
The Absence of Retroelement Activity Is Characteristic for Childhood Acute Leukemias and Adult Acute Lymphoblastic Leukemia

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
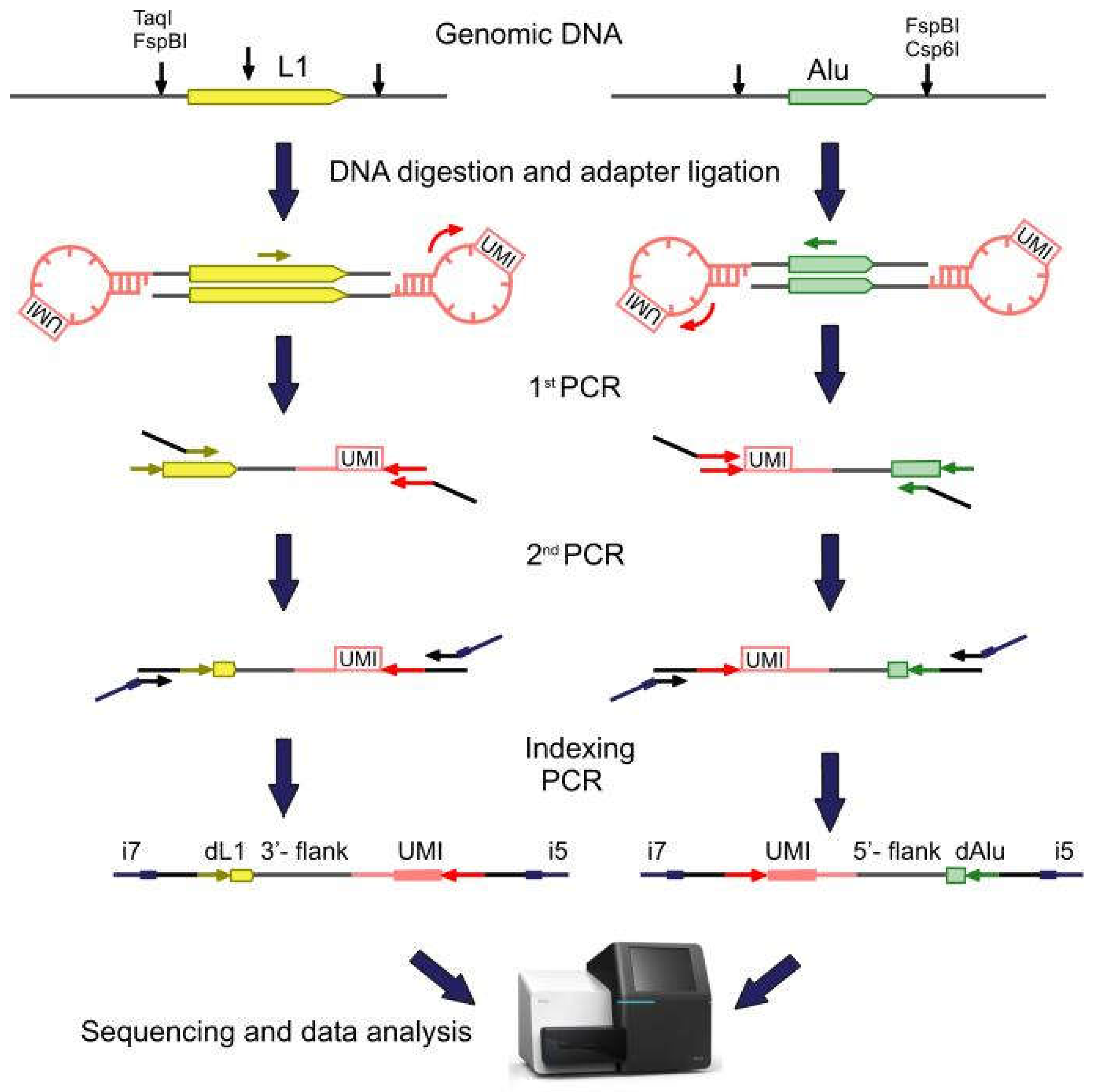
2.1. Library Preparation, Sequencing, and Data Analysis
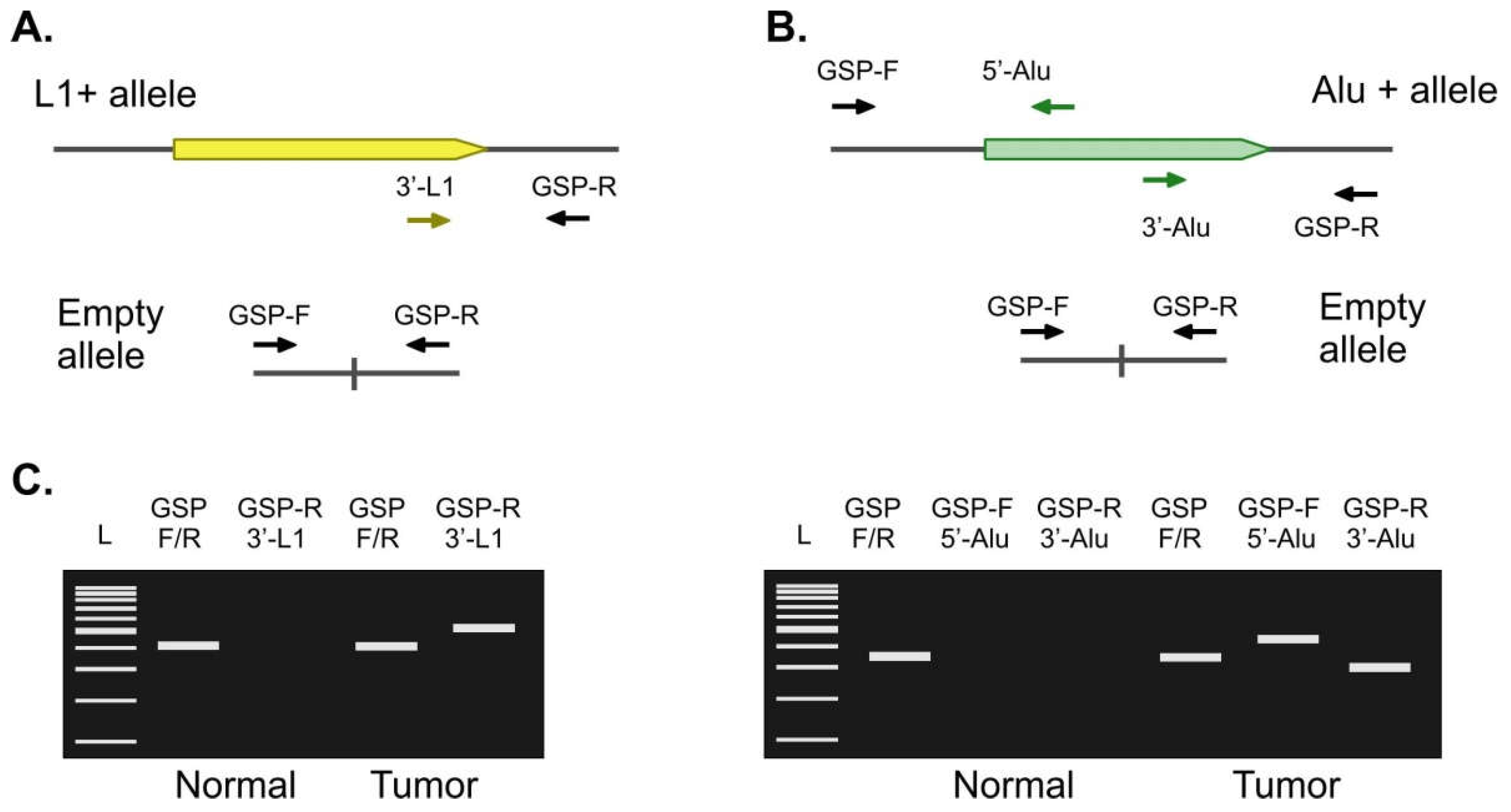
2.2. Validation of Candidate Tumor-Specific Insertions
2.3. Evaluation of the Method Sensitivity
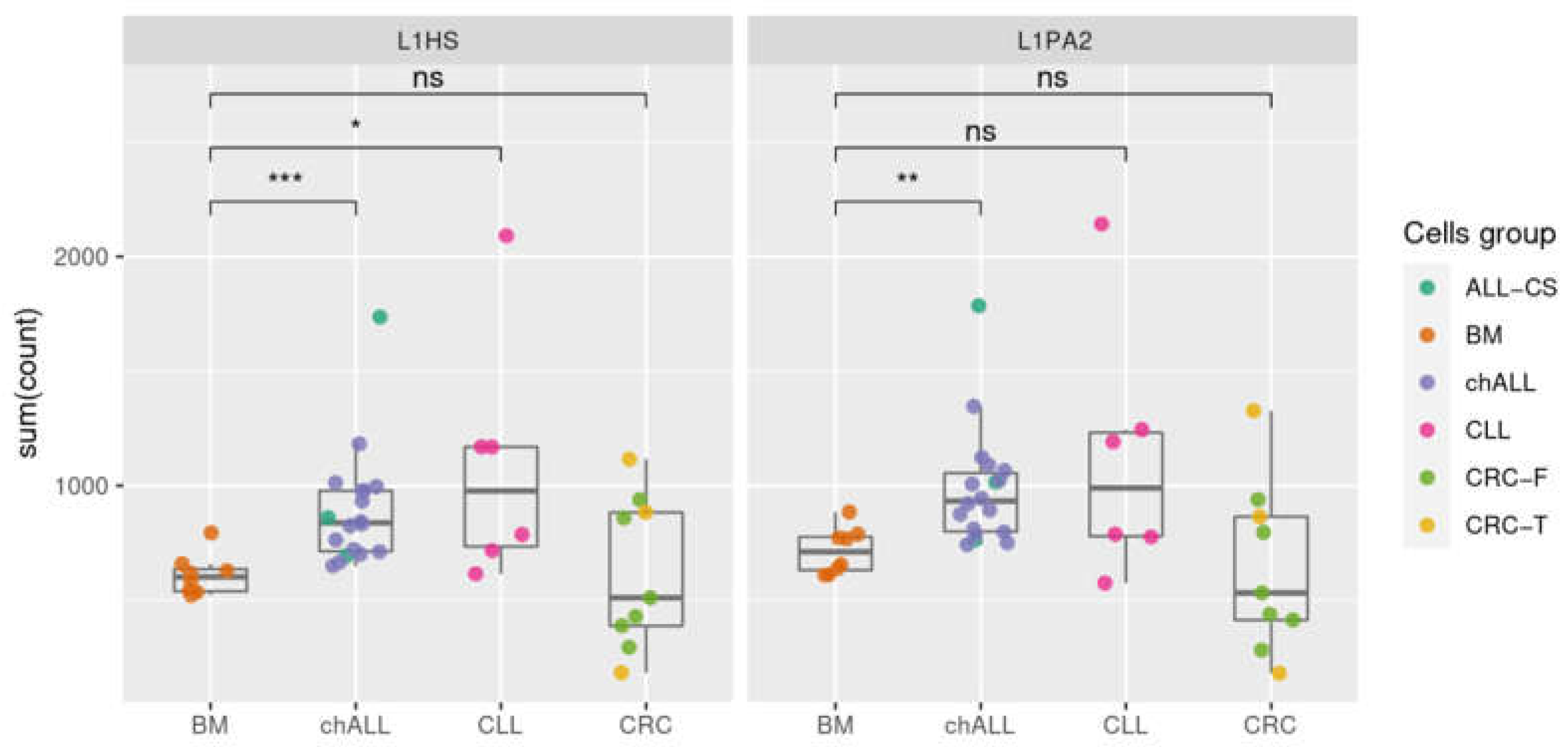
2.4. Transcription of Active L1 Retroelements in Leukemia
3. Discussion
4. Materials and Methods
4.1. Sample Collection and DNA Isolation
4.2. Library Preparation and Sequencing
4.3. Validation of RE Insertions by Locus-Specific PCR
4.4. Evaluation of Method Sensitivity
4.5. Transcriptome Sequencing and Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.; Smit, A.; Boissinot, S. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res. 2006, 16, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Penzkofer, T.; Jager, M.; Figlerowicz, M.; Badge, R.; Mundlos, S.; Robinson, P.N.; Zemojtel, T. L1Base 2: More retrotransposition-active LINE-1s, more mammalian genomes. Nucleic Acids Res. 2017, 45, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, H.H., Jr.; Moran, J.V. Mobile DNA in Health and Disease. N. Engl. J. Med. 2017, 377, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belancio, V.P.; Roy-Engel, A.M.; Deininger, P.L. All y’all need to know ‘bout retroelements in cancer. Semin. Cancer Biol. 2010, 20, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Bundo, M.; Toyoshima, M.; Okada, Y.; Akamatsu, W.; Ueda, J.; Nemoto-Miyauchi, T.; Sunaga, F.; Toritsuka, M.; Ikawa, D.; Kakita, A.; et al. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 2014, 81, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Martin, B.; Alvarez, E.G.; Baez-Ortega, A.; Zamora, J.; Supek, F.; Demeulemeester, J.; Santamarina, M.; Ju, Y.S.; Temes, J.; Garcia-Souto, D.; et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet. 2020, 52, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Tubio, J.M.; Li, Y.; Ju, Y.S.; Martincorena, I.; Cooke, S.L.; Tojo, M.; Gundem, G.; Pipinikas, C.P.; Zamora, J.; Raine, K.; et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 2014, 345, 1251343. [Google Scholar] [CrossRef] [Green Version]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulpa, D.A.; Moran, J.V. Cis-preferential LINE-1 reverse transcriptase activity in ribonucleoprotein particles. Nat. Struct. Mol. Biol. 2006, 13, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Helman, E.; Lawrence, M.S.; Stewart, C.; Sougnez, C.; Getz, G.; Meyerson, M. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014, 24, 1053–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J., 3rd; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feusier, J.; Watkins, W.S.; Thomas, J.; Farrell, A.; Witherspoon, D.J.; Baird, L.; Ha, H.; Xing, J.; Jorde, L.B. Pedigree-based estimation of human mobile element retrotransposition rates. Genome Res. 2019, 29, 1567–1577. [Google Scholar] [CrossRef] [Green Version]
- Goubert, C.; Thomas, J.; Payer, L.M.; Kidd, J.M.; Feusier, J.; Watkins, W.S.; Burns, K.H.; Jorde, L.B.; Feschotte, C. TypeTE: A tool to genotype mobile element insertions from whole genome resequencing data. Nucleic Acids Res. 2020, 48, e36. [Google Scholar] [CrossRef] [Green Version]
- Komkov, A.Y.; Minervina, A.A.; Nugmanov, G.A.; Saliutina, M.V.; Khodosevich, K.V.; Lebedev, Y.B.; Mamedov, I.Z. An advanced enrichment method for rare somatic retroelement insertions sequencing. Mob. DNA 2018, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.W.; Ha, H.; Lin, T.; Sun, N.; Burns, K.H.; Xing, J. Integrated Mobile Element Scanning (ME-Scan) method for identifying multiple types of polymorphic mobile element insertions. Mob. DNA 2020, 11, 12. [Google Scholar] [CrossRef]
- Mamedov, I.Z.; Arzumanyan, E.S.; Amosova, A.L.; Lebedev, Y.B.; Sverdlov, E.D. Whole-genome experimental identification of insertion/deletion polymorphisms of interspersed repeats by a new general approach. Nucleic Acids Res. 2005, 33, e16. [Google Scholar] [CrossRef] [Green Version]
- Shukla, R.; Upton, K.R.; Munoz-Lopez, M.; Gerhardt, D.J.; Fisher, M.E.; Nguyen, T.; Brennan, P.M.; Baillie, J.K.; Collino, A.; Ghisletti, S.; et al. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 2013, 153, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Solyom, S.; Ewing, A.D.; Rahrmann, E.P.; Doucet, T.; Nelson, H.H.; Burns, M.B.; Harris, R.S.; Sigmon, D.F.; Casella, A.; Erlanger, B.; et al. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 2012, 22, 2328–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upton, K.R.; Gerhardt, D.J.; Jesuadian, J.S.; Richardson, S.R.; Sanchez-Luque, F.J.; Bodea, G.O.; Ewing, A.D.; Salvador-Palomeque, C.; van der Knaap, M.S.; Brennan, P.M.; et al. Ubiquitous l1 mosaicism in hippocampal neurons. Cell 2015, 161, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Evrony, G.D.; Lee, E.; Park, P.J.; Walsh, C.A. Resolving rates of mutation in the brain using single-neuron genomics. Elife 2016, 5, e12966. [Google Scholar] [CrossRef] [PubMed]
- Komkov, A.Y.; Urazbakhtin, S.Z.; Saliutina, M.V.; Komech, E.A.; Shelygin, Y.A.; Nugmanov, G.A.; Shubin, V.P.; Smirnova, A.O.; Bobrov, M.Y.; Tsukanov, A.S.; et al. SeqURE—A new copy-capture based method for sequencing of unknown Retroposition events. Mob. DNA 2020, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Kurnosov, A.A.; Ustyugova, S.V.; Nazarov, V.I.; Minervina, A.A.; Komkov, A.Y.; Shugay, M.; Pogorelyy, M.V.; Khodosevich, K.V.; Mamedov, I.Z.; Lebedev, Y.B. The evidence for increased L1 activity in the site of human adult brain neurogenesis. PLoS ONE 2015, 10, e0117854. [Google Scholar] [CrossRef]
- Kivioja, T.; Vaharautio, A.; Karlsson, K.; Bonke, M.; Enge, M.; Linnarsson, S.; Taipale, J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 2012, 9, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Nugmanov, G.A.; Komkov, A.Y.; Saliutina, M.V.; Minervina, A.A.; Lebedev, Y.B.; Mamedov, I.Z. A Pipeline for the Error-free Identification of Somatic Alu Insertions in High-throughput Sequencing Data. Mol. Biol. 2019, 53, 138–146. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Gerousi, M.; Psomopoulos, F.; Kotta, K.; Tsagiopoulou, M.; Stavroyianni, N.; Anagnostopoulos, A.; Anastasiadis, A.; Gkanidou, M.; Kotsianidis, I.; Ntoufa, S.; et al. The Calcitriol/Vitamin D Receptor System Regulates Key Immune Signaling Pathways in Chronic Lymphocytic Leukemia. Cancers 2021, 13, 285. [Google Scholar] [CrossRef]
- Cajuso, T.; Sulo, P.; Tanskanen, T.; Katainen, R.; Taira, A.; Hanninen, U.A.; Kondelin, J.; Forsstrom, L.; Valimaki, N.; Aavikko, M.; et al. Retrotransposon insertions can initiate colorectal cancer and are associated with poor survival. Nat. Commun. 2019, 10, 4022. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Nishisho, I.; Horii, A.; Miyoshi, Y.; Utsunomiya, J.; Kinzler, K.W.; Vogelstein, B.; Nakamura, Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992, 52, 643–645. [Google Scholar] [PubMed]
- Cuellar, T.L.; Herzner, A.M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardeljan, D.; Steranka, J.P.; Liu, C.; Li, Z.; Taylor, M.S.; Payer, L.M.; Gorbounov, M.; Sarnecki, J.S.; Deshpande, V.; Hruban, R.H.; et al. Cell fitness screens reveal a conflict between LINE-1 retrotransposition and DNA replication. Nat. Struct. Mol. Biol. 2020, 27, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Den Nijs, J.I.; Gonggrijp, H.S.; Augustinus, E.; Leeksma, C.H. Hot bands: A simple G-banding method for leukemic metaphases. Cancer Genet. Cytogenet. 1985, 15, 373–374. [Google Scholar] [CrossRef]
- Gabert, J.; Beillard, E.; van der Velden, V.H.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cave, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef]
- Meyer, C.; Schneider, B.; Reichel, M.; Angermueller, S.; Strehl, S.; Schnittger, S.; Schoch, C.; Jansen, M.W.; van Dongen, J.J.; Pieters, R.; et al. Diagnostic tool for the identification of MLL rearrangements including unknown partner genes. Proc. Natl. Acad. Sci. USA 2005, 102, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afrin, S.; Zhang, C.R.C.; Meyer, C.; Stinson, C.L.; Pham, T.; Bruxner, T.J.C.; Venn, N.C.; Trahair, T.N.; Sutton, R.; Marschalek, R.; et al. Targeted Next-Generation Sequencing for Detecting MLL Gene Fusions in Leukemia. Mol. Cancer Res. 2018, 16, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Lopes, B.A.; Caye-Eude, A.; Cave, H.; Arfeuille, C.; Cuccuini, W.; Sutton, R.; Venn, N.C.; Oh, S.H.; Tsaur, G.; et al. Human MLL/KMT2A gene exhibits a second breakpoint cluster region for recurrent MLL-USP2 fusions. Leukemia 2019, 33, 2306–2340. [Google Scholar] [CrossRef]
- Jansen, M.W.; van der Velden, V.H.; van Dongen, J.J. Efficient and easy detection of MLL-AF4, MLL-AF9 and MLL-ENL fusion gene transcripts by multiplex real-time quantitative RT-PCR in TaqMan and LightCycler. Leukemia 2005, 19, 2016–2018. [Google Scholar] [CrossRef] [Green Version]
- Blagodatskikh, K.A.; Kramarov, V.M.; Barsova, E.V.; Garkovenko, A.V.; Shcherbo, D.S.; Shelenkov, A.A.; Ustinova, V.V.; Tokarenko, M.R.; Baker, S.C.; Kramarova, T.V.; et al. Improved DOP-PCR (iDOP-PCR): A robust and simple WGA method for efficient amplification of low copy number genomic DNA. PLoS ONE 2017, 12, e0184507. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Thierry-Mieg, J.; Thierry-Mieg, D.; Busby, B.; Madden, T.L. Magic-BLAST, an accurate RNA-seq aligner for long and short reads. BMC Bioinform. 2019, 20, 405. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Number | Age (Median) | Gender (Male/Female) | Percent of Malignant Cells in the Tumor Sample (Median) |
|---|---|---|---|---|
| Pediatric T-ALL | 13 | 1–15 (10) | 8/5 | 41–95 (87.5) |
| Pediatric B-ALL | 31 | 1–16 (4) | 16/15 | 42–86 (77.5) |
| Pediatric AML | 14 | 1–15 (7) | 6/8 | 55–90 (74) |
| Adult T-ALL | 4 | 29–70 (54) | 2/2 | 75–96 (86) |
| Adult B-ALL | 10 | 19–51 (26) | 7/3 | 23–86 (72) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urazbakhtin, S.; Smirnova, A.; Volakhava, A.; Zerkalenkova, E.; Salyutina, M.; Doubek, M.; Jelinkova, H.; Khudainazarova, N.; Volchkov, E.; Belyaeva, L.; et al. The Absence of Retroelement Activity Is Characteristic for Childhood Acute Leukemias and Adult Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2022, 23, 1756. https://doi.org/10.3390/ijms23031756
Urazbakhtin S, Smirnova A, Volakhava A, Zerkalenkova E, Salyutina M, Doubek M, Jelinkova H, Khudainazarova N, Volchkov E, Belyaeva L, et al. The Absence of Retroelement Activity Is Characteristic for Childhood Acute Leukemias and Adult Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2022; 23(3):1756. https://doi.org/10.3390/ijms23031756
Chicago/Turabian StyleUrazbakhtin, Shamil, Anastasia Smirnova, Anastasiya Volakhava, Elena Zerkalenkova, Maria Salyutina, Michael Doubek, Hana Jelinkova, Nelly Khudainazarova, Egor Volchkov, Laima Belyaeva, and et al. 2022. "The Absence of Retroelement Activity Is Characteristic for Childhood Acute Leukemias and Adult Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 23, no. 3: 1756. https://doi.org/10.3390/ijms23031756
APA StyleUrazbakhtin, S., Smirnova, A., Volakhava, A., Zerkalenkova, E., Salyutina, M., Doubek, M., Jelinkova, H., Khudainazarova, N., Volchkov, E., Belyaeva, L., Komech, E., Pavlova, S., Lebedev, Y., Plevova, K., Olshanskaya, Y., Komkov, A., & Mamedov, I. (2022). The Absence of Retroelement Activity Is Characteristic for Childhood Acute Leukemias and Adult Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences, 23(3), 1756. https://doi.org/10.3390/ijms23031756