
Diet-Induced Metabolic Dysfunction of Hypothalamic Nutrient Sensing in Rodents

 and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
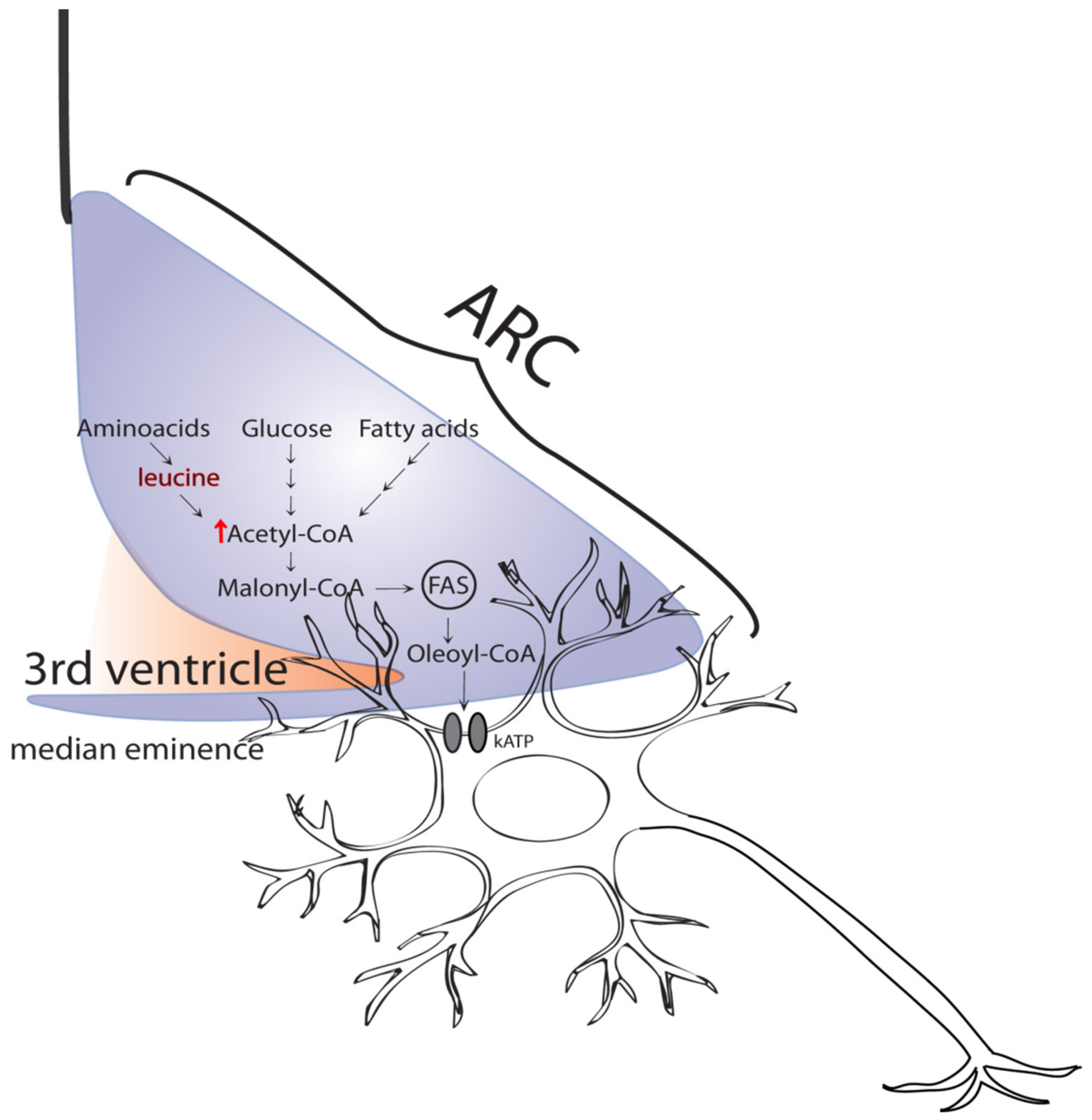
2. Role of the Hypothalamus in the Control of Glycemia
3. Circulating Amino Acids in the Regulation of Glucose Production
4. Physiological Relevance of Metabolic Nutrient Sensing
5. Disruption of Central Glucose Regulation by High-Fat Diets
6. Conclusions
7. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedman, J.M. A war on obesity, not the obese. Science 2003, 299, 856–858. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.O.; Wyatt, H.R.; Reed, G.W.; Peters, J.C. Obesity and the environment: Where do we go from here? Science 2003, 299, 853–855. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. S1), S81–S90. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.K.; Chari, M.; Lam, T.K. CNS regulation of glucose homeostasis. Physiology 2009, 24, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Arble, D.M.; Sandoval, D.A. CNS control of glucose metabolism: Response to environmental challenges. Front. Neurosci. 2013, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruud, J.; Steculorum, S.M.; Bruning, J.C. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat. Commun. 2017, 8, 15259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bich, L.; Mossio, M.; Soto, A.M. Glycemia Regulation: From Feedback Loops to Organizational Closure. Front. Physiol. 2020, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Jais, A.; Bruning, J.C. Arcuate Nucleus-Dependent Regulation of Metabolism-Pathways to Obesity and Diabetes Mellitus. Endocr. Rev. 2022, 43, 314–328. [Google Scholar] [CrossRef]
- Obici, S.; Rossetti, L. Minireview: Nutrient sensing and the regulation of insulin action and energy balance. Endocrinology 2003, 144, 5172–5178. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.K.; Pocai, A.; Gutierrez-Juarez, R.; Obici, S.; Bryan, J.; Aguilar-Bryan, L.; Schwartz, G.J.; Rossetti, L. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat. Med. 2005, 11, 320–327. [Google Scholar] [CrossRef]
- Ciofi, P.; Garret, M.; Lapirot, O.; Lafon, P.; Loyens, A.; Prevot, V.; Levine, J.E. Brain-endocrine interactions: A microvascular route in the mediobasal hypothalamus. Endocrinology 2009, 150, 5509–5519. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Cha, S.H.; Chohnan, S.; Lane, M.D. Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc. Natl. Acad. Sci. USA 2003, 100, 12624–12629. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.K.; Kurowski, T.G.; Ruderman, N.B. A malonyl-CoA fuel-sensing mechanism in muscle: Effects of insulin, glucose, and denervation. Am. J. Physiol. 1995, 269, E283–E289. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Saha, A.K.; Kraegen, E.W. Minireview: Malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology 2003, 144, 5166–5171. [Google Scholar] [CrossRef] [Green Version]
- Obici, S.; Feng, Z.; Morgan, K.; Stein, D.; Karkanias, G.; Rossetti, L. Central administration of oleic acid inhibits glucose production and food intake. Diabetes 2002, 51, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Obici, S.; Feng, Z.; Arduini, A.; Conti, R.; Rossetti, L. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat. Med. 2003, 9, 756–761. [Google Scholar] [CrossRef]
- Ruderman, N.; Prentki, M. AMP kinase and malonyl-CoA: Targets for therapy of the metabolic syndrome. Nat. Rev. Drug Discov. 2004, 3, 340–351. [Google Scholar] [CrossRef]
- Saggerson, D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu. Rev. Nutr. 2008, 28, 253–272. [Google Scholar] [CrossRef]
- Lam, T.K.; Gutierrez-Juarez, R.; Pocai, A.; Rossetti, L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science 2005, 309, 943–947. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.K.; Schwartz, G.J.; Rossetti, L. Hypothalamic sensing of fatty acids. Nat. Neurosci. 2005, 8, 579–584. [Google Scholar] [CrossRef]
- Pocai, A.; Lam, T.K.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic K(ATP) channels control hepatic glucose production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef]
- Su, Y.; Lam, T.K.; He, W.; Pocai, A.; Bryan, J.; Aguilar-Bryan, L.; Gutierrez-Juarez, R. Hypothalamic leucine metabolism regulates liver glucose production. Diabetes 2012, 61, 85–93. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Lam, T.K.; Obici, S.; Rossetti, L. Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat. Neurosci. 2006, 9, 227–233. [Google Scholar] [CrossRef]
- Pocai, A.; Obici, S.; Schwartz, G.J.; Rossetti, L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005, 1, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Arrieta-Cruz, I.; Su, Y.; Knight, C.M.; Lam, T.K.; Gutierrez-Juarez, R. Evidence for a role of proline and hypothalamic astrocytes in the regulation of glucose metabolism in rats. Diabetes 2013, 62, 1152–1158. [Google Scholar] [CrossRef] [Green Version]
- Arrieta-Cruz, I.; Su, Y.; Gutierrez-Juarez, R. Suppression of Endogenous Glucose Production by Isoleucine and Valine and Impact of Diet Composition. Nutrients 2016, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Nakamura, Y.; Inaba, Y.; Matsumoto, M.; Kido, Y.; Asahara, S.; Matsuda, T.; Watanabe, H.; Maeda, A.; Inagaki, F.; et al. Histidine augments the suppression of hepatic glucose production by central insulin action. Diabetes 2013, 62, 2266–2277. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Park, S.J.; Lee, H.; Siddiqi, F.; Lee, J.E.; Menzies, F.M.; Rubinsztein, D.C. Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A. Cell Metab. 2019, 29, 192–201.e197. [Google Scholar] [CrossRef] [Green Version]
- Guo, K.; Yu, Y.H.; Hou, J.; Zhang, Y. Chronic leucine supplementation improves glycemic control in etiologically distinct mouse models of obesity and diabetes mellitus. Nutr. Metab. 2010, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Macotela, Y.; Emanuelli, B.; Bång, A.M.; Espinoza, D.O.; Boucher, J.; Beebe, K.; Gall, W.; Kahn, C.R. Dietary leucine—An environmental modifier of insulin resistance acting on multiple levels of metabolism. PLoS ONE 2011, 6, e21187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.H.; Fletcher, P.J.; Anderson, G.H. Extracellular amino acid profiles in the paraventricular nucleus of the rat hypothalamus are influenced by diet composition. Brain Res. 2001, 892, 320–328. [Google Scholar] [CrossRef]
- Lin, H.V.; Plum, L.; Ono, H.; Gutierrez-Juarez, R.; Shanabrough, M.; Borok, E.; Horvath, T.L.; Rossetti, L.; Accili, D. Divergent regulation of energy expenditure and hepatic glucose production by insulin receptor in agouti-related protein and POMC neurons. Diabetes 2010, 59, 337–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konner, A.C.; Janoschek, R.; Plum, L.; Jordan, S.D.; Rother, E.; Ma, X.; Xu, C.; Enriori, P.; Hampel, B.; Barsh, G.S.; et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007, 5, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Borgmann, D.; Ciglieri, E.; Biglari, N.; Brandt, C.; Cremer, A.L.; Backes, H.; Tittgemeyer, M.; Wunderlich, F.T.; Bruning, J.C.; Fenselau, H. Gut-brain communication by distinct sensory neurons differently controls feeding and glucose metabolism. Cell Metab. 2021, 33, 1466–1482.e7. [Google Scholar] [CrossRef]
- Branstrom, R.; Aspinwall, C.A.; Valimaki, S.; Ostensson, C.G.; Tibell, A.; Eckhard, M.; Brandhorst, H.; Corkey, B.E.; Berggren, P.O.; Larsson, O. Long-chain CoA esters activate human pancreatic beta-cell KATP channels: Potential role in Type 2 diabetes. Diabetologia 2004, 47, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Eaton, S.B.; Konner, M. Paleolithic nutrition. A consideration of its nature and current implications. N. Engl. J. Med. 1985, 312, 283–289. [Google Scholar] [CrossRef]
- Hill, J.O.; Peters, J.C. Environmental contributions to the obesity epidemic. Science 1998, 280, 1371–1374. [Google Scholar] [CrossRef]
- Morgan, K.; Obici, S.; Rossetti, L. Hypothalamic responses to long-chain fatty acids are nutritionally regulated. J. Biol. Chem. 2004, 279, 31139–31148. [Google Scholar] [CrossRef] [Green Version]
- Pocai, A.; Lam, T.K.; Obici, S.; Gutierrez-Juarez, R.; Muse, E.D.; Arduini, A.; Rossetti, L. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J. Clin. Invest. 2006, 116, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.K.; Gutierrez-Juarez, R.; Pocai, A.; Bhanot, S.; Tso, P.; Schwartz, G.J.; Rossetti, L. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat. Med. 2007, 13, 171–180. [Google Scholar] [CrossRef]
- Zhao, W.T.; Luo, Y.; Zhang, Y.; Zhou, Y.; Zhao, T.T. High protein diet is of benefit for patients with type 2 diabetes: An updated meta-analysis. Medicine 2018, 97, e13149. [Google Scholar] [CrossRef] [PubMed]
- Skytte, M.J.; Samkani, A.; Petersen, A.D.; Thomsen, M.N.; Astrup, A.; Chabanova, E.; Frystyk, J.; Holst, J.J.; Thomsen, H.S.; Madsbad, S.; et al. A carbohydrate-reduced high-protein diet improves HbA(1c) and liver fat content in weight stable participants with type 2 diabetes: A randomised controlled trial. Diabetologia 2019, 62, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Nan, F.; Wang, L.Y.; Jiang, H.; Chen, W.; Jiang, Y. Effects of high-protein diet on glycemic control, insulin resistance and blood pressure in type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. 2020, 39, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, Y.; Hu, W.; Liang, Y.; Zheng, L.; Zheng, J.; Wang, B.; Guo, X. Different Effects of Leucine Supplementation and/or Exercise on Systemic Insulin Sensitivity in Mice. Front. Endocrinol. 2021, 12, 651303. [Google Scholar] [CrossRef]
- Cavaliere, G.; Viggiano, E.; Trinchese, G.; De Filippo, C.; Messina, A.; Monda, V.; Valenzano, A.; Cincione, R.I.; Zammit, C.; Cimmino, F.; et al. Long Feeding High-Fat Diet Induces Hypothalamic Oxidative Stress and Inflammation, and Prolonged Hypothalamic AMPK Activation in Rat Animal Model. Front. Physiol. 2018, 9, 818. [Google Scholar] [CrossRef]
- McLean, F.H.; Campbell, F.M.; Langston, R.F.; Sergi, D.; Resch, C.; Grant, C.; Morris, A.C.; Mayer, C.D.; Williams, L.M. A high-fat diet induces rapid changes in the mouse hypothalamic proteome. Nutr. Metab. 2019, 16, 26. [Google Scholar] [CrossRef]
- Santos, L.S.; Cordeiro, G.S.; Matos, R.J.B.; Perez, G.S.; Silva, R.T.; Boaventura, G.T.; Barreto-Medeiros, J.M. High-fat diet promotes hypothalamic inflammation in animal models: A systematic review. Nutr. Rev. 2021, 80, 392–399. [Google Scholar] [CrossRef]
- Ren, H.; Vieira-de-Abreu, A.; Yan, S.; Reilly, A.M.; Chan, O.; Accili, D. Altered Central Nutrient Sensing in Male Mice Lacking Insulin Receptors in Glut4-Expressing Neurons. Endocrinology 2019, 160, 2038–2048. [Google Scholar] [CrossRef] [Green Version]
- Reilly, A.M.; Zhou, S.; Panigrahi, S.K.; Yan, S.; Conley, J.M.; Sheets, P.L.; Wardlaw, S.L.; Ren, H. Gpr17 deficiency in POMC neurons ameliorates the metabolic derangements caused by long-term high-fat diet feeding. Nutr. Diabetes 2019, 9, 29. [Google Scholar] [CrossRef]
- Knutson, M.D.; Leeuwenburgh, C. Resveratrol and novel potent activators of SIRT1: Effects on aging and age-related diseases. Nutr. Rev. 2008, 66, 591–596. [Google Scholar] [CrossRef]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Ramadori, G.; Gautron, L.; Fujikawa, T.; Vianna, C.R.; Elmquist, J.K.; Coppari, R. Central administration of resveratrol improves diet-induced diabetes. Endocrinology 2009, 150, 5326–5333. [Google Scholar] [CrossRef]
- Knight, C.M.; Gutierrez-Juarez, R.; Lam, T.K.; Arrieta-Cruz, I.; Huang, L.; Schwartz, G.; Barzilai, N.; Rossetti, L. Mediobasal Hypothalamic SIRT1 Is Essential for Resveratrol’s Effects on Insulin Action in Rats. Diabetes 2011, 60, 2691–2700. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrieta-Cruz, I.; Torres-Ávila, B.S.; Martínez-Coria, H.; López-Valdés, H.E.; Gutiérrez-Juárez, R. Diet-Induced Metabolic Dysfunction of Hypothalamic Nutrient Sensing in Rodents. Int. J. Mol. Sci. 2022, 23, 3958. https://doi.org/10.3390/ijms23073958
Arrieta-Cruz I, Torres-Ávila BS, Martínez-Coria H, López-Valdés HE, Gutiérrez-Juárez R. Diet-Induced Metabolic Dysfunction of Hypothalamic Nutrient Sensing in Rodents. International Journal of Molecular Sciences. 2022; 23(7):3958. https://doi.org/10.3390/ijms23073958
Chicago/Turabian StyleArrieta-Cruz, Isabel, Blanca Samara Torres-Ávila, Hilda Martínez-Coria, Héctor Eduardo López-Valdés, and Roger Gutiérrez-Juárez. 2022. "Diet-Induced Metabolic Dysfunction of Hypothalamic Nutrient Sensing in Rodents" International Journal of Molecular Sciences 23, no. 7: 3958. https://doi.org/10.3390/ijms23073958