
In Silico Drug Repositioning to Target the SARS-CoV-2 Main Protease as Covalent Inhibitors Employing a Combined Structure-Based Virtual Screening Strategy of Pharmacophore Models and Covalent Docking

 ,
,  , , and
, , and
Abstract
:1. Introduction
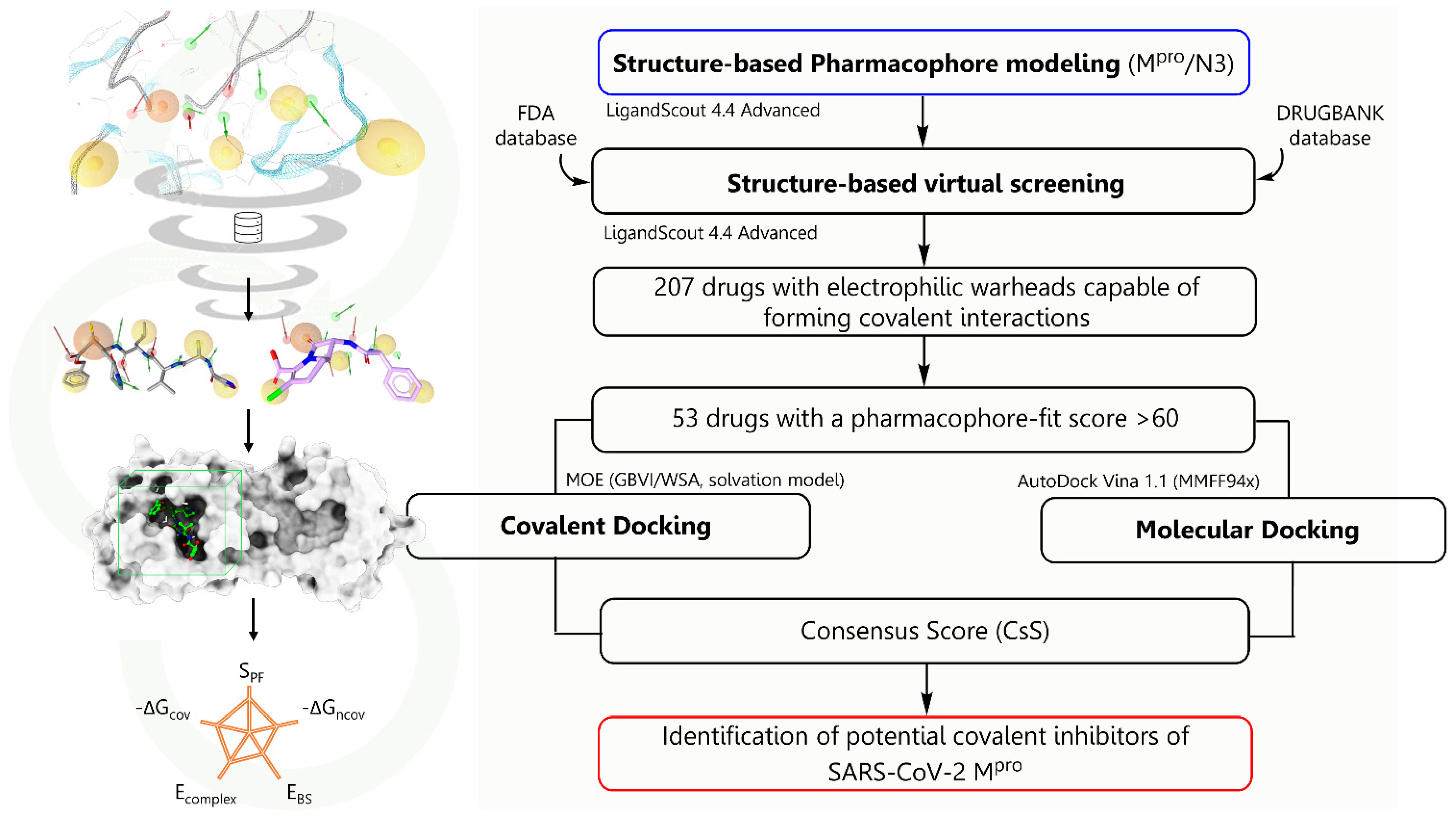
2. Results and Discussion
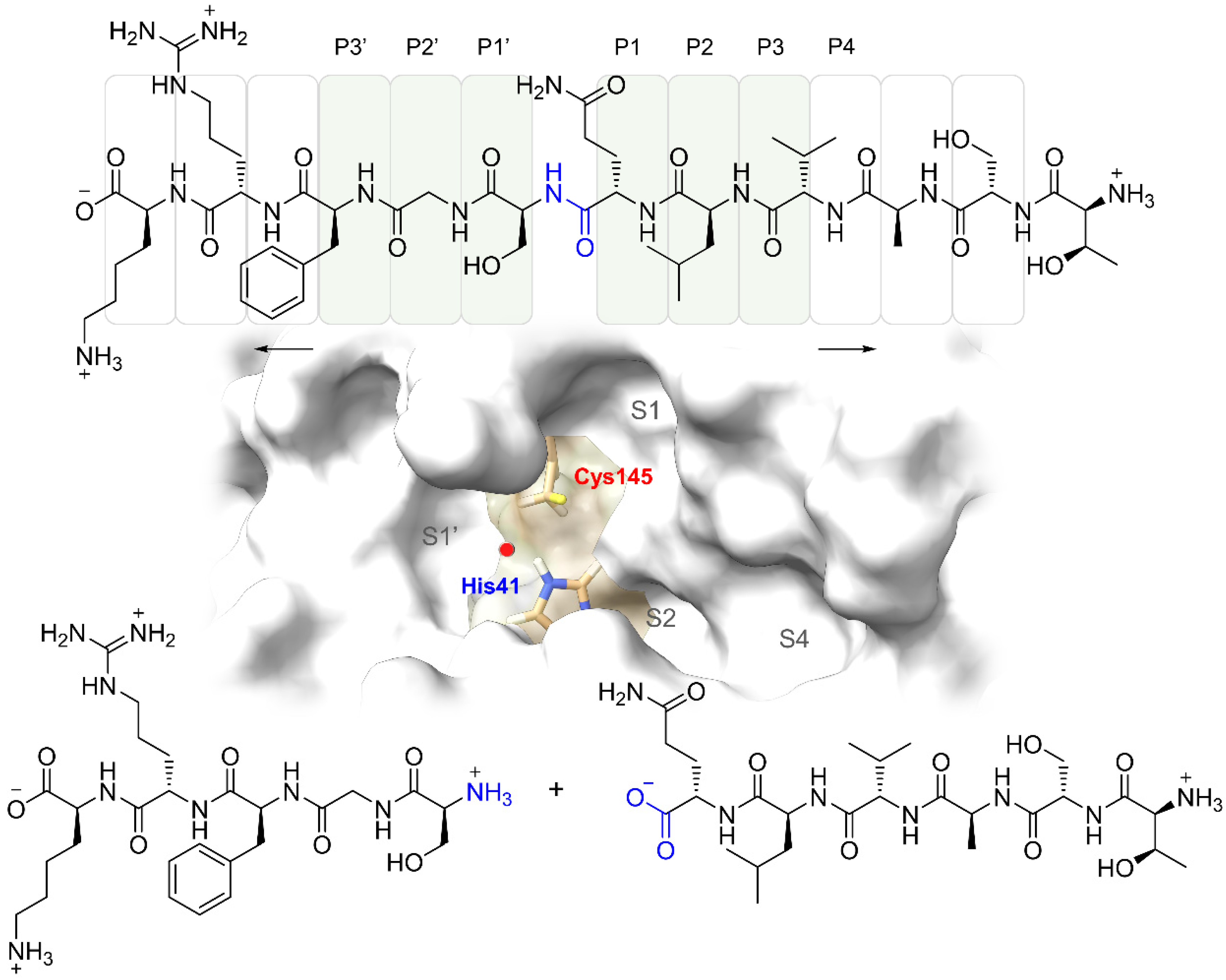
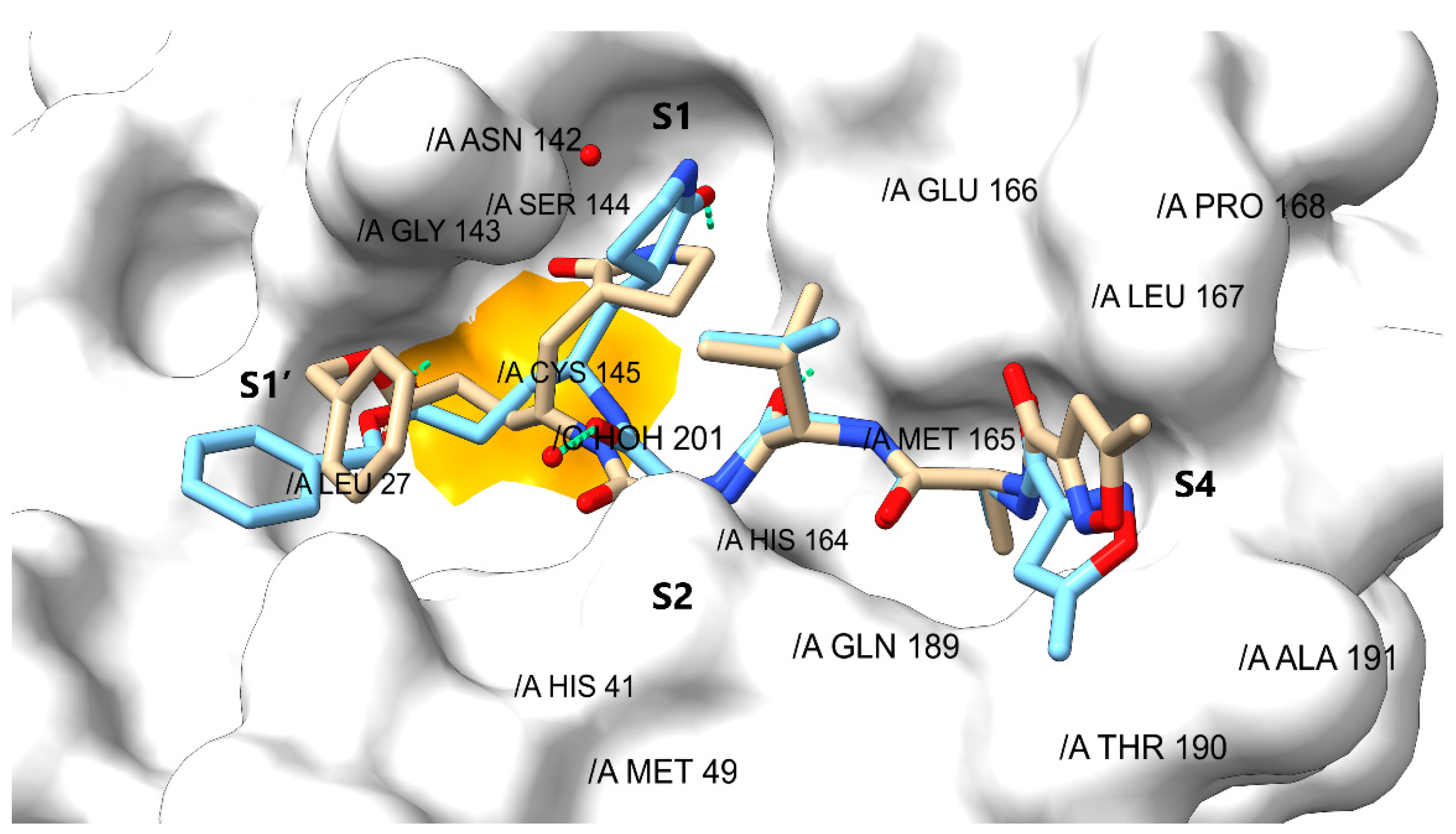
2.1. Structure-Based Pharmacophoric Modeling
2.2. Virtual Screening on the Structure-Based Pharmacophoric Map
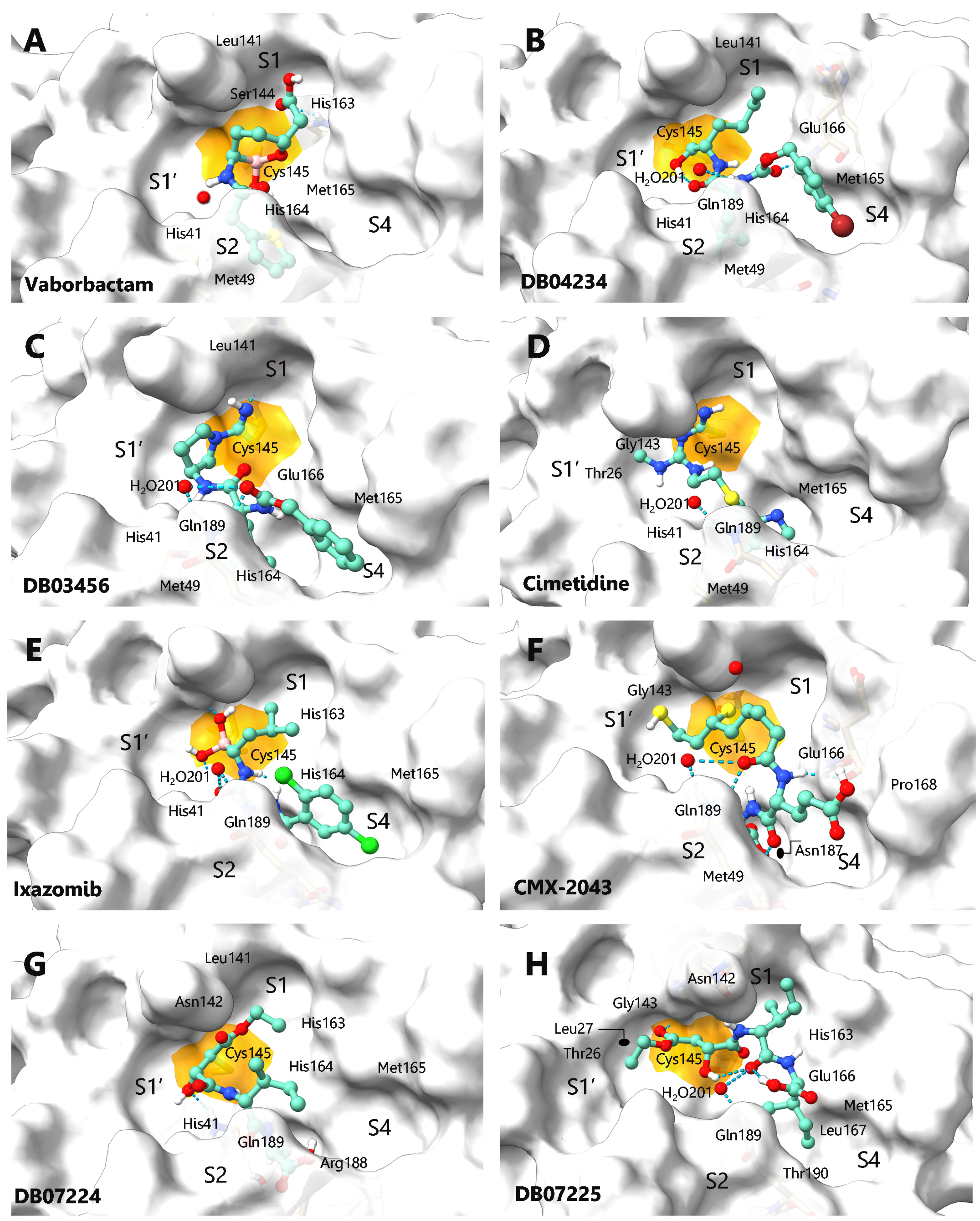
2.3. Validation and Covalent Docking Dependent Virtual Screening
3. Materials and Methods
3.1. Drug Database
3.2. Structure-Based (SB) Pharmacophore Model
3.3. Pharmacophore Model-Based Virtual Screening
3.4. Covalent Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OMS, Coronavirus Disease 2019 (COVID-19) Situation Report. 2021. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---28-december-2021 (accessed on 30 December 2021).
- Zappulli, V.; Ferro, S.; Bonsembiante, F.; Brocca, G.; Calore, A.; Cavicchioli, L.; Centelleghe, C.; Corazzola, G.; De Vreese, S.; Gelain, M.E.; et al. Pathology of coronavirus infections: A review of lesions in animals in the one-health perspective. Animals 2020, 10, 2377. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Peng, W.; Zhu, Y.; Lu, S.; Zhou, M.; Lin, W.; Wu, W.; Huang, S.; Jiang, L.; Luo, X.; et al. Recent progress in understanding 2019 novel coronavirus (SARS-CoV-2) associated with human respiratory disease: Detection, mechanisms and treatment. Int. J. Antimicrob. Agents. 2020, 55, 105950. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Li, D.; Tong, W.; Shi, T.; Ning, B. Biochemical features, and mutations of key proteins in SARS-CoV-2 and their impacts on RNA therapeutics. Biochem. Pharmacol. 2021, 189, 114424. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.T.H.; Moesser, M.A.; Walters, R.K.; Malla, T.R.; Twidale, R.M.; John, T.; Deeks, H.M.; Johnston-Wood, T.; Mikhailov, V.; Sessions, R.B.; et al. Discovery of SARS-CoV-2 Mpro peptide inhibitors from modelling substrate and ligand binding. Chem. Sci. 2021, 12, 13686–13703. [Google Scholar] [CrossRef]
- Petri, L.; Egyed, A.; Bajusz, D.; Imre, T.; Hetényi, A.; Martinek, T.; Ábrányi-Balogh, P.; Keserű, G.M. An electrophilic warhead library for mapping the reactivity and accessibility of tractable cysteines in protein kinases. Eur. J. Med. Chem. 2020, 207, 112836. [Google Scholar] [CrossRef]
- Shen, X.; Wu, C.; Lei, M.; Yan, Q.; Zhang, H.; Zhang, L.; Wang, X.; Yang, Y.; Li, J.; Zhu, Y.; et al. Anti-tumor activity of a novel proteasome inhibitor D395 against multiple myeloma and its lower cardiotoxicity compared with carfilzomib. Cell Death Dis. 2021, 12, 429. [Google Scholar] [CrossRef]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2S,3R)-N-((S)-3-(Cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar] [CrossRef]
- Mathew, G.E.; Oh, J.M.; Mohan, K.; Tengli, A.; Mathew, B.; Kim, H. Development of methylthiosemicarbazones as new reversible monoamine oxidase-B inhibitors for the treatment of Parkinson’s disease. J. Biomol. Struct. Dyn. 2021, 39, 4786–4794. [Google Scholar] [CrossRef]
- Soubhye, J.; Van Antwerpen, P.; Dufrasne, F. A patent review of myeloperoxidase inhibitors for treating chronic inflammatory syndromes (focus on cardiovascular diseases, 2013–2019). Expert Opin. Ther. Pat. 2020, 30, 595–608. [Google Scholar] [CrossRef]
- Adhikari, A.A.; Seegar, T.; Ficarro, S.B.; McCurry, M.D.; Ramachandran, D.; Yao, L.; Chaudhari, S.N.; Ndousse-Fetter, S.; Banks, A.S.; Marto, J.A.; et al. Development of a covalent inhibitor of gut bacterial bile salt hydrolases. Nat. Chem. Biol. 2020, 16, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ye, X.; Yang, X.; Cai, Y.; Wang, S.; Tang, J.; Sachdeva, M.; Qian, Y.; Hu, W.; Leeds, J.A.; et al. Discovery of novel antibiotics as covalent inhibitors of fatty acid synthesis. ACS Chem. Biol. 2020, 15, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Mirzaie, S.; Abdi, F.; GhavamiNejad, A.; Lu, B.; Wu, X.Y. Covalent antiviral agents. In Antiviral Drug Discovery and Development; Liu, X., Zhan, P., Menéndez-Arias, L., Poongavanam, V., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2021; Volume 1322. [Google Scholar] [CrossRef]
- Gehringer, M. Covalent inhibitors: Back on track? Future Med. Chem. 2020, 12, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Amendola, G.; Ettari, R.; Previti, S.; Di Chio, C.; Messere, A.; Di Maro, S.; Hammerschmidt, S.J.; Zimmer, C.; Zimmermann, R.A.; Schirmeister, T.; et al. Lead discovery of SARS-CoV-2 main protease inhibitors through covalent docking-based virtual screening. J. Chem. Inf. Model. 2021, 61, 2062–2073. [Google Scholar] [CrossRef] [PubMed]
- Awoonor-Williams, E.; Abu-Saleh, A.A.A. Covalent and non-covalent binding free energy calculations for peptidomimetic inhibitors of SARS-CoV-2 main protease. Phys. Chem. Chem. Phys. 2021, 23, 6746–6757. [Google Scholar] [CrossRef] [PubMed]
- A Post-Exposure Prophylaxis Study of PF-07321332/Ritonavir in Adult Household Contacts of an Individual With Symptomatic COVID-19. NCT05047601. NIH U.S. National Library of Medicine. Available online: https://www.clinicaltrials.gov/ct2/show/NCT05047601 (accessed on 27 September 2021).
- Mondal, D.; Warshel, A. Exploring the mechanism of covalent inhibition: Simulating the binding free energy of α-ketoamide inhibitors of the main protease of SARS-CoV-2. Biochemistry 2020, 59, 4601–4608. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.H.; Turki, T. A new advanced in silico drug discovery method for novel coronavirus (SARS-CoV-2) with tensor decomposition-based unsupervised feature extraction. PLoS ONE 2020, 15, e0238907. [Google Scholar] [CrossRef]
- Luo, H.; Li, M.; Yang, M.; Wu, F.X.; Li, Y.; Wang, J. Biomedical data and computational models for drug repositioning: A comprehensive review. Brief Bioinform. 2021, 22, 1604–1619. [Google Scholar] [CrossRef]
- Yousefi, H.; Mashouri, L.; Okpechi, S.C.; Alahari, N.; Alahari, S.K. Repurposing existing drugs for the treatment of COVID-19/SARS-CoV-2 infection: A review describing drug mechanisms of action. Biochem. Pharmacol. 2021, 183, 114296. [Google Scholar] [CrossRef]
- Serafim, M.S.M.; Gertrudes, J.C.; Costa, D.M.A.; Oliveira, P.R.; Maltarollo, V.G.; Honorio, K.M. Knowing and combating the enemy: A brief review on SARS-CoV-2 and computational approaches applied to the discovery of drug candidates. Biosci. Rep. 2021, 41, BSR20202616. [Google Scholar] [CrossRef] [PubMed]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste makes waste: A critical review of docking-based virtual screening in drug repurposing for SARS-CoV-2 main protease (M-pro) inhibition. Med. Res. Rev. 2021, 42, 744–769. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Mekonnen, D.; Mohammed, A.; Shi, R.; Jin, T. Potency, safety, and pharmacokinetic profiles of potential inhibitors targeting SARS-CoV-2 main protease. Front. Pharmacol. 2021, 11, 630500. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Verma, A.; Bihani, S.; Burli, A.; Mantri, K.; Srivastava, S. Proteomics advances towards developing SARS-CoV-2 therapeutics using in silico drug repurposing approaches. Drug. Discov. Today. Technol. 2021, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Günther, S.; Reinke, P.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef]
- Ferreira, G.M.; Kronenberger, T.; Tonduru, A.K.; Hirata, R.D.C.; Hirata, M.H.; Poso, A. SARS-CoV-2 Mpro conformational changes induced by covalently bound ligands. J. Biomol. Struct. Dyn. 2021, 15, 1–11. [Google Scholar] [CrossRef]
- Mitra, K.; Ghanta, P.; Acharya, S.; Chakrapani, G.; Ramaiah, B.; Doble, M. Dual inhibitors of SARS-CoV-2 proteases: Pharmacophore and molecular dynamics based drug repositioning and phytochemical leads. J. Biomol. Struct. Dyn. 2021, 39, 6324–6337. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Arafa, R.K. On the search for COVID-19 therapeutics: Identification of potential SARS-CoV-2 main protease inhibitors by virtual screening, pharmacophore modeling and molecular dynamics. J. Biomol. Struct. Dyn. 2021, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Battisti, V.; Wieder, O.; Garon, A.; Seidel, T.; Urban, E.; Langer, T.A. Computational approach to identify potential novel inhibitors against the coronavirus SARS-CoV-2. Mol. Inform. 2020, 39, e2000090. [Google Scholar] [CrossRef] [PubMed]
- Balaramnavar, V.M.; Ahmad, K.; Saeed, M.; Ahmad, I.; Kamal, M.; Jawed, T. Pharmacophore-based approaches in the rational repurposing technique for FDA approved drugs targeting SARS-CoV-2 Mpro. RSC Adv. 2020, 10, 40264–40275. [Google Scholar] [CrossRef]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci. Rep. 2020, 10, 12493. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, H.; Shen, C.; Hu, X.; Gao, J.; Li, D.; Cao, D.; Hou, T. Combined strategies in structure-based virtual screening. Phys. Chem. Chem. Phys. 2020, 22, 3149–3159. [Google Scholar] [CrossRef]
- Jiang, Z.; Xu, J.; Yan, A.; Wang, L. A comprehensive comparative assessment of 3D molecular similarity tools in ligand-based virtual screening. Brief. Bioinform. 2021, 22, bbab231. [Google Scholar] [CrossRef]
- Becker, D.; Kaczmarska, Z.; Arkona, C.; Schulz, R.; Tauber, C.; Wolber, G.; Hilgenfeld, R.; Coll, M.; Rademann, J. Irreversible inhibitors of the 3C protease of Coxsackie virus through templated assembly of protein-binding fragments. Nat. Commun. 2016, 7, 12761. [Google Scholar] [CrossRef] [Green Version]
- Schulz, R.; Atef, A.; Becker, D.; Gottschalk, F.; Tauber, C.; Wagner, S.; Arkona, C.; Abdel-Hafez, A.A.; Farag, H.H.; Rademann, J.; et al. Phenylthiomethyl ketone-based fragments show selective and irreversible inhibition of enteroviral 3C proteases. J Med. Chem. 2018, 61, 1218–1230. [Google Scholar] [CrossRef]
- Alamri, M.A.; Tahir, U.; Qamar, M.; Afzal, O.; Alabbas, A.B.; Riadi, Y.; Alqahtani, S.M. Discovery of anti-MERS-CoV small covalent inhibitors through pharmacophore modeling, covalent docking and molecular dynamics simulation. J. Mol. Liq. 2021, 330, 115699. [Google Scholar] [CrossRef]
- Luo, D.; Tong, J.B.; Zhang, X.; Xiao, X.C.; Bian, S. Computational strategies towards developing novel SARS-CoV-2 Mpro inhibitors against COVID-19. J. Mol. Struct. 2021, 1247, 131378. [Google Scholar] [CrossRef]
- Pathak, N.; Chen, Y.T.; Hsu, Y.C.; Hsu, N.Y.; Kuo, C.J.; Tsai, H.P.; Kang, J.J.; Huang, C.H.; Chang, S.Y.; Chang, Y.H.; et al. Uncovering flexible active site conformations of SARS-CoV-2 3CL proteases through protease pharmacophore clusters and COVID-19 drug repurposing. ACS Nano. 2021, 15, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Su, H.; Zhao, W.; Xie, H.; Shao, Q.; Xu, Y. What coronavirus 3C-like protease tells us: From structure, substrate selectivity, to inhibitor design. Med. Res. Rev. 2021, 41, 1965–1998. [Google Scholar] [CrossRef] [PubMed]
- Palacio-Rodríguez, K.; Lans, I.; Cavasotto, C.N.; Cossio, P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 2019, 9, 5142. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Li, Y.S.; Zeng, R.; Liu, F.L.; Luo, R.H.; Huang, C.; Wang, Y.F.; Zhang, J.; Quan, B.; Shen, C.; et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Arafet, K.; Serrano-Aparicio, N.; Lodola, A.; Mulholland, A.J.; González, F.V.; Świderek, K.; Moliner, V. Mechanism of inhibition of SARS-CoV-2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 2020, 12, 1433–1444. [Google Scholar] [CrossRef]
- Vega-Valdez, I.R.; Rosalez Melvin, N.; Santiago-Quintana, J.M.; Farfán-García, E.D.; Soriano-Ursúa, M.A. Docking simulations exhibit bortezomib and other boron-containing peptidomimetics as potential inhibitors of SARS-CoV-2 main protease. Curr. Chem. Biol. 2020, 14, 279–288. [Google Scholar] [CrossRef]
- Pan, X.; Li, X.; Ning, S.; Zhi, H. Inferring SARS-CoV-2 functional genomics from viral transcriptome with identification of potential antiviral drugs and therapeutic targets. Cell Biosci. 2021, 11, 171. [Google Scholar] [CrossRef]
- Graham, B.J.; Windsor, I.W.; Gold, B.; Raines, R.T. Boronic acid with high oxidative stability and utility in biological contexts. Proc. Natl. Acad. Sci. USA 2021, 118, e2013691118. [Google Scholar] [CrossRef]
- Tsivkovski, R.; Lomovskaya, O. Biochemical activity of vaborbactam. Antimicrob. Agents Chemother. 2020, 64, e01935-19. [Google Scholar] [CrossRef] [Green Version]
- Gossen, J.; Albani, S.; Hanke, A.; Joseph, B.P.; Bergh, C.; Kuzikov, M.; Costanzi, E.; Manelfi, C.; Storici, P.; Gribbon, P.; et al. A blueprint for high affinity SARS-CoV-2 Mpro inhibitors from activity-based compound library screening guided by analysis of protein dynamics. ACS Pharmacol. Transl. Sci. 2021, 4, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Fuhler, G.M.; Pan, Y. Could histamine H1 receptor antagonists be used for treating COVID-19? Int. J. Mol. Sci. 2021, 22, 5672. [Google Scholar] [CrossRef] [PubMed]
- Effect of H2 Receptor Antagonist and Proton Pump Inhibitor on the Positivity Rates and Clinical Outcomes of COVID-19. NCT04834752. NIH U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT04834752 (accessed on 28 September 2021).
- Shitrit, A.; Zaidman, D.; Kalid, O.; Bloch, I.; Doron, D.; Yarnizky, T.; Buch, I.; Segev, I.; Ben-Zeev, E.; Segev, E.; et al. Conserved interactions required for inhibition of the main protease of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Sci. Rep. 2020, 10, 20808. [Google Scholar] [CrossRef] [PubMed]
- Bicalutamide to Block TMPRSS2 in Males with COVID-19 Infection. NCT04509999. NIH U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT04509999 (accessed on 28 September 2021).
- Leach, D.A.; Mohr, A.; Giotis, E.S.; Cil, E.; Isac, A.M.; Yates, L.L.; Barclay, W.S.; Zwacka, R.M.; Bevan, C.L.; Brooke, G.N. The antiandrogen enzalutamide downregulates TMPRSS2 and reduces cellular entry of SARS-CoV-2 in human lung cells. Nat. Commun. 2021, 12, 4068. [Google Scholar] [CrossRef] [PubMed]
- Unni, S.; Aouti, S.; Thiyagarajan, S.; Padmanabhan, B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020, 45, 130. [Google Scholar] [CrossRef]
- Malla, T.R.; Tumber, A.; John, T.; Brewitz, L.; Strain-Damerell, C.; Owen, C.D.; Lukacik, P.; Chan, H.T.H.; Maheswaran, P.; Salah, E.; et al. Mass spectrometry reveals potential of β-lactams as SARS-CoV-2 Mpro inhibitors. Chem. Commun. 2021, 57, 1430–1433. [Google Scholar] [CrossRef] [PubMed]
- Chedid, M.; Waked, R.; Haddad, E.; Chetata, N.; Saliba, G.; Choucair, J. Antibiotics in treatment of COVID-19 complications: A review of frequency, indications, and efficacy. J. Infect. Public Health 2021, 14, 570–576. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL Proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, eabl4784. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell. 2021, 22, 1–5. [Google Scholar] [CrossRef]
- Study of PF-07321332 in Healthy Participants. NCT04756531. NIH U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT04756531 (accessed on 19 October 2021).
- Patel, S.; Homaei, A.; El-Seedi, H.R.; Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 2018, 105, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Yang, W.L.; Yang, F.Y.; Zhang, L.; Huang, W.J.; Hou, W.; Fan, C.F.; Jin, R.H.; Feng, Y.M.; Wang, Y.C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal. Transduct. Target Ther. 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Luo, S.; Libby, P.; Shi, G.P. Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef] [PubMed]
- Bollavaram, K.; Leeman, T.H.; Lee, M.W.; Kulkarni, A.; Upshaw, S.G.; Yang, J.; Song, H.; Platt, M.O. Multiple sites on SARS-CoV-2 spike protein are susceptible to proteolysis by cathepsins B., K., L., S., and V. Protein Sci. 2021, 30, 1131–1143. [Google Scholar] [CrossRef]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020, 6, eabe0751. [Google Scholar] [CrossRef]
- Costanzi, E.; Kuzikov, M.; Esposito, F.; Albani, S.; Demitri, N.; Giabbai, B.; Camasta, M.; Tramontano, E.; Rossetti, G.; Zaliani, A.; et al. Structural and biochemical analysis of the dual inhibition of MG-132 against SARS-CoV-2 main protease (Mpro/3CLpro) and human cathepsin-L. Int. J. Mol. Sci. 2021, 22, 11779. [Google Scholar] [CrossRef]
- Poli, G.; Seidel, T.; Langer, T. Conformational sampling of small molecules with iCon: Performance Assessment in comparison with OMEGA. Front. Chem. 2018, 6, 229. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. Ligand scout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Wojciechowski, M.; Lesyng, B. Generalized born model: Analysis refinement, and applications to proteins. J. Phys. Chem. B 2004, 108, 18368–18376. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided. Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naim, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Interactions a | SPF b | EBE c | EBS d | EC e | SBA f | ΔGncov g | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T25 | M49 | C145 | H164 | M165 | E166 | L167 | Q189 | T190 | A191 | H20 | |||||||||
| INHIBITOR N3 | H | H | HBA | CI | HBD | H | HBD | HBA | H | HBD | HBD | H | HBA | 106.93 | 5034.60 | 713.40 | 5815.39 | −34.11 | −7.70 |
| INHIBITOR N1 | H | HBA | CI | HBD | H | HBD | HBA | H | HBD | HBD | H | HBA | 106.90 | 3132.36 | 642.09 | 3819.69 | −21.22 | −6.80 | |
| INHIBITOR N9 | H | CI | HBD | H | HBD | HBA | H | HBD | HBD | HBA | 96.23 | 2216.40 | 540.26 | 2796.46 | −15.40 | −6.30 | |||
| INHIBITOR I2 | H | CI | HBD | H | HBD | HBA | H | HBD | HBA | 86.48 | 1112.63 | 617.64 | 1770.67 | −9.61 | −7.00 | ||||
| IXAZOMIB | H | CI | HBD | H | HBD | HBA | H | HBD | HBA | 83.90 | 170.89 | 469.81 | 674.98 | −3.22 | −6.70 | ||||
| CALPAIN INH | H | CI | HBD | HBD | HBA | HBD | HBA | 77.81 | 630.46 | 457.86 | 1127.00 | −22.19 | −5.10 | ||||||
| DB08119 | H | HBA | CI | HBD | HBA | HBD | HBA | 77.80 | 2071.81 | 491.17 | 2619.91 | −27.30 | −6.20 | ||||||
| DB03984 | H | HBA | CI | HBD | HBA | HBD | HBA | 76.82 | 2337.41 | 522.87 | 2930.08 | −21.93 | −6.70 | ||||||
| DB07224 | H | HBA | CI | HBD | HBA | HBD | HBA | 76.48 | 156.99 | 476.63 | 659.17 | −15.30 | −6.10 | ||||||
| DB07225 | H | H | HBA | CI | HBD | HBA | HBD | HBA | 76.48 | 204.10 | 479.96 | 705.99 | −1.17 | −5.80 | |||||
| AMPRENAVIR | H | HBA | CI | HBD | H | H | HBD | HBA | 76.23 | 679.32 | 593.57 | 1324.18 | −20.21 | −7.80 | |||||
| OPROZOMIB | H | CI | HBD | HBA | HBD | HBD | HBA | 76.12 | 2033.90 | 577.90 | 2696.86 | −19.37 | −6.30 | ||||||
| CARFILZOMIB | H | CI | HBD | HBD | HBA | HBD | HBA | 76.03 | 5948.19 | 756.99 | 6849.93 | −36.01 | −8.20 | ||||||
| DB03456 | H | HBA | CI | HBD | HBA | HBD | HBA | 75.87 | 103.11 | 461.78 | 584.84 | −14.91 | −6.80 | ||||||
| DB07299 | H | CI | HBD | H | HBD | HBA | H | HBA | 75.65 | 398.77 | 580.32 | 934.01 | 2.54 | −6.50 | |||||
| DB03767 | H | HBA | CI | HBD | HBA | HBD | HBA | 75.52 | 504.95 | 523.68 | 1074.14 | −18.57 | −6.60 | ||||||
| DB04234 | H | H | CI | HBD | HBA | HBD | HBA | 75.34 | 62.13 | 457.82 | 544.49 | −3.05 | −6.10 | ||||||
| BOCEPREVIR | H | CI | HBD | H | HBD | H | HBA | 75.28 | 266.36 | 474.85 | 824.16 | −9.45 | −5.40 | ||||||
| VABORBACTAM | HBA | CI | HBD | H | HBA | H | HBD | HBA | 73.71 | −10.58 | 457.61 | 453.04 | −6.40 | −6.50 | |||||
| DB07749 | H | CI | HBD | HBA | HBD | HBA | 67.84 | 2577.53 | 485.06 | 3106.40 | −17.85 | −6.10 | |||||||
| CEFTAROLINE | H | H | HBA | CI | HBD | HBA | HBA | 66.62 | 2556.58 | 698.58 | 3252.33 | 17.64 | −7.10 | ||||||
| TELAPREVIR | CI | HBD | HBD | HBA | HBD | HBA | 66.43 | 4479.26 | 576.38 | 5196.13 | −21.93 | −6.20 | |||||||
| DB07160 | H | HBA | CI | HBD | HBA | 66.40 | 924.43 | 487.56 | 1420.27 | −19.95 | −5.20 | ||||||||
| PREDNISONE | H | HBA | CI | HBD | HBD | HBA | 66.34 | 1095.73 | 551.05 | 1738.53 | −3.08 | −7.30 | |||||||
| DARUNAVIR | H | HBA | CI | HBD | H | HBA | H | HBD | HBA | 66.32 | 340.83 | 471.19 | 861.32 | −30.86 | −7.50 | ||||
| PREXASERTIB | HBA | CI | H | HBA | H | HBD | HBA | 66.27 | 339.96 | 514.87 | 921.30 | −16.68 | −7.50 | ||||||
| TEMOCILLIN | H | HBA | CI | HBD | HBA | HBA | 66.11 | 1131.23 | 475.67 | 1660.75 | −11.36 | −6.80 | |||||||
| CIMETIDINE | CI | HBD | H | HBA | H | HBD | HBA | 65.99 | 120.44 | 459.12 | 557.49 | −10.99 | −5.30 | ||||||
| SCOPOLAMINE | H | HBA | CI | HBA | HBD | HBA | 65.98 | 122.54 | 459.65 | 646.78 | −6.35 | −6.30 | |||||||
| FLUOXYMESTERONE | H | HBA | CI | HBA | HBD | HBA | 65.98 | 1110.31 | 519.19 | 1733.89 | −20.21 | −7.20 | |||||||
| METHSCOPOLAMINE | H | HBA | CI | HBA | HBD | HBA | 65.98 | 1476.45 | 510.42 | 2070.99 | −17.74 | −7.00 | |||||||
| DB08614 | H | CI | HBD | HBD | HBA | HBA | 65.93 | 623.04 | 492.22 | 1151.02 | −19.01 | −7.30 | |||||||
| FLOVAGATRAN | CI | HBD | H | HBD | HBA | H | HBA | 65.86 | 1325.16 | 516.40 | 1892.44 | −7.04 | −7.00 | ||||||
| FELYPRESSIN | H | HBA | CI | H | HBD | H | HBA | 65.83 | 9699.06 | 993.86 | 10,838.66 | −19.45 | −6.80 | ||||||
| DB07987 | H | CI | HBD | HBA | HBD | HBD | 65.72 | 1777.82 | 554.85 | 2342.50 | −18.83 | −6.00 | |||||||
| AZTREONAM | HBA | CI | HBD | HBD | HBA | 65.62 | 944.90 | 570.26 | 1482.14 | 5.99 | −6.80 | ||||||||
| PREDNISOLONE | H | HBA | CI | HBD | HBD | HBA | 65.59 | 1507.06 | 506.38 | 2120.64 | −16.09 | −7.80 | |||||||
| RIOCIGUAT | H | HBA | CI | HBD | H | H | HBA | 65.57 | 348.80 | 529.93 | 955.36 | −10.18 | −8.30 | ||||||
| TIOTROPIUM | H | HBA | CI | HBA | HBD | HBA | 65.57 | 858.19 | 513.34 | 1466.31 | −7.52 | −7.00 | |||||||
| CMX−2043 | HBA | CI | H | HBD | HBA | H | HBD | 65.45 | 156.96 | 472.39 | 634.24 | −13.38 | −5.90 | ||||||
| GAXILOSE | CI | HBD | HBD | HBA | HBD | HBA | 65.45 | 2334.44 | 578.92 | 2999.85 | 15.21 | −6.50 | |||||||
| CINOLAZEPAM | H | HBA | CI | HBA | HBD | HBA | 65.40 | 2514.84 | 846.67 | 3451.69 | −15.19 | −7.40 | |||||||
| Mdl 101,146 | H | HBA | CI | H | HBA | H | HBA | 65.33 | 4065.30 | 624.83 | 4813.17 | −16.43 | −7.20 | ||||||
| FOSAMPRENAVIR | H | HBA | CI | HBD | H | H | HBA | 65.33 | 6894.18 | 648.29 | 7505.59 | −10.23 | −7.20 | ||||||
| BICALUTAMIDE | H | HBA | CI | HBD | HBA | HBD | 65.19 | 376.99 | 497.65 | 932.83 | −15.10 | −7.40 | |||||||
| DB04293 | H | H | HBA | CI | HBD | HBA | 65.17 | 152.09 | 483.24 | 704.77 | 0.79 | −7.30 | |||||||
| ATAZANAVIR | H | CI | HBD | HBD | HBA | HBA | 65.17 | 7923.60 | 860.74 | 8900.26 | −28.37 | −5.40 | |||||||
| BMS−488043 | H | H | HBA | CI | HBD | HBA | HBA | 65.15 | 1800.92 | 537.19 | 2446.41 | −6.45 | −7.90 | ||||||
| DB04232 | H | H | HBA | CI | HBD | HBA | 65.10 | 1768.70 | 529.64 | 2367.43 | −1.12 | −6.90 | |||||||
| CEPHALOGLYCIN | H | HBA | CI | HBD | HBD | HBA | 64.48 | 3432.94 | 596.57 | 4091.77 | −8.54 | −7.60 | |||||||
| MUPIROCIN | H | HBA | CI | HBA | HBD | HBA | 63.87 | 4899.57 | 820.50 | 5761.68 | −13.58 | −6.70 | |||||||
| CEFDITOREN | H | H | CI | HBD | HBA | HBA | 63.78 | 3465.77 | 799.57 | 4352.81 | −3.02 | −6.50 | |||||||
| CABAZITAXEL | H | CI | HBD | H | HBA | H | HBD | HBA | 63.67 | 7750.80 | 1172.22 | 9132.50 | −23.63 | −6.70 | |||||
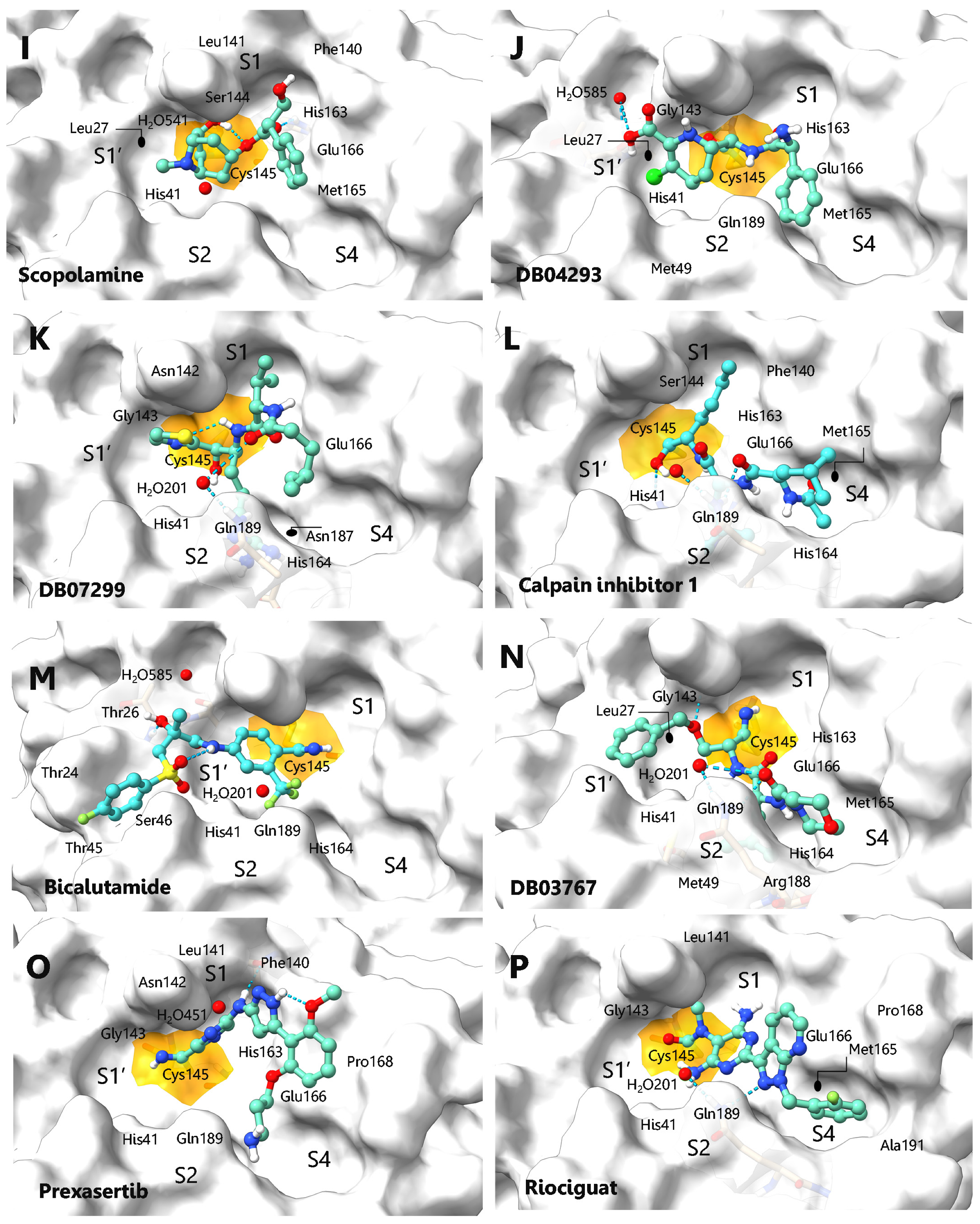
| Drug | CsScore | SPF a | ΔGncov b | ΔGcov c | Interactions | |
|---|---|---|---|---|---|---|
| Ref | INHIBITOR N3 | 412.82 | 106.93 | −7.7 | −10.1 | Leu27, His41, Met49, Asn142, Gly143, Ser144, Cys145, His164, Met165, Glu166, Leu167, Pro168, Gln 189, Thr190, Ala191, H2O201 |
| (A) | VABORBACTAM | 436.08 | 73.71 | −6.5 | −5.9 | His41, Met 49, Leu141, Ser144, Cys145, His163, His164, Met165 |
| (B) | DB04234 | 487.59 | 75.34 | −6.1 | −7.3 | His41, Met 49, Leu141, Cys145, His164, Met165, Glu166, Gln 189, H2O201 |
| (C) | DB03456 | 496.79 | 75.87 | −6.8 | −6.1 | His41, Met 49, Leu141, Cys145, His164, Met165, Glu166, Gln189, H2O201 |
| (D) | CIMETIDINE | 503.00 | 65.99 | −5.3 | −5.3 | Thr26, His41, Met49, Gly143, Cys145, His164, Met165, Gln189, H2O201 |
| (E) | IXAZOMIB | 521.07 | 83.90 | −6.7 | −5.9 | His41, Cys145, His163, His164, Met165, Leu167, Gln189, Thr190, H2O201 |
| (F) | CMX-2043 | 523.19 | 65.45 | −5.9 | −6.8 | His41, Met 49, Gly143, Glu166, Pro168, Asp187, Gln189, H2O201 |
| (G) | DB07224 | 529.37 | 76.48 | −6.1 | −6.6 | His41, Leu141, Asn142, Cys145, His163, His164, Met165, Arg188, Gln189, H2O201 |
| (H) | DB07225 | 540.42 | 76.48 | −5.8 | −6.6 | Leu27, Thr26, Asn142, Gly143, Cys145, His163, Met165, Glu166, Leu167, Thr190 |
| (I) | SCOPOLAMINE | 558.11 | 65.98 | −6.3 | −5.6 | Leu27, His41, Phe140, Leu141, Ser144, Cys145, His163, Met165, Glu166, H2O541 |
| (J) | DB04293 | 596.86 | 65.17 | −7.3 | −6.2 | Thr26, His41, Met49, Gly143, Cys145, His163, Met165, Glu166, Gln189, H2O585. |
| (K) | DB07299 | 621.90 | 75.65 | −6.5 | −7.3 | His41, Asn142, Gly143, Cys145, His164, Met165, Glu166, Asp187, Gln189, H2O201 |
| (L) | CALPAIN INH-1 | 626.71 | 77.81 | −5.1 | −7.4 | His41, Phe140, Ser144, Cys145, His163, His164, Met165, Glu166, Gln189 |
| (M) | BICALUTAMIDE | 678.69 | 65.19 | −7.4 | −5.5 | Thr24, Thr26, His41, Thr45, Ser46, Cys145, His164, Gln189, H2O201, H2O585 |
| (N) | DB03767 | 682.37 | 75.52 | −6.6 | −7.2 | Leu27, His41, Met49, Gly143, Cys145, His163, His164, Met165, Glu166, Arg188, Gln189, H2O201 |
| (O) | PREXASERTIB | 687.56 | 66.27 | −7.5 | −5.3 | His41, Phe140, Leu141, Asn142, Gly143, Cys145, His163, Glu166, Pro168, Gln189 H2O541 |
| (P) | RIOCIGUAT | 717.46 | 65.57 | −8.3 | −5.7 | His41, Leu141, Gly143, Cys145, Met165, Glu166, Pro168, Gln189, Ala191, H2O201 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Mendoza, L.H.; Mendoza-Figueroa, H.L.; García-Vázquez, J.B.; Correa-Basurto, J.; García-Machorro, J. In Silico Drug Repositioning to Target the SARS-CoV-2 Main Protease as Covalent Inhibitors Employing a Combined Structure-Based Virtual Screening Strategy of Pharmacophore Models and Covalent Docking. Int. J. Mol. Sci. 2022, 23, 3987. https://doi.org/10.3390/ijms23073987
Vázquez-Mendoza LH, Mendoza-Figueroa HL, García-Vázquez JB, Correa-Basurto J, García-Machorro J. In Silico Drug Repositioning to Target the SARS-CoV-2 Main Protease as Covalent Inhibitors Employing a Combined Structure-Based Virtual Screening Strategy of Pharmacophore Models and Covalent Docking. International Journal of Molecular Sciences. 2022; 23(7):3987. https://doi.org/10.3390/ijms23073987
Chicago/Turabian StyleVázquez-Mendoza, Luis Heriberto, Humberto L. Mendoza-Figueroa, Juan Benjamín García-Vázquez, José Correa-Basurto, and Jazmín García-Machorro. 2022. "In Silico Drug Repositioning to Target the SARS-CoV-2 Main Protease as Covalent Inhibitors Employing a Combined Structure-Based Virtual Screening Strategy of Pharmacophore Models and Covalent Docking" International Journal of Molecular Sciences 23, no. 7: 3987. https://doi.org/10.3390/ijms23073987