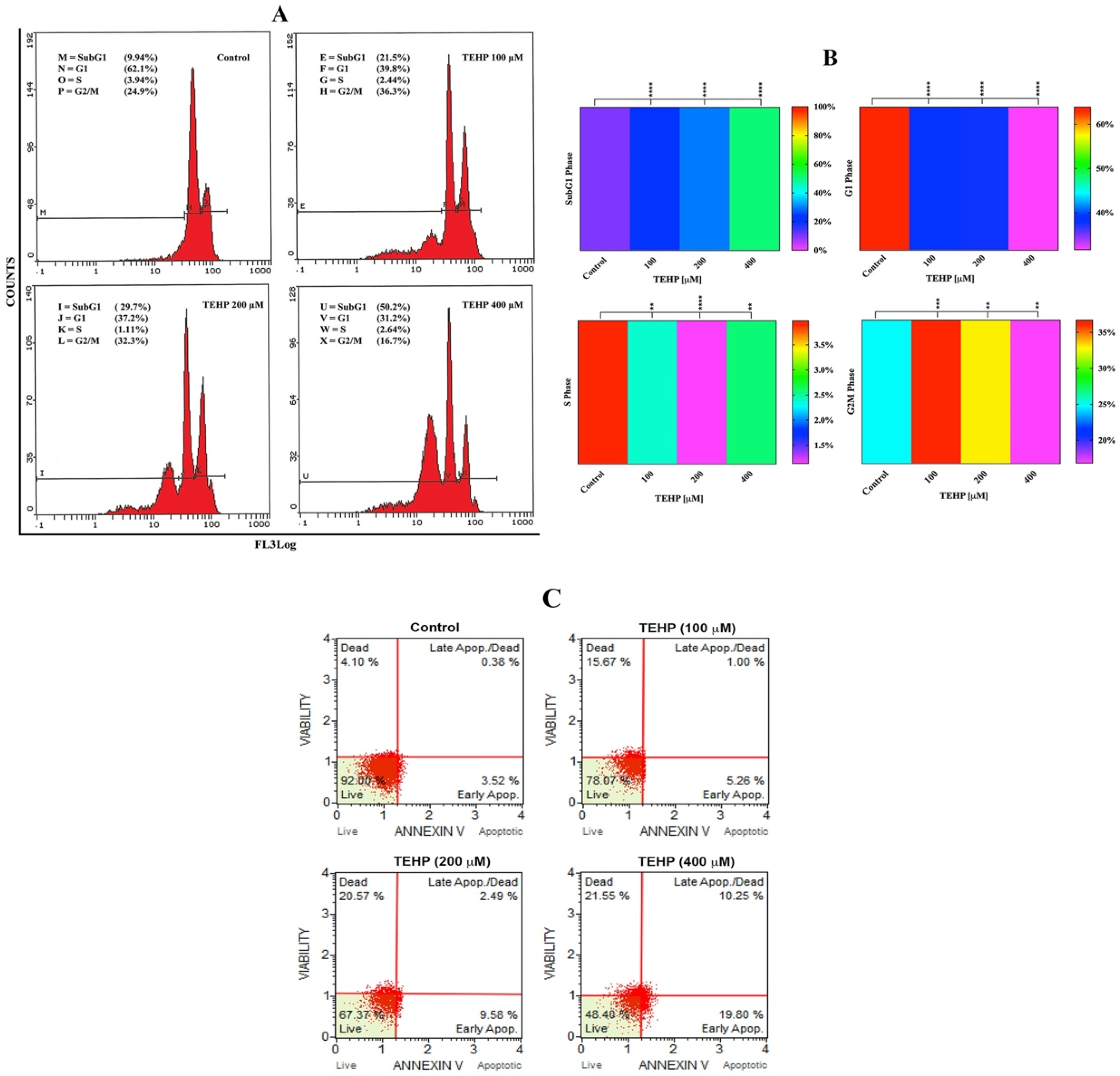
3.4. TEHP Trigger Apoptosis in HepG2 Cells
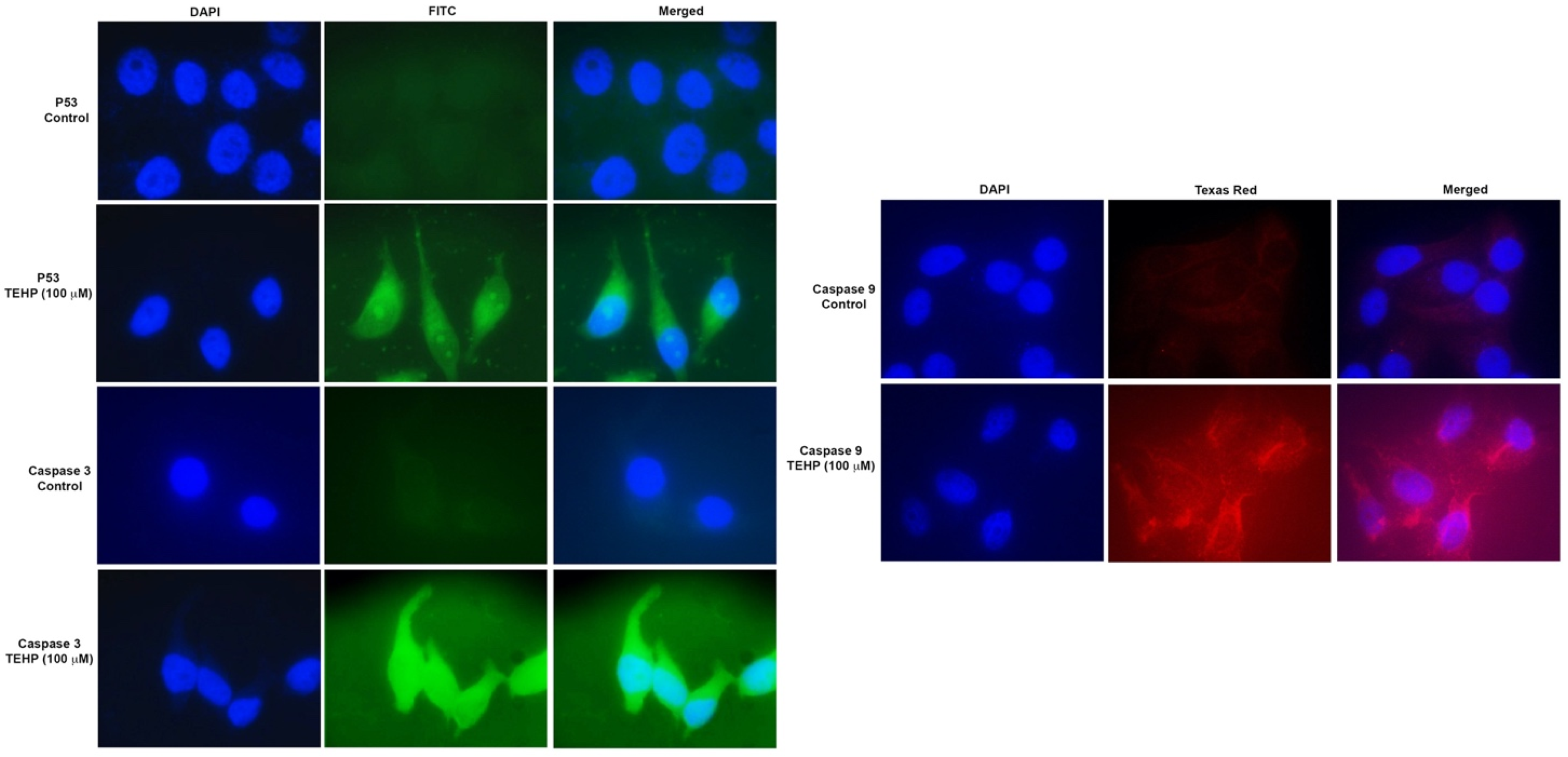
3.5. Translational Activation of Apoptotic Proteins
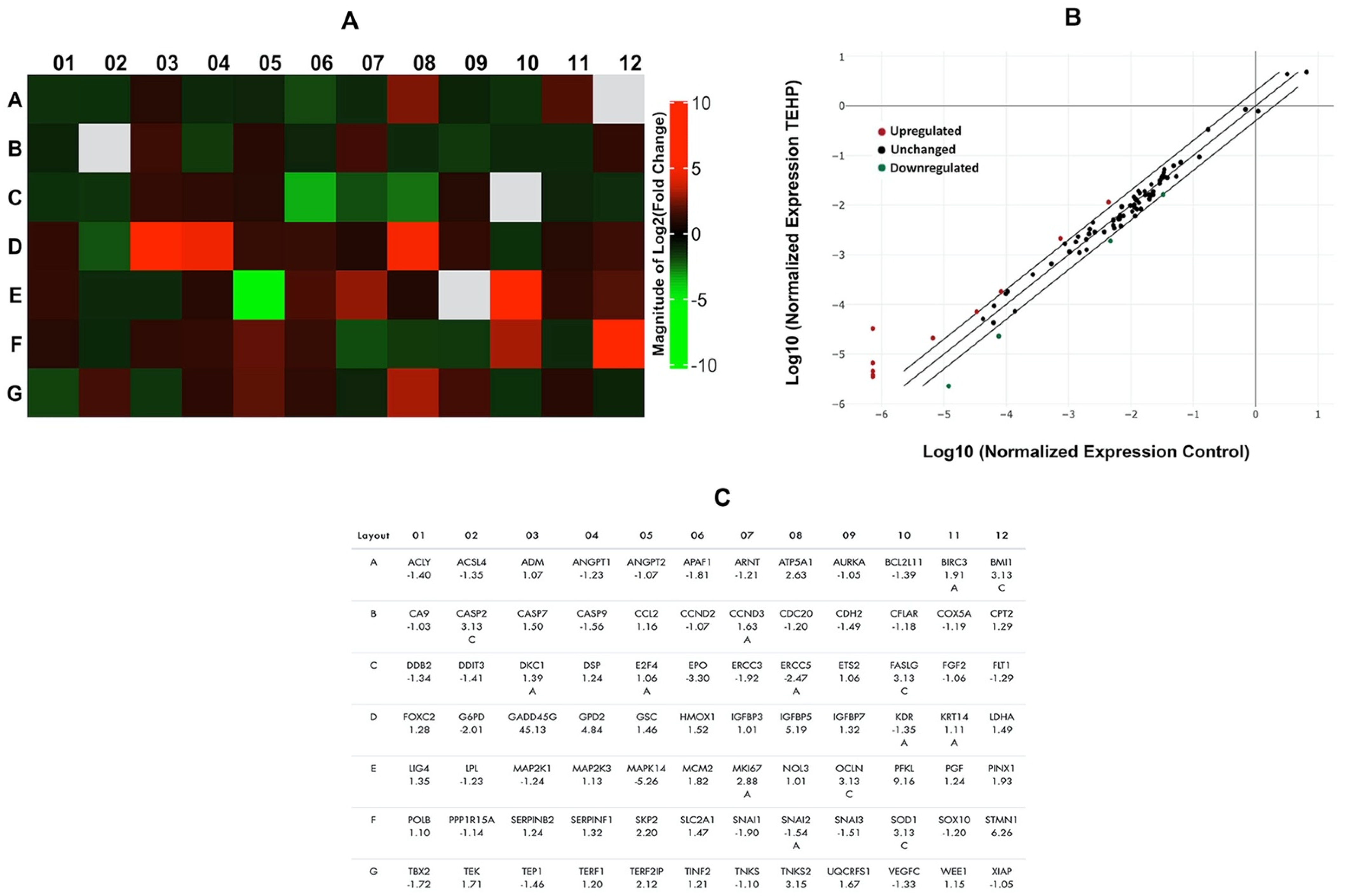
3.6. Activation of Cancer Pathway Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flameretardants-Online. The Flame Retardants Market. Available online: https://www.flameretardants-online.com/flame-retardants/market (accessed on 31 January 2022).
- Wang, X.; Zhu, Q.; Yan, X.; Wang, Y.; Liao, C.; Jiang, G. A review of organophosphate flame retardants and plasticizers in the environment: Analysis, occurrence and risk assessment. Sci. Total Environ. 2020, 731, 139071. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yin, L.; Wen, X.; Jiang, C.; Long, Y.; Zhang, J.; Liu, R. Organophosphate Esters in China: Fate, Occurrence, and Human Exposure. Toxics 2021, 9, 310. [Google Scholar] [CrossRef] [PubMed]
- Marklund, A.; Andersson, B.; Haglund, P. Organophosphorus flame retardants and plasticizers in air from various indoor environments. J. Environ. Monit. 2005, 7, 814–819. [Google Scholar] [CrossRef]
- Van den Eede, N.; Dirtu, A.C.; Neels, H.; Covaci, A. Analytical developments and preliminary assessment of human exposure to organophosphate flame retardants from indoor dust. Environ. Int. 2011, 37, 454–461. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, W.; Kannan, K.; Moon, H.B. Occurrence and exposure assessment of organophosphate flame retardants (OPFRs) through the consumption of drinking water in Korea. Water Res. 2016, 103, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.J.; Wang, Y.; Li, W.; Kannan, K. Occurrence of and human exposure to organophosphate flame retardants/plasticizers in indoor air and dust from various microenvironments in the United States. Environ. Int. 2019, 125, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, H.; Zhu, H.; Yao, Y.; Chen, H.; Ren, C.; Wu, F.; Kannan, K. Occurrence and distribution of organophosphate flame retardants (OPFRs) in soil and outdoor settled dust from a multi-waste recycling area in China. Sci. Total Environ. 2018, 625, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Yadav, I.C.; Devi, N.L.; Li, J.; Zhang, G.; Covaci, A. Concentration and spatial distribution of organophosphate esters in the soil-sediment profile of Kathmandu Valley, Nepal: Implication for risk assessment. Sci. Total Environ. 2018, 613–614, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Alhakamy, N.A.; Ismail, I.M.I.; Nazar, E.; Summan, A.S.; Shah Eqani, S.A.; Malarvannan, G. Exposure to Phthalate and Organophosphate Esters via Indoor Dust and PM10 Is a Cause of Concern for the Exposed Saudi Population. Int. J. Environ. Res. Public Health 2021, 18, 2125. [Google Scholar] [CrossRef]
- He, C.; Toms, L.M.L.; Thai, P.; Van den Eede, N.; Wang, X.; Li, Y.; Baduel, C.; Harden, F.A.; Heffernan, A.L.; Hobson, P. Urinary metabolites of organophosphate esters: Concentrations and age trends in Australian children. Environ. Int. 2018, 111, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Bai, X.Y.; Lu, S.Y.; Zhang, B.; Xie, L.; Zheng, H.C.; Jiang, Y.C.; Zhou, M.Z.; Zhou, Z.Q.; Song, S.M. Urinary metabolites of organophosphate flame retardants in China: Health risk from tris (2-chloroethyl) phosphate (TCEP) exposure. Environ. Int. 2018, 121, 1363–1371. [Google Scholar] [CrossRef]
- Caballero-Gallardo, K.; Olivero-Verbel, J.; Corada-Fernández, C.; Lara-Martín, P.A.; Juan-García, A. Emerging contaminants and priority substances in marine sediments from Cartagena Bay and the Grand Marsh of Santa Marta (Ramsar site), Colombia. Environ. Monit. Assess. 2021, 193, 596. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.; Chen, D.; Sun, J.; Greig, D.J.; Wu, Y. Characterization of brominated, chlorinated, and phosphate flame retardants in San Francisco Bay, an urban estuary. Sci. Total Environ. 2019, 652, 212–223. [Google Scholar] [CrossRef]
- Meng, Y.; Xu, X.; Niu, D.; Xu, Y.; Qiu, Y.; Zhu, Z.; Zhang, H.; Yin, D. Organophosphate flame retardants induce oxidative stress and Chop/Caspase 3-related apoptosis via Sod1/p53/Map3k6/Fkbp5 in NCI-1975 cells. Sci. Total Environ. 2022, 819, 153160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lu, M.; Dong, X.; Wang, C.; Zhang, C.; Liu, W.; Zhao, M. Potential Estrogenic Effects of Phosphorus-Containing Flame Retardants. Environ. Sci. Technol. 2014, 48, 6995–7001. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Fang, J.; Shi, Y.; Tang, S.; Dong, H.; Liu, Y.; Deng, F.; Giesy, J.P.; Godri Pollitt, K.J.; Cai, Y.; et al. Exposure to organophosphate esters in elderly people: Relationships of OPE body burdens with indoor air and dust concentrations and food consumption. Environ. Int. 2021, 157, 106803. [Google Scholar] [CrossRef]
- Kucharska, A.; Cequier, E.; Thomsen, C.; Becher, G.; Covaci, A.; Voorspoels, S. Assessment of human hair as an indicator of exposure to organophosphate flame retardants. Case study on a Norwegian mother–child cohort. Environ. Int. 2015, 83, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Poma, G.; Sales, C.; Bruyland, B.; Christia, C.; Goscinny, S.; Van Loco, J.; Covaci, A. Occurrence of Organophosphorus Flame Retardants and Plasticizers (PFRs) in Belgian Foodstuffs and Estimation of the Dietary Exposure of the Adult Population. Environ. Sci. Technol. 2018, 52, 2331–2338. [Google Scholar] [CrossRef]
- Zhang, X.; Zou, W.; Mu, L.; Chen, Y.; Ren, C.; Hu, X.; Zhou, Q. Rice ingestion is a major pathway for human exposure to organophosphate flame retardants (OPFRs) in China. J. Hazard. Mater. 2016, 318, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Gbadamosi, M.R.; Abdallah, M.A.E.; Harrad, S. A critical review of human exposure to organophosphate esters with a focus on dietary intake. Sci. Total Environ. 2021, 771, 144752. [Google Scholar] [CrossRef]
- National Toxicology Program. NTP Toxicology and Carcinogenesis Studies of Tris (2-ethylhexyl)phosphate (CAS No. 78-42-2) In F344/N Rats and B6C3F1 Mice (Gavage Studies). Natl. Toxicol. Program Tech. Rep. Ser. 1984, 274, 1–178. Available online: https://https://pubmed.ncbi.nlm.nih.gov/12748680/ (accessed on 2 April 2022).
- MacFarland, H.N.; Punte, C.L. Toxicological Studies on Tri-(2-Ethylhexyl)-Phosphate. Arch. Environ. Health Int. J. 1966, 13, 13–20. [Google Scholar] [CrossRef]
- Pelletier, G.; Rigden, M.; Wang, G.S.; Caldwell, D.; Siddique, S.; Leingartner, K.; Kosarac, I.; Cakmak, S.; Kubwabo, C. Comparison of tris(2-ethylhexyl) phosphate and di(2-ethylhexyl) phosphoric acid toxicities in a rat 28-day oral exposure study. J. Appl. Toxicol. JAT 2020, 40, 600–618. [Google Scholar] [CrossRef] [Green Version]
- Gerets, H.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.; Dhalluin, S.; Atienzar, F. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef] [Green Version]
- Reistad, T.; Mariussen, E.; Ring, A.; Fonnum, F. In vitro toxicity of tetrabromobisphenol-A on cerebellar granule cells: Cell death, free radical formation, calcium influx and extracellular glutamate. Toxicol. Sci. 2007, 96, 268–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronborg, T.M.; Hansen, J.F.; Rasmussen, Å.K.; Vorkamp, K.; Nielsen, C.H.; Frederiksen, M.; Hofman-Bang, J.; Hahn, C.H.; Ramhøj, L.; Feldt-Rasmussen, U. The flame retardant DE-71 (a mixture of polybrominated diphenyl ethers) inhibits human differentiated thyroid cell function in vitro. PLoS ONE 2017, 12, e0179858. [Google Scholar] [CrossRef] [Green Version]
- Saquib, Q.; Siddiqui, M.A.; Ahmad, J.; Ansari, S.M.; Al-Wathnani, H.A.; Rensing, C. 6-OHBDE-47 induces transcriptomic alterations of CYP1A1, XRCC2, HSPA1A, EGR1 genes and trigger apoptosis in HepG2 cells. Toxicology 2018, 400, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Al-Salem, A.M.; Saquib, Q.; Siddiqui, M.A.; Ahmad, J.; Wahab, R.; Al-Khedhairy, A.A. Organophosphorus flame retardant (tricresyl phosphate) trigger apoptosis in HepG2 cells: Transcriptomic evidence on activation of human cancer pathways. Chemosphere 2019, 237, 124519. [Google Scholar] [CrossRef]
- Saquib, Q.; Al-Khedhairy, A.A.; Al-Arifi, S.; Dhawan, A.; Musarrat, J. Assessment of methyl thiophanate–Cu (II) induced DNA damage in human lymphocytes. Toxicol. In Vitro 2009, 23, 848–854. [Google Scholar] [CrossRef]
- Fernández-Arribas, J.; Moreno, T.; Bartrolí, R.; Eljarrat, E. COVID-19 face masks: A new source of human and environmental exposure to organophosphate esters. Environ. Int. 2021, 154, 106654. [Google Scholar] [CrossRef] [PubMed]
- LeCluyse, E.L. Human hepatocyte culture systems for the in vitro evaluation of cytochrome P450 expression and regulation. Eur. J. Pharm. Sci. 2001, 13, 343–368. [Google Scholar] [CrossRef]
- Li, A.P. Human hepatocytes: Isolation, cryopreservation and applications in drug development. Chem.-Biol. Interact. 2007, 168, 16–29. [Google Scholar] [CrossRef]
- Roymans, D.; Van Looveren, C.; Leone, A.; Parker, J.B.; McMillian, M.; Johnson, M.D.; Koganti, A.; Gilissen, R.; Silber, P.; Mannens, G.; et al. Determination of cytochrome P450 1A2 and cytochrome P450 3A4 induction in cryopreserved human hepatocytes. Biochem. Pharmacol. 2004, 67, 427–437. [Google Scholar] [CrossRef]
- Javitt, N.B. Hep G2 cells as a resource for metabolic studies: Lipoprotein, cholesterol, and bile acids. FASEB J. 1990, 4, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L.; Fracanzani, A.L.; Gatti, S.; Cairo, G.; Fargion, S. Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am. J. Pathol. 2008, 172, 738–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, M.T.; Tolosa, L.; Gómez-Lechón, M.J. Culture and Functional Characterization of Human Hepatoma HepG2 Cells. In Protocols in In Vitro Hepatocyte Research; Vinken, M., Rogiers, V., Eds.; Springer: New York, NY, USA, 2015; pp. 77–93. [Google Scholar] [CrossRef]
- Gomez-Lechon, J.M.; Donato, T.M.; Lahoz, A.; Castell, V.J. Cell Lines: A Tool for In Vitro Drug Metabolism Studies. Curr. Drug Metab. 2008, 9, 1–11. [Google Scholar] [CrossRef]
- Liu, C.L.; Lim, Y.-P.; Hu, M.L. Fucoxanthin attenuates rifampin-induced cytochrome P450 3A4 (CYP3A4) and multiple drug resistance 1 (MDR1) gene expression through pregnane X receptor (PXR)-mediated pathways in human hepatoma HepG2 and colon adenocarcinoma LS174T cells. Mar. Drugs 2012, 10, 242–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirona, R.G.; Lee, W.; Leake, B.F.; Lan, L.B.; Cline, C.B.; Lamba, V.; Parviz, F.; Duncan, S.A.; Inoue, Y.; Gonzalez, F.J.; et al. The orphan nuclear receptor HNF4α determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat. Med. 2003, 9, 220–224. [Google Scholar] [CrossRef]
- Chen, Z.; Xie, J.; Li, Q.; Hu, K.; Yang, Z.; Yu, H.; Liu, Y. Human CYP enzyme-activated clastogenicity of 2-ethylhexyl diphenyl phosphate (a flame retardant) in mammalian cells. Environ. Pollut. 2021, 285, 117527. [Google Scholar] [CrossRef] [PubMed]
- Wilkening, S.; Stahl, F.; Bader, A. Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metab. Dispos. 2003, 31, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Xiang, P.; He, R.W.; Liu, R.Y.; Li, K.; Gao, P.; Cui, X.Y.; Li, H.; Liu, Y.; Ma, L.Q. Cellular responses of normal (HL-7702) and cancerous (HepG2) hepatic cells to dust extract exposure. Chemosphere 2018, 193, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Saquib, Q.; Siddiqui, M.A.; Ahmed, J.; Al-Salim, A.; Ansari, S.M.; Faisal, M.; Al-Khedhairy, A.A.; Musarrat, J.; AlWathnani, H.A.; Alatar, A.A. Hazards of low dose flame-retardants (BDE-47 and BDE-32): Influence on transcriptome regulation and cell death in human liver cells. J. Hazard. Mater. 2016, 308, 37–49. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M.K. Mechanisms of Cell Death in Oxidative Stress. Antioxid. Redox Signal. 2006, 9, 49–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Duan, X.M.; Liu, L.H.; Fang, Y.Q.; Tan, Y. Carboxyfluorescein Diacetate Succinimidyl Ester Fluorescent Dye for Cell Labeling. Acta Biochim. Biophys. Sin. 2005, 37, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Al-Salem, A.M.; Saquib, Q.; Siddiqui, M.A.; Ahmad, J.; Al-Khedhairy, A.A. Tris(2-chloroethyl) Phosphate (TCEP) Elicits Hepatotoxicity by Activating Human Cancer Pathway Genes in HepG2 Cells. Toxics 2020, 8, 109. [Google Scholar] [CrossRef]
- Norbury, C.J.; Zhivotovsky, B. DNA damage-induced apoptosis. Oncogene 2004, 23, 2797–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtgraaf, J.H.; Versmissen, J.; van der Giessen, W.J. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc. Revasc. Med. 2006, 7, 165–172. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, R.; Giesy, J.P.; Li, Y.; Wang, P. Tris (1,3-dichloro-2-propyl) phosphate treatment induces DNA damage, cell cycle arrest and apoptosis in murine RAW264.7 macrophages. J. Toxicol. Sci. 2019, 44, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, M.S.; Fornace, A.J., Jr. Role of p53 family members in apoptosis. J. Cell. Physiol. 2000, 182, 171–181. [Google Scholar] [CrossRef]
- El-Deiry, W.S. Regulation of p53 downstream genes. Semin. Cancer Biol. 1998, 8, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potokar, M.; Milisav, I.; Kreft, M.; Stenovec, M.; Zorec, R. Apoptosis triggered redistribution of caspase-9 from cytoplasm to mitochondria. FEBS Lett. 2003, 544, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.E.; de Vasconcellos, J.F.; Sarkar, D.; Libermann, T.A.; Fisher, P.B.; Zerbini, L.F. GADD45 proteins: Central players in tumorigenesis. Curr. Mol. Med. 2012, 12, 634–651. [Google Scholar] [CrossRef]
- Walsh, C.S.; Ogawa, S.; Karahashi, H.; Scoles, D.R.; Pavelka, J.C.; Tran, H.; Miller, C.W.; Kawamata, N.; Ginther, C.; Dering, J.; et al. ERCC5 is a novel biomarker of ovarian cancer prognosis. J. Clin. Oncol. 2008, 26, 2952–2958. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; de Vries, M.C.; Smeitink, J.A. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 2013, 136, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiński, M.M.; Sauer, S.W.; Kamiński, M.; Opp, S.; Ruppert, T.; Grigaravičius, P.; Grudnik, P.; Gröne, H.-J.; Krammer, P.H.; Gülow, K. T cell Activation Is Driven by an ADP-Dependent Glucokinase Linking Enhanced Glycolysis with Mitochondrial Reactive Oxygen Species Generation. Cell Rep. 2012, 2, 1300–1315. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhang, W.; Zhang, N.; Zhou, Y.; Hu, G.; Xue, M.; Liu, J.; Li, Y. Down-regulation of insulin-like growth factor binding protein 5 is involved in intervertebral disc degeneration via the ERK signalling pathway. J. Cell. Mol. Med. 2019, 23, 6368–6377. [Google Scholar] [CrossRef]
- Gleason, C.E.; Ning, Y.; Cominski, T.P.; Gupta, R.; Kaestner, K.H.; Pintar, J.E.; Birnbaum, M.J. Role of Insulin-Like Growth Factor-Binding Protein 5 (IGFBP5) in Organismal and Pancreatic β-Cell Growth. Mol. Endocrinol. 2010, 24, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Fang, W.; Zhang, G.; Jia, L.; Lei, W.; Jia, R.; Li, T. Studies on Cataclastic Lithofacies and Copper Metallogenic Regularity for the Glutenite-type Sareke Copper deposit, Xinjiang. Acta Geol. Sin.-Engl. Ed. 2017, 91, 210–211. [Google Scholar] [CrossRef] [Green Version]
- Marshman, E.; Green, K.A.; Flint, D.J.; White, A.; Streuli, C.H.; Westwood, M. Insulin-like growth factor binding protein 5 and apoptosis in mammary epithelial cells. J. Cell Sci. 2003, 116, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochrie, J.D.; Phillips, K.; Tonner, E.; Flint, D.J.; Allan, G.J.; Price, N.C.; Beattie, J. Insulin-like growth factor binding protein (IGFBP)-5 is upregulated during both differentiation and apoptosis in primary cultures of mouse mammary epithelial cells. J. Cell. Physiol. 2006, 207, 471–479. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, Y.; Cai, Y.; Liu, R.; Lu, M.; Li, T.; Fu, Y.; Guo, M.; Huang, H.; Ou, Y.; et al. A20 targets PFKL and glycolysis to inhibit the progression of hepatocellular carcinoma. Cell Death Dis. 2020, 11, 89. [Google Scholar] [CrossRef]
- Igea, A.; Nebreda, A.R. The Stress Kinase p38α as a Target for Cancer Therapy. Cancer Res. 2015, 75, 3997–4002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yu, X.; Yu, H.; Liu, B.; Zhang, Z.; Kong, C.; Li, Z. Knockdown of MAPK14 inhibits the proliferation and migration of clear cell renal cell carcinoma by downregulating the expression of CDC25B. Cancer Med. 2020, 9, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Hu, J.; Shang, Y.; Zhong, Y.F.; Yu, Z.; An, J. Tributylphosphate (TBP) and tris (2-butoxyethyl) phosphate (TBEP) induced apoptosis and cell cycle arrest in HepG2 cells. Toxicol. Res. 2017, 6, 902–911. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Hu, J.; Shang, Y.; Zhong, Y.; Zhang, X.; Yu, Z. The cytotoxicity of organophosphate flame retardants on HepG2, A549 and Caco-2 cells. J. Environ. Sci. Health A Toxic Hazard. Subst. Environ. Eng. 2016, 51, 980–988. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Olive Tail Moment (Arbitrary Unit) | Tail Intensity (%) |
|---|---|---|
| Control | 0.65 ± 0.09 | 3.23 ± 0.33 |
| EMS (2 mM) | 12.26 ± 1.88 ** | 34.21 ± 2.65 ** |
| TEHP (µM) | ||
| 100 | 3.04 ± 0.41 ** | 10.54 ± 1.39 ** |
| 200 | 6.08 ± 1.40 ** | 14.27 ± 0.92 ** |
| 400 | 8.96 ± 0.92 ** | 20.23 ± 1.66 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saquib, Q.; Al-Salem, A.M.; Siddiqui, M.A.; Ansari, S.M.; Zhang, X.; Al-Khedhairy, A.A. Cyto-Genotoxic and Transcriptomic Alterations in Human Liver Cells by Tris (2-Ethylhexyl) Phosphate (TEHP): A Putative Hepatocarcinogen. Int. J. Mol. Sci. 2022, 23, 3998. https://doi.org/10.3390/ijms23073998
Saquib Q, Al-Salem AM, Siddiqui MA, Ansari SM, Zhang X, Al-Khedhairy AA. Cyto-Genotoxic and Transcriptomic Alterations in Human Liver Cells by Tris (2-Ethylhexyl) Phosphate (TEHP): A Putative Hepatocarcinogen. International Journal of Molecular Sciences. 2022; 23(7):3998. https://doi.org/10.3390/ijms23073998
Chicago/Turabian StyleSaquib, Quaiser, Abdullah M. Al-Salem, Maqsood A. Siddiqui, Sabiha M. Ansari, Xiaowei Zhang, and Abdulaziz A. Al-Khedhairy. 2022. "Cyto-Genotoxic and Transcriptomic Alterations in Human Liver Cells by Tris (2-Ethylhexyl) Phosphate (TEHP): A Putative Hepatocarcinogen" International Journal of Molecular Sciences 23, no. 7: 3998. https://doi.org/10.3390/ijms23073998