
SH2 Domains: Folding, Binding and Therapeutical Approaches

 , , , ,
, , , ,
Abstract
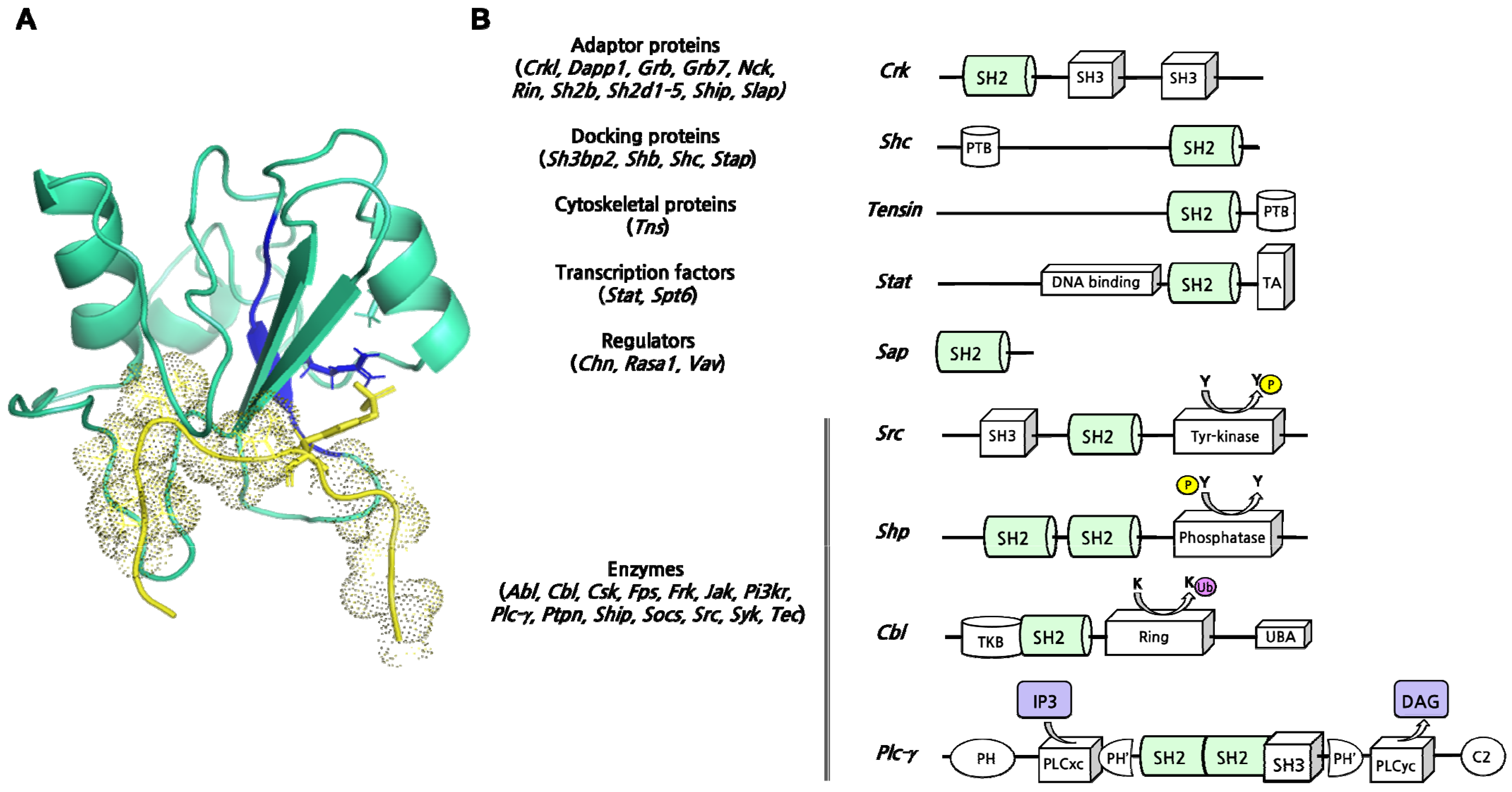
:1. SH2 Domains Are the Archetypical “Readers” of Phosphorylated Tyrosines
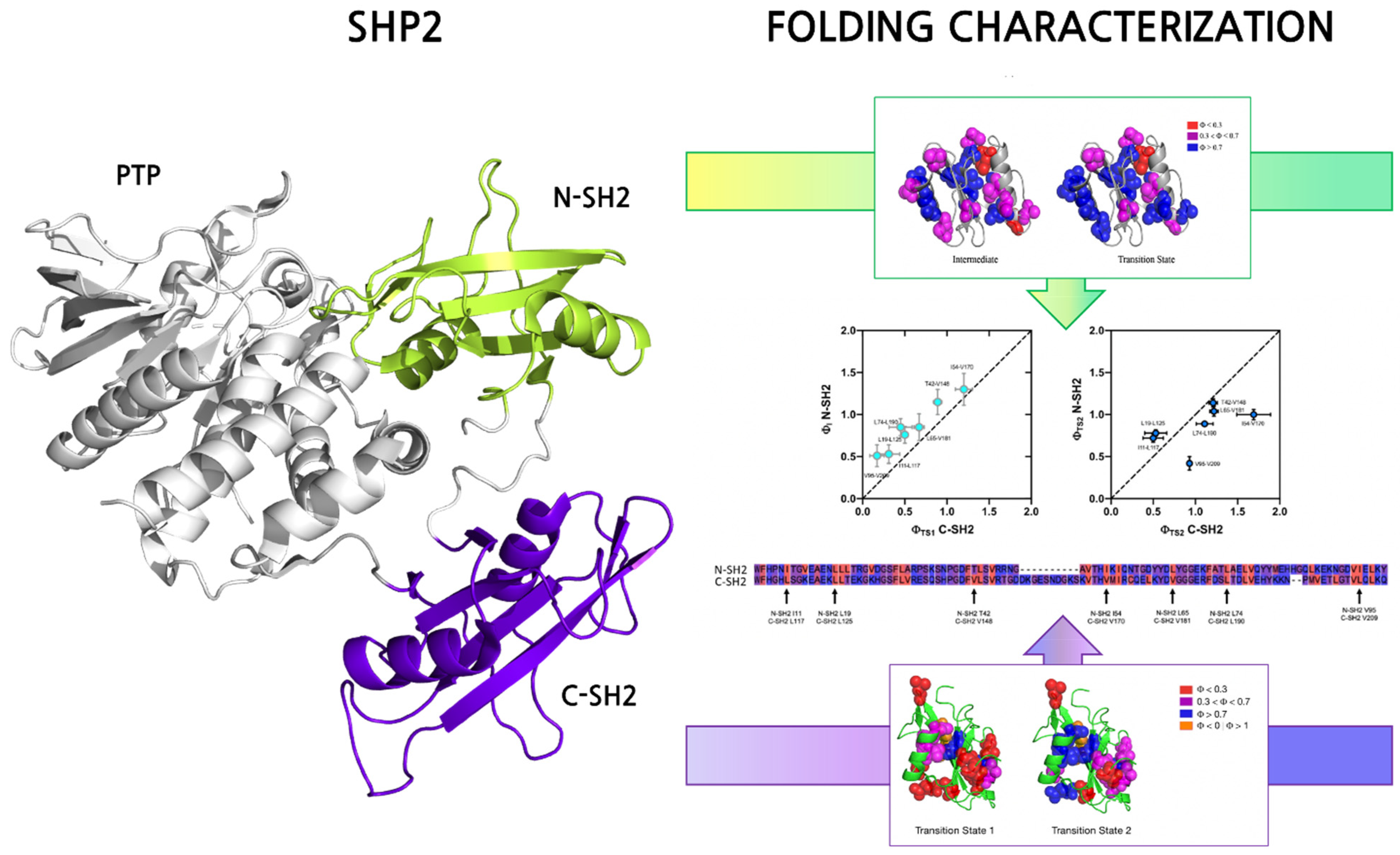
2. Folding Properties of SH2 Domains
3. SH2 Domain Binding Properties
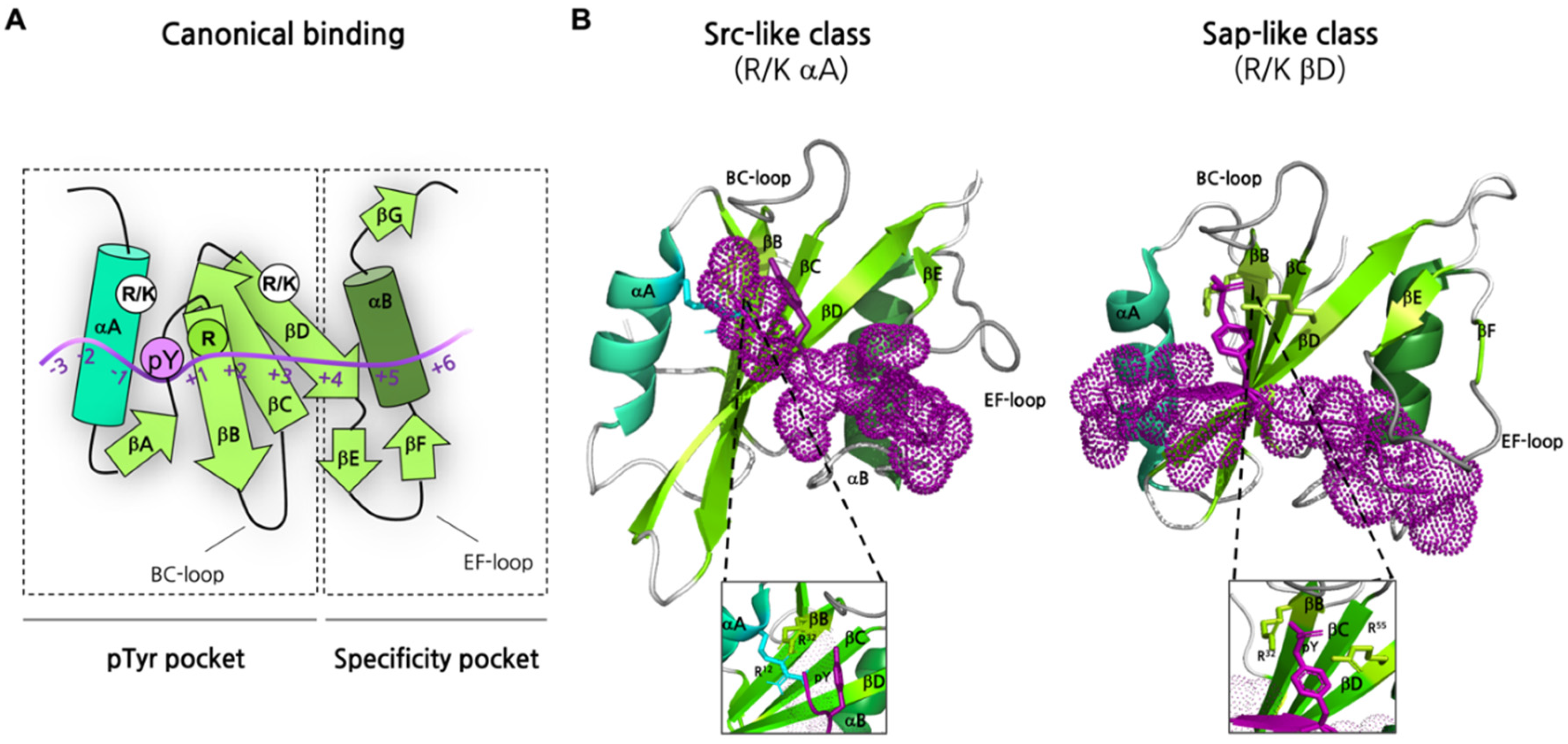
3.1. Defining the Structural Determinants of Recognition and Specificity
3.2. Biophysical Characterization of SH2 Binding
3.3. Non-Canonical SH2 Binding
3.4. Biomolecular Condensate Formation Mediated by SH2 Domains
4. The Pathological Role of SH2-Containing Proteins
4.1. Noonan Syndrome: A Genetic Disorder Due to Mutations Affecting SH2 Domains from SHP2
4.2. SH2-Containing Proteins and Immunodeficiency Disorders
4.3. SH2-Containing Proteins and Cancer
5. Pharmacological Strategies Aimed to Regulate/Inhibit SH2 Domains
5.1. Peptidomimetics and Small Molecules Inhibitors
5.2. Allosteric Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pawson, T.; Nash, P. Assembly of Cell Regulatory Systems through Protein Interaction Domains. Science 2003, 300, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, T. Tyrosine Phosphorylation: Thirty Years and Counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Kavanaugh, W.M.; Turck, C.W.; Williams, L.T. PTB Domain Binding to Signaling Proteins through a Sequence Motif Containing Phosphotyrosine. Science 1995, 268, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Benes, C.H.; Wu, N.; Elia, A.E.H.; Dharia, T.; Cantley, L.C.; Soltoff, S.P. The C2 Domain of PKCdelta Is a Phosphotyrosine Binding Domain. Cell 2005, 121, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, M.; Chow, S.Y.; Yusoff, P.; Seetharaman, J.; Ng, C.; Sinniah, S.; Koh, X.W.; Asgar, N.F.M.; Li, D.; Yim, D.; et al. Structure of a Novel Phosphotyrosine-Binding Domain in Hakai That Targets E-Cadherin. EMBO J. 2012, 31, 1308–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.A.; Shah, E.; Jablonowski, K.; Stergachis, A.; Engelmann, B.; Nash, P.D. The SH2 Domain–Containing Proteins in 21 Species Establish the Provenance and Scope of Phosphotyrosine Signaling in Eukaryotes. Sci. Signal. 2011, 4, ra83. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, M.; Gelb, B.D. Noonan Syndrome and Related Disorders: Genetics and Pathogenesis. Annu. Rev. Genom. Hum. Genet. 2005, 6, 45–68. [Google Scholar] [CrossRef]
- Keilhack, H.; David, F.S.; McGregor, M.; Cantley, L.C.; Neel, B.G. Diverse Biochemical Properties of Shp2 Mutants. Implications for Disease Phenotypes. J. Biol. Chem. 2005, 280, 30984–30993. [Google Scholar] [CrossRef] [Green Version]
- Niihori, T.; Aoki, Y.; Ohashi, H.; Kurosawa, K.; Kondoh, T.; Ishikiriyama, S.; Kawame, H.; Kamasaki, H.; Yamanaka, T.; Takada, F.; et al. Functional Analysis of PTPN11/SHP-2 Mutants Identified in Noonan Syndrome and Childhood Leukemia. J. Hum. Genet. 2005, 50, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 Tyrosine Phosphatase as an Intracellular Target of Helicobacter Pylori CagA Protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef]
- Poy, F.; Yaffe, M.B.; Sayos, J.; Saxena, K.; Morra, M.; Sumegi, J.; Cantley, L.C.; Terhorst, C.; Eck, M.J. Crystal Structures of the XLP Protein SAP Reveal a Class of SH2 Domains with Extended, Phosphotyrosine-Independent Sequence Recognition. Mol. Cell 1999, 4, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Waksman, G.; Kominos, D.; Robertson, S.C.; Pant, N.; Baltimore, D.; Birge, R.B.; Cowburn, D.; Hanafusa, H.; Mayer, B.J.; Overduin, M.; et al. Crystal Structure of the Phosphotyrosine Recognition Domain SH2 of V-Src Complexed with Tyrosine-Phosphorylated Peptides. Nature 1992, 358, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Waksman, G.; Shoelson, S.E.; Pant, N.; Cowburn, D.; Kuriyan, J. Binding of a High Affinity Phosphotyrosyl Peptide to the Src SH2 Domain: Crystal Structures of the Complexed and Peptide-Free Forms. Cell 1993, 72, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.J.; Spencer, J.; Clarke, A.R. An Integrated Kinetic Analysis of Intermediates and Transition States in Protein Folding Reactions. J. Mol. Biol. 1995, 253, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Gianni, S.; Ivarsson, Y.; Jemth, P.; Brunori, M.; Travaglini-Allocatelli, C. Identification and Characterization of Protein Folding Intermediates. Biophys. Chem. 2007, 128, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, D.; Troilo, F.; Toto, A.; Travaglini-Allocatelli, C.; Brunori, M.; Gianni, S. Mechanism of Folding and Binding of the N-Terminal SH2 Domain from SHP2. J. Phys. Chem. B 2018, 122, 11108–11114. [Google Scholar] [CrossRef]
- Fersht, A.R.; Matouschek, A.; Serrano, L. The Folding of an Enzyme. I. Theory of Protein Engineering Analysis of Stability and Pathway of Protein Folding. J. Mol. Biol. 1992, 224, 771–782. [Google Scholar] [CrossRef]
- Fersht, A.R.; Sato, S. Phi-Value Analysis and the Nature of Protein-Folding Transition States. Proc. Natl. Acad. Sci. USA 2004, 101, 7976–7981. [Google Scholar] [CrossRef] [Green Version]
- Visconti, L.; Malagrinò, F.; Gianni, S.; Toto, A. Structural Characterization of an On-Pathway Intermediate and Transition State in the Folding of the N-Terminal SH2 Domain from SHP2. FEBS J. 2019, 286, 4769–4777. [Google Scholar] [CrossRef]
- Toto, A.; Malagrinò, F.; Nardella, C.; Pennacchietti, V.; Pagano, L.; Santorelli, D.; Diop, A.; Gianni, S. Characterization of Early and Late Transition States of the Folding Pathway of a SH2 Domain. Protein Sci. Publ. Protein Soc. 2022, 31, e4332. [Google Scholar] [CrossRef]
- Wildes, D.; Anderson, L.M.; Sabogal, A.; Marqusee, S. Native State Energetics of the Src SH2 Domain: Evidence for a Partially Structured State in the Denatured Ensemble. Protein Sci. Publ. Protein Soc. 2006, 15, 1769–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visconti, L.; Malagrinò, F.; Toto, A.; Gianni, S. The Kinetics of Folding of the NSH2 Domain from P85. Sci. Rep. 2019, 9, 4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troilo, F.; Malagrinò, F.; Visconti, L.; Toto, A.; Gianni, S. The Effect of Proline Cis-Trans Isomerization on the Folding of the C-Terminal SH2 Domain from P85. Int. J. Mol. Sci. 2019, 21, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardella, C.; Toto, A.; Santorelli, D.; Pagano, L.; Diop, A.; Pennacchietti, V.; Pietrangeli, P.; Marcocci, L.; Malagrinò, F.; Gianni, S. Folding and Binding Mechanisms of the SH2 Domain from Crkl. Biomolecules 2022, 12, 1014. [Google Scholar] [CrossRef]
- Nardella, C.; Malagrinò, F.; Pagano, L.; Rinaldo, S.; Gianni, S.; Toto, A. Determining Folding and Binding Properties of the C-Terminal SH2 Domain of SHP2. Protein Sci. Publ. Protein Soc. 2021, 30, 2385–2395. [Google Scholar] [CrossRef]
- Ivarsson, Y.; Jemth, P. Affinity and Specificity of Motif-Based Protein-Protein Interactions. Curr. Opin. Struct. Biol. 2019, 54, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, G.; Keating, A.E. Protein Binding Specificity versus Promiscuity. Curr. Opin. Struct. Biol. 2011, 21, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, G. Kinetic Studies of Protein-Protein Interactions. Curr. Opin. Struct. Biol. 2002, 12, 41–47. [Google Scholar] [CrossRef]
- Gianni, S.; Jemth, P. How Fast Is Protein-Ligand Association? Trends Biochem. Sci. 2017, 42, 847–849. [Google Scholar] [CrossRef]
- Koch, C.A.; Anderson, D.; Moran, M.F.; Ellis, C.; Pawson, T. SH2 and SH3 Domains: Elements That Control Interactions of Cytoplasmic Signaling Proteins. Science 1991, 252, 668–674. [Google Scholar] [CrossRef]
- Pawson, T.; Gish, G.D.; Nash, P. SH2 Domains, Interaction Modules and Cellular Wiring. Trends Cell Biol. 2001, 11, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Song, J. Structural Insight into the Binding Diversity between the Tyr-Phosphorylated Human EphrinBs and Nck2 SH2 Domain. J. Biol. Chem. 2005, 280, 19205–19212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, T.; Huang, H.; Zhao, B.; Li, L.; Liu, H.; Voss, C.K.; Wu, C.; Schiller, M.R.; Li, S.S.-C. Loops Govern SH2 Domain Specificity by Controlling Access to Binding Pockets. Sci. Signal. 2010, 3, ra34. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Mayer, B.J. The SH2 Domain: Versatile Signaling Module and Pharmaceutical Target. Biochim. Biophys. Acta 2005, 1747, 1–25. [Google Scholar] [CrossRef]
- Kaneko, T.; Joshi, R.; Feller, S.M.; Li, S.S. Phosphotyrosine Recognition Domains: The Typical, the Atypical and the Versatile. Cell Commun. Signal. CCS 2012, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Mayer, B.J.; Jackson, P.K.; Baltimore, D. The Noncatalytic Src Homology Region 2 Segment of Abl Tyrosine Kinase Binds to Tyrosine-Phosphorylated Cellular Proteins with High Affinity. Proc. Natl. Acad. Sci. USA 1991, 88, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.J.; Pendergast, A.M.; Havlik, M.H.; Puil, L.; Pawson, T.; Witte, O.N. A Limited Set of SH2 Domains Binds BCR through a High-Affinity Phosphotyrosine-Independent Interaction. Mol. Cell. Biol. 1992, 12, 5087–5093. [Google Scholar] [CrossRef]
- Liu, B.A.; Jablonowski, K.; Raina, M.; Arcé, M.; Pawson, T.; Nash, P.D. The Human and Mouse Complement of SH2 Domain Proteins-Establishing the Boundaries of Phosphotyrosine Signaling. Mol. Cell 2006, 22, 851–868. [Google Scholar] [CrossRef]
- Diella, F.; Haslam, N.; Chica, C.; Budd, A.; Michael, S.; Brown, N.P.; Trave, G.; Gibson, T.J. Understanding Eukaryotic Linear Motifs and Their Role in Cell Signaling and Regulation. Front. Biosci. J. Virtual Libr. 2008, 13, 6580–6603. [Google Scholar] [CrossRef] [Green Version]
- Neduva, V.; Russell, R.B. Peptides Mediating Interaction Networks: New Leads at Last. Curr. Opin. Biotechnol. 2006, 17, 465–471. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Toto, A.; Malagrinò, F.; Visconti, L.; Troilo, F.; Pagano, L.; Brunori, M.; Jemth, P.; Gianni, S. Templated Folding of Intrinsically Disordered Proteins. J. Biol. Chem. 2020, 295, 6586–6593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.; Uversky, V.N.; Chen, Z.; Dunker, A.K.; Obradovic, Z. Short Linear Motifs Recognized by SH2, SH3 and Ser/Thr Kinase Domains Are Conserved in Disordered Protein Regions. BMC Genom. 2008, 9 (Suppl. S2), S26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marasco, M.; Carlomagno, T. Specificity and Regulation of Phosphotyrosine Signaling through SH2 Domains. J. Struct. Biol. X 2020, 4, 100026. [Google Scholar] [CrossRef]
- Songyang, Z.; Shoelson, S.E.; McGlade, J.; Olivier, P.; Pawson, T.; Bustelo, X.R.; Barbacid, M.; Sabe, H.; Hanafusa, H.; Yi, T. Specific Motifs Recognized by the SH2 Domains of Csk, 3BP2, Fps/Fes, GRB-2, HCP, SHC, Syk, and Vav. Mol. Cell. Biol. 1994, 14, 2777–2785. [Google Scholar] [CrossRef]
- Liu, X.; Brodeur, S.R.; Gish, G.; Songyang, Z.; Cantley, L.C.; Laudano, A.P.; Pawson, T. Regulation of C-Src Tyrosine Kinase Activity by the Src SH2 Domain. Oncogene 1993, 8, 1119–1126. [Google Scholar]
- Ladbury, J.E.; Lemmon, M.A.; Zhou, M.; Green, J.; Botfield, M.C.; Schlessinger, J. Measurement of the Binding of Tyrosyl Phosphopeptides to SH2 Domains: A Reappraisal. Proc. Natl. Acad. Sci. USA 1995, 92, 3199–3203. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.A.; Engelmann, B.W.; Nash, P.D. The Language of SH2 Domain Interactions Defines Phosphotyrosine-Mediated Signal Transduction. FEBS Lett. 2012, 586, 2597–2605. [Google Scholar] [CrossRef] [Green Version]
- Grucza, R.A.; Bradshaw, J.M.; Fütterer, K.; Waksman, G. SH2 Domains: From Structure to Energetics, a Dual Approach to the Study of Structure-Function Relationships. Med. Res. Rev. 1999, 19, 273–293. [Google Scholar] [CrossRef]
- Cantley, L.C.; Auger, K.R.; Carpenter, C.; Duckworth, B.; Graziani, A.; Kapeller, R.; Soltoff, S. Oncogenes and Signal Transduction. Cell 1991, 64, 281–302. [Google Scholar] [CrossRef]
- Songyang, Z.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J. SH2 Domains Recognize Specific Phosphopeptide Sequences. Cell 1993, 72, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Li, S.S.-C.; Harper, J.W.; Songyang, Z. An Oriented Peptide Array Library (OPAL) Strategy to Study Protein-Protein Interactions. J. Biol. Chem. 2004, 279, 8802–8807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Li, L.; Wu, C.; Schibli, D.; Colwill, K.; Ma, S.; Li, C.; Roy, P.; Ho, K.; Songyang, Z.; et al. Defining the Specificity Space of the Human SRC Homology 2 Domain. Mol. Cell. Proteom. MCP 2008, 7, 768–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Huang, H.; Voss, C.; Kaneko, T.; Qin, W.T.; Sidhu, S.; Li, S.S.-C. Surface Loops in a Single SH2 Domain Are Capable of Encoding the Spectrum of Specificity of the SH2 Family. Mol. Cell. Proteom. MCP 2019, 18, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Eck, M.J.; Shoelson, S.E.; Harrison, S.C. Recognition of a High-Affinity Phosphotyrosyl Peptide by the Src Homology-2 Domain of P56lck. Nature 1993, 362, 87–91. [Google Scholar] [CrossRef]
- Ogura, K.; Tsuchiya, S.; Terasawa, H.; Yuzawa, S.; Hatanaka, H.; Mandiyan, V.; Schlessinger, J.; Inagaki, F. Solution Structure of the SH2 Domain of Grb2 Complexed with the Shc-Derived Phosphotyrosine-Containing Peptide. J. Mol. Biol. 1999, 289, 439–445. [Google Scholar] [CrossRef]
- Rahuel, J.; Gay, B.; Erdmann, D.; Strauss, A.; Garcia-Echeverría, C.; Furet, P.; Caravatti, G.; Fretz, H.; Schoepfer, J.; Grütter, M.G. Structural Basis for Specificity of Grb2-SH2 Revealed by a Novel Ligand Binding Mode. Nat. Struct. Biol. 1996, 3, 586–589. [Google Scholar] [CrossRef]
- Zhao, H.; Naganathan, S.; Beckett, D. Thermodynamic and Structural Investigation of Bispecificity in Protein-Protein Interactions. J. Mol. Biol. 2009, 389, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Akiva, E.; Itzhaki, Z.; Margalit, H. Built-in Loops Allow Versatility in Domain-Domain Interactions: Lessons from Self-Interacting Domains. Proc. Natl. Acad. Sci. USA 2008, 105, 13292–13297. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, S.S.; Fellouse, F.A. Synthetic Therapeutic Antibodies. Nat. Chem. Biol. 2006, 2, 682–688. [Google Scholar] [CrossRef]
- Hiipakka, M.; Saksela, K. Versatile Retargeting of SH3 Domain Binding by Modification of Non-Conserved Loop Residues. FEBS Lett. 2007, 581, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.-C. Specificity and Versatility of SH3 and Other Proline-Recognition Domains: Structural Basis and Implications for Cellular Signal Transduction. Biochem. J. 2005, 390, 641–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visconti, L.; Malagrinò, F.; Pagano, L.; Toto, A. Understanding the Mechanism of Recognition of Gab2 by the N-SH2 Domain of SHP2. Life 2020, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Visconti, L.; Toto, A.; Jarvis, J.A.; Troilo, F.; Malagrinò, F.; De Simone, A.; Gianni, S. Demonstration of Binding Induced Structural Plasticity in a SH2 Domain. Front. Mol. Biosci. 2020, 7, 89. [Google Scholar] [CrossRef]
- Toto, A.; Malagrinò, F.; Visconti, L.; Troilo, F.; Gianni, S. Unveiling the Molecular Basis of the Noonan Syndrome-Causing Mutation T42A of SHP2. Int. J. Mol. Sci. 2020, 21, 461. [Google Scholar] [CrossRef] [Green Version]
- Jaber Chehayeb, R.; Boggon, T.J. SH2 Domain Binding: Diverse FLVRs of Partnership. Front. Endocrinol. 2020, 11, 575220. [Google Scholar] [CrossRef]
- Jaber Chehayeb, R.; Wang, J.; Stiegler, A.L.; Boggon, T.J. The GTPase-Activating Protein P120RasGAP Has an Evolutionarily Conserved “FLVR-Unique” SH2 Domain. J. Biol. Chem. 2020, 295, 10511–10521. [Google Scholar] [CrossRef]
- Wallweber, H.J.A.; Tam, C.; Franke, Y.; Starovasnik, M.A.; Lupardus, P.J. Structural Basis of Recognition of Interferon-α Receptor by Tyrosine Kinase 2. Nat. Struct. Mol. Biol. 2014, 21, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK-Receptor Interactions. Front. Endocrinol. 2017, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wlodawer, A.; Lubkowski, J. Crystal Structure of a Complex of the Intracellular Domain of Interferon λ Receptor 1 (IFNLR1) and the FERM/SH2 Domains of Human JAK1. J. Mol. Biol. 2016, 428, 4651–4668. [Google Scholar] [CrossRef]
- Hwang, P.M.; Li, C.; Morra, M.; Lillywhite, J.; Muhandiram, D.R.; Gertler, F.; Terhorst, C.; Kay, L.E.; Pawson, T.; Forman-Kay, J.D.; et al. A “Three-Pronged” Binding Mechanism for the SAP/SH2D1A SH2 Domain: Structural Basis and Relevance to the XLP Syndrome. EMBO J. 2002, 21, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Piraner, D.; Gorenstein, N.M.; Geahlen, R.L.; Beth Post, C. Differential Recognition of Syk-Binding Sites by Each of the Two Phosphotyrosine-Binding Pockets of the Vav SH2 Domain. Biopolymers 2013, 99, 897–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groesch, T.D.; Zhou, F.; Mattila, S.; Geahlen, R.L.; Post, C.B. Structural Basis for the Requirement of Two Phosphotyrosine Residues in Signaling Mediated by Syk Tyrosine Kinase. J. Mol. Biol. 2006, 356, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Hatada, M.H.; Lu, X.; Laird, E.R.; Green, J.; Morgenstern, J.P.; Lou, M.; Marr, C.S.; Phillips, T.B.; Ram, M.K.; Theriault, K. Molecular Basis for Interaction of the Protein Tyrosine Kinase ZAP-70 with the T-Cell Receptor. Nature 1995, 377, 32–38. [Google Scholar] [CrossRef]
- Hu, J.; Liu, J.; Ghirlando, R.; Saltiel, A.R.; Hubbard, S.R. Structural Basis for Recruitment of the Adaptor Protein APS to the Activated Insulin Receptor. Mol. Cell 2003, 12, 1379–1389. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Fuxreiter, M. Protein Interactions in Liquid-Liquid Phase Separation. J. Mol. Biol. 2022, 434, 167388. [Google Scholar] [CrossRef]
- Alberti, S.; Dormann, D. Liquid-Liquid Phase Separation in Disease. Annu. Rev. Genet. 2019, 53, 171–194. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Brangwynne, C.P. Liquid Phase Condensation in Cell Physiology and Disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Horvath, A.; Miskei, M.; Ambrus, V.; Vendruscolo, M.; Fuxreiter, M. Sequence-Based Prediction of Protein Binding Mode Landscapes. PLoS Comput. Biol. 2020, 16, e1007864. [Google Scholar] [CrossRef] [PubMed]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual Specificity Kinase DYRK3 Couples Stress Granule Condensation/Dissolution to MTORC1 Signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berchtold, D.; Battich, N.; Pelkmans, L. A Systems-Level Study Reveals Regulators of Membrane-Less Organelles in Human Cells. Mol. Cell 2018, 72, 1035–1049.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-W.; Nocka, L.M.; Stinger, B.L.; DeGrandchamp, J.B.; Lew, L.J.N.; Alvarez, S.; Phan, H.T.; Kondo, Y.; Kuriyan, J.; Groves, J.T. A Two-Component Protein Condensate of the EGFR Cytoplasmic Tail and Grb2 Regulates Ras Activation by SOS at the Membrane. Proc. Natl. Acad. Sci. USA 2022, 119, e2122531119. [Google Scholar] [CrossRef]
- Chardin, P.; Camonis, J.H.; Gale, N.W.; van Aelst, L.; Schlessinger, J.; Wigler, M.H.; Bar-Sagi, D. Human Sos1: A Guanine Nucleotide Exchange Factor for Ras That Binds to GRB2. Science 1993, 260, 1338–1343. [Google Scholar] [CrossRef]
- Ding, C.-B.; Yu, W.-N.; Feng, J.-H.; Luo, J.-M. Structure and Function of Gab2 and Its Role in Cancer (Review). Mol. Med. Rep. 2015, 12, 4007–4014. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Yu, D.-H.; Shen, R.; Feng, G.-S. Gab2, a New Pleckstrin Homology Domain-Containing Adapter Protein, Acts to Uncouple Signaling from ERK Kinase to Elk-1. J. Biol. Chem. 1999, 274, 19649–19654. [Google Scholar] [CrossRef] [Green Version]
- Noonan, J.A. Hypertelorism with Turner Phenotype. A New Syndrome with Associated Congenital Heart Disease. Am. J. Dis. Child. 1960 1968, 116, 373–380. [Google Scholar] [CrossRef]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan Syndrome. Lancet Lond. Engl. 2013, 381, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Bobone, S.; Pannone, L.; Biondi, B.; Solman, M.; Flex, E.; Canale, V.C.; Calligari, P.; De Faveri, C.; Gandini, T.; Quercioli, A.; et al. Targeting Oncogenic Src Homology 2 Domain-Containing Phosphatase 2 (SHP2) by Inhibiting Its Protein-Protein Interactions. J. Med. Chem. 2021, 64, 15973–15990. [Google Scholar] [CrossRef]
- Calligari, P.; Santucci, V.; Stella, L.; Bocchinfuso, G. Discriminating between Competing Models for the Allosteric Regulation of Oncogenic Phosphatase SHP2 by Characterizing Its Active State. Comput. Struct. Biotechnol. J. 2021, 19, 6125–6139. [Google Scholar] [CrossRef] [PubMed]
- LaRochelle, J.R.; Fodor, M.; Vemulapalli, V.; Mohseni, M.; Wang, P.; Stams, T.; LaMarche, M.J.; Chopra, R.; Acker, M.G.; Blacklow, S.C. Structural Reorganization of SHP2 by Oncogenic Mutations and Implications for Oncoprotein Resistance to Allosteric Inhibition. Nat. Commun. 2018, 9, 4508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentires-Alj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.; Bardelli, A.; Sugarbaker, D.J.; et al. Activating Mutations of the Noonan Syndrome-Associated SHP2/PTPN11 Gene in Human Solid Tumors and Adult Acute Myelogenous Leukemia. Cancer Res. 2004, 64, 8816–8820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.; Kalaitzidis, D.; Neel, B.G. The Tyrosine Phosphatase Shp2 (PTPN11) in Cancer. Cancer Metastasis Rev. 2008, 27, 179–192. [Google Scholar] [CrossRef]
- Martinelli, S.; Torreri, P.; Tinti, M.; Stella, L.; Bocchinfuso, G.; Flex, E.; Grottesi, A.; Ceccarini, M.; Palleschi, A.; Cesareni, G.; et al. Diverse Driving Forces Underlie the Invariant Occurrence of the T42A, E139D, I282V and T468M SHP2 Amino Acid Substitutions Causing Noonan and LEOPARD Syndromes. Hum. Mol. Genet. 2008, 17, 2018–2029. [Google Scholar] [CrossRef] [Green Version]
- Cunnick, J.M.; Dorsey, J.F.; Munoz-Antonia, T.; Mei, L.; Wu, J. Requirement of SHP2 Binding to Grb2-Associated Binder-1 for Mitogen-Activated Protein Kinase Activation in Response to Lysophosphatidic Acid and Epidermal Growth Factor. J. Biol. Chem. 2000, 275, 13842–13848. [Google Scholar] [CrossRef] [Green Version]
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.K.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonilla, F.A.; et al. Newborn Screening for Severe Combined Immunodeficiency in 11 Screening Programs in the United States. JAMA 2014, 312, 729. [Google Scholar] [CrossRef]
- Yan, Q.; Barros, T.; Visperas, P.R.; Deindl, S.; Kadlecek, T.A.; Weiss, A.; Kuriyan, J. Structural Basis for Activation of ZAP-70 by Phosphorylation of the SH2-Kinase Linker. Mol. Cell. Biol. 2013, 33, 2188–2201. [Google Scholar] [CrossRef] [Green Version]
- Candotti, F.; Oakes, S.A.; Johnston, J.A.; Giliani, S.; Schumacher, R.F.; Mella, P.; Fiorini, M.; Ugazio, A.G.; Badolato, R.; Notarangelo, L.D.; et al. Structural and Functional Basis for JAK3-Deficient Severe Combined Immunodeficiency. Blood 1997, 90, 3996–4003. [Google Scholar] [CrossRef] [Green Version]
- Sharifinejad, N.; Jamee, M.; Zaki-Dizaji, M.; Lo, B.; Shaghaghi, M.; Mohammadi, H.; Jadidi-Niaragh, F.; Shaghaghi, S.; Yazdani, R.; Abolhassani, H.; et al. Clinical, Immunological, and Genetic Features in 49 Patients With ZAP-70 Deficiency: A Systematic Review. Front. Immunol. 2020, 11, 831. [Google Scholar] [CrossRef]
- Väliaho, J.; Smith, C.I.E.; Vihinen, M. BTKbase: The Mutation Database for X-Linked Agammaglobulinemia. Hum. Mutat. 2006, 27, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Teng, Y.; Pan, J.; Shi, Q.; Liu, Y.; Zhang, F.; Liang, D.; Li, Z.; Wu, L. Identification of Four Novel Mutations in BTK from Six Chinese Families with X-Linked Agammaglobulinemia. Clin. Chim. Acta Int. J. Clin. Chem. 2022, 531, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.-R.; Pai, M.-T.; Wu, C.-W.; Cheng, J.-W.; Lung, F.-D.T.; Roller, P.P.; Lei, B.; Wei, C.-J.; Tu, S.-C.; Chen, S.-H.; et al. Stability and Peptide Binding Specificity of Btk SH2 Domain: Molecular Basis for X-Linked Agammaglobulinemia. Protein Sci. 2000, 9, 2377–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, D.P.; Lamontanara, A.J.; La Sala, G.; Jeong, S.; Sohn, Y.-K.; Panjkovich, A.; Georgeon, S.; Kükenshöner, T.; Marcaida, M.J.; Pojer, F.; et al. Btk SH2-Kinase Interface Is Critical for Allosteric Kinase Activation and Its Targeting Inhibits B-Cell Neoplasms. Nat. Commun. 2020, 11, 2319. [Google Scholar] [CrossRef]
- Available online: https://www.cancer.org/cancer/prostate-cancer/about/key-statistics.html (accessed on 15 November 2022).
- Claessens, F.; Helsen, C.; Prekovic, S.; Van den Broeck, T.; Spans, L.; Van Poppel, H.; Joniau, S. Emerging Mechanisms of Enzalutamide Resistance in Prostate Cancer. Nat. Rev. Urol. 2014, 11, 712–716. [Google Scholar] [CrossRef]
- Tang, Q.; Fang, J.; Lai, W.; Hu, Y.; Liu, C.; Hu, X.; Song, C.; Cheng, T.; Liu, R.; Huang, X. Hippo Pathway Monomerizes STAT3 to Regulate Prostate Cancer Growth. Cancer Sci. 2022, 113, 2753–2762. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. Immunity, Inflammation and Cancer: A Leading Role for Adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef]
- Matsuda, T.; Oritani, K. Possible Therapeutic Applications of Targeting STAP Proteins in Cancer. Biol. Pharm. Bull. 2021, 44, 1810–1818. [Google Scholar] [CrossRef]
- Kitai, Y.; Iwakami, M.; Saitoh, K.; Togi, S.; Isayama, S.; Sekine, Y.; Muromoto, R.; Kashiwakura, J.-I.; Yoshimura, A.; Oritani, K.; et al. STAP-2 Protein Promotes Prostate Cancer Growth by Enhancing Epidermal Growth Factor Receptor Stabilization. J. Biol. Chem. 2017, 292, 19392–19399. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Song, G.; Zhang, S.; Chen, J.; Hu, X.; Zhu, H.; Jia, X.; Li, Z.; Song, W.; Chen, J.; et al. Asialoglycoprotein Receptor 1 Functions as a Tumor Suppressor in Liver Cancer via Inhibition of STAT3. Cancer Res. 2022, 82, 3987–4000. [Google Scholar] [CrossRef]
- Melvin, W.J.; Audu, C.O.; Davis, F.M.; Sharma, S.B.; Joshi, A.; DenDekker, A.; Wolf, S.; Barrett, E.; Mangum, K.; Zhou, X.; et al. Coronavirus Induces Diabetic Macrophage-Mediated Inflammation via SETDB2. Proc. Natl. Acad. Sci. USA 2021, 118, e2101071118. [Google Scholar] [CrossRef]
- Ren, R. Mechanisms of BCR–ABL in the Pathogenesis of Chronic Myelogenous Leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jang, H.; Zhang, M.; Tsai, C.-J.; Maloney, R.; Nussinov, R. The Structural Basis of BCR-ABL Recruitment of GRB2 in Chronic Myelogenous Leukemia. Biophys. J. 2022, 121, 2251–2265. [Google Scholar] [CrossRef] [PubMed]
- Ruschmann, J.; Ho, V.; Antignano, F.; Kuroda, E.; Lam, V.; Ibaraki, M.; Snyder, K.; Kim, C.; Flavell, R.A.; Kawakami, T.; et al. Tyrosine Phosphorylation of SHIP Promotes Its Proteasomal Degradation. Exp. Hematol. 2010, 38, 392–402.e1. [Google Scholar] [CrossRef]
- Amin, H.; Hoshino, K.; Yang, H.; Lin, Q.; Lai, R.; Garcia-Manero, G. Decreased Expression Level of SH2 Domain-Containing Protein Tyrosine Phosphatase-1 (Shp1) Is Associated with Progression of Chronic Myeloid Leukaemia. J. Pathol. 2007, 212, 402–410. [Google Scholar] [CrossRef]
- Toda, J.; Ichii, M.; Oritani, K.; Shibayama, H.; Tanimura, A.; Saito, H.; Yokota, T.; Motooka, D.; Okuzaki, D.; Kitai, Y.; et al. Signal-Transducing Adapter Protein-1 Is Required for Maintenance of Leukemic Stem Cells in CML. Oncogene 2020, 39, 5601–5615. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Wasik, M.A.; Slupianek, A.; Salomoni, P.; Kitamura, T.; Calabretta, B.; Skorski, T. Signal Transducer and Activator of Transcription (STAT)5 Activation by BCR/ABL Is Dependent on Intact Src Homology (SH)3 and SH2 Domains of BCR/ABL and Is Required for Leukemogenesis. J. Exp. Med. 1999, 189, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Elliott, T.S.; Slowey, A.; Ye, Y.; Conway, S.J. The Use of Phosphate Bioisosteres in Medicinal Chemistry and Chemical Biology. MedChemComm 2012, 3, 735. [Google Scholar] [CrossRef]
- Domchek, S.M.; Auger, K.R.; Chatterjee, S.; Burke, T.R.; Shoelson, S.E. Inhibition of SH2 Domain/Phosphoprotein Association by a Nonhydrolyzable Phosphonopeptide. Biochemistry 1992, 31, 9865–9870. [Google Scholar] [CrossRef]
- Furet, P.; Caravatti, G.; Denholm, A.A.; Faessler, A.; Fretz, H.; García-Echeverría, C.; Gay, B.; Irving, E.; Press, N.J.; Rahuel, J.; et al. Structure-Based Design and Synthesis of Phosphinate Isosteres of Phosphotyrosine for Incorporation in Grb2-SH2 Domain Inhibitors. Part 1. Bioorg. Med. Chem. Lett. 2000, 10, 2337–2341. [Google Scholar] [CrossRef]
- Walker, C.V.; Caravatti, G.; Denholm, A.A.; Egerton, J.; Faessler, A.; Furet, P.; García-Echeverría, C.; Gay, B.; Irving, E.; Jones, K.; et al. Structure-Based Design and Synthesis of Phosphinate Isosteres of Phosphotyrosine for Incorporation in Grb2-SH2 Domain Inhibitors. Part 2. Bioorg. Med. Chem. Lett. 2000, 10, 2343–2346. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Liao, W.S.-L.; McMurray, J.S. Synthesis of Phosphatase-Stable, Cell-Permeable Peptidomimetic Prodrugs That Target the SH2 Domain of Stat3. Org. Lett. 2009, 11, 3394–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.K.; Gao, F.; Lu, Z.; Ren, Z.; Ramesh, R.; Birtwistle, J.S.; Kaluarachchi, K.K.; Chen, X.; Bast, R.C.; Liao, W.S.; et al. Potent and Selective Phosphopeptide Mimetic Prodrugs Targeted to the Src Homology 2 (SH2) Domain of Signal Transducer and Activator of Transcription 3. J. Med. Chem. 2011, 54, 3549–3563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.S.; Mandal, P.K.; Liao, W.S.; Klostergaard, J.; Robertson, F.M. The Consequences of Selective Inhibition of Signal Transducer and Activator of Transcription 3 (STAT3) Tyrosine705 Phosphorylation by Phosphopeptide Mimetic Prodrugs Targeting the Src Homology 2 (SH2) Domain. JAK-STAT 2012, 1, 263–347. [Google Scholar] [CrossRef] [Green Version]
- Kulathu, Y.; Grothe, G.; Reth, M. Autoinhibition and Adapter Function of Syk. Immunol. Rev. 2009, 232, 286–299. [Google Scholar] [CrossRef]
- Dekker, F.J.; de Mol, N.J.; Fischer, M.J.E.; Liskamp, R.M.J. Amino Propynyl Benzoic Acid Building Block in Rigid Spacers of Divalent Ligands Binding to the Syk SH2 Domains with Equally High Affinity as the Natural Ligand. Bioorg. Med. Chem. Lett. 2003, 13, 1241–1244. [Google Scholar] [CrossRef]
- Kuil, J.; van Wandelen, L.T.M.; de Mol, N.J.; Liskamp, R.M.J. A Photoswitchable ITAM Peptidomimetic: Synthesis and Real Time Surface Plasmon Resonance (SPR) Analysis of the Effects of Cis-Trans Isomerization on Binding. Bioorg. Med. Chem. 2008, 16, 1393–1399. [Google Scholar] [CrossRef]
- Kuil, J.; van Wandelen, L.T.M.; de Mol, N.J.; Liskamp, R.M.J. Switching between Low and High Affinity for the Syk Tandem SH2 Domain by Irradiation of Azobenzene Containing ITAM Peptidomimetics. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2009, 15, 685–691. [Google Scholar] [CrossRef]
- Oligino, L.; Lung, F.D.; Sastry, L.; Bigelow, J.; Cao, T.; Curran, M.; Burke, T.R.; Wang, S.; Krag, D.; Roller, P.P.; et al. Nonphosphorylated Peptide Ligands for the Grb2 Src Homology 2 Domain. J. Biol. Chem. 1997, 272, 29046–29052. [Google Scholar] [CrossRef] [Green Version]
- Quartararo, J.S.; Eshelman, M.R.; Peraro, L.; Yu, H.; Baleja, J.D.; Lin, Y.-S.; Kritzer, J.A. A Bicyclic Peptide Scaffold Promotes Phosphotyrosine Mimicry and Cellular Uptake. Bioorg. Med. Chem. 2014, 22, 6387–6391. [Google Scholar] [CrossRef] [Green Version]
- Quartararo, J.S.; Wu, P.; Kritzer, J.A. Peptide Bicycles That Inhibit the Grb2 SH2 Domain. Chembiochem Eur. J. Chem. Biol. 2012, 13, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Cerulli, R.A.; Kritzer, J.A. Phosphotyrosine Isosteres: Past, Present and Future. Org. Biomol. Chem. 2020, 18, 583–605. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Eckols, T.K.; Xu, X.; Kasembeli, M.M.; Chen, Y.; Adachi, M.; Song, Y.; Mo, Q.; Lai, S.Y.; Tweardy, D.J. Small-Molecule Inhibition of STAT3 in Radioresistant Head and Neck Squamous Cell Carcinoma. Oncotarget 2016, 7, 26307–26330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbi-Verge, C.; Marinelli, F.; Zafra-Ruano, A.; Ruiz-Sanz, J.; Luque, I.; Faraldo-Gómez, J.D. Two-State Dynamics of the SH3-SH2 Tandem of Abl Kinase and the Allosteric Role of the N-Cap. Proc. Natl. Acad. Sci. USA 2013, 110, E3372–E3380. [Google Scholar] [CrossRef] [Green Version]
- Asmamaw, M.D.; Shi, X.-J.; Zhang, L.-R.; Liu, H.-M. A Comprehensive Review of SHP2 and Its Role in Cancer. Cell. Oncol. Dordr. 2022, 45, 729–753. [Google Scholar] [CrossRef]
- Chen, Y.-N.P.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric Inhibition of SHP2 Phosphatase Inhibits Cancers Driven by Receptor Tyrosine Kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Xie, J.; Si, X.; Gu, S.; Wang, M.; Shen, J.; Li, H.; Shen, J.; Li, D.; Fang, Y.; Liu, C.; et al. Allosteric Inhibitors of SHP2 with Therapeutic Potential for Cancer Treatment. J. Med. Chem. 2017, 60, 10205–10219. [Google Scholar] [CrossRef]
- LaMarche, M.J.; Acker, M.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.H.-T.; Chen, Y.-N.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 13578–13594. [Google Scholar] [CrossRef]
- Vainonen, J.P.; Momeny, M.; Westermarck, J. Druggable Cancer Phosphatases. Sci. Transl. Med. 2021, 13, eabe2967. [Google Scholar] [CrossRef]
- Wu, X.; Xu, G.; Li, X.; Xu, W.; Li, Q.; Liu, W.; Kirby, K.A.; Loh, M.L.; Li, J.; Sarafianos, S.G.; et al. Small Molecule Inhibitor That Stabilizes the Autoinhibited Conformation of the Oncogenic Tyrosine Phosphatase SHP2. J. Med. Chem. 2019, 62, 1125–1137. [Google Scholar] [CrossRef]
- Fodor, M.; Price, E.; Wang, P.; Lu, H.; Argintaru, A.; Chen, Z.; Glick, M.; Hao, H.-X.; Kato, M.; Koenig, R.; et al. Dual Allosteric Inhibition of SHP2 Phosphatase. ACS Chem. Biol. 2018, 13, 647–656. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Human Genes | Protein Short Names | SH2 Domains | Molecular Functions | Uniprot Entry |
|---|---|---|---|---|---|
| ABL | ABL1, ABL2 | Abl-1, Abl-2 | 1 | Enzyme (Tyrosine-kinase) | P00519, P42684 |
| CBL | CBL, CBLB, CBLC | Cbl, Cbl-b, Cbl-c | 1 | Enzyme (E3 ubiquitin-protein ligase) | P22681, Q13191, Q9ULV8 |
| CHN | CHN1, CHN2 | Chin-1, Chin-2 | 1 | Regulator (GTPase activity activator) | P15882, P52757 |
| CRK | CRK, CRKL | Crk, Crkl | 1 | Adaptor protein | P46108, P46109 |
| CSK | CSK, MATK | Csk; Ctk | 1 | Enzyme (Tyrosine-kinase) | P41240, P42679 |
| DAPP1 | DAPP1 | Dapp-1 | 1 | Adaptor protein | Q9UN19 |
| FPS | FPS (FES), FER | Fps (Fes), Fer | 1 | Enzyme (Tyrosine-kinase) | P07332, P16591 |
| FRK | FRK, BRK, SRMS | Frk, Brk-1, Srms | 1 | Enzyme (Tyrosine-kinase) | P42685, Q8WUW1, Q9H3Y6 |
| GRB | GRB2, GADS, GRAP | Grb2, Gads, Grap | 1 | Adaptor protein | P62993, O75791, Q13588 |
| GRB7 | GRB7, GRB10, GRB14 | Grb7, Grb10, Grb14 | 1 | Adaptor protein | Q14451, P0CE43, Q14449 |
| JAK | TYK2, JAK1, JAK2, JAK3 | Tyk2, Jak1, Jak2, Jak3 | 1, atypical | Enzyme (Tyrosine-kinase) | P29597, P23458, O60674, P52333 |
| NCK | NCK1, NCK2 | Nck1, Nck2 | 1 | Adaptor protein | P16333, O43639 |
| PI3KR | PIK3R1, PIK3R2, PIK3R3 | p85A, p85B, p55G | 2 | Enzyme (1-phosphatidylinositol-3-kinase) | P27986, O00459, Q92569 |
| PLCg | PLCG1, PLCG2 | Plcg1, Plcg2 | 2 | Enzyme (phosphatidylinositol phospholipase) | P19174, P16885, |
| PTPN | PTPN6, PTPN11 | Shp2 (Ptp) | 2 | Enzyme (Tyrosine phosphatase) | Q06124 |
| RASA1 | RASA1 | Gap (RasGap, p120RasGap) | 2 | Regulator (GTPase activity inhibitor) | P20936 |
| RIN | RIN1, RIN2, RIN3 | Rin1, Rin2, Rin3 | 1 | Regulator (Ras effector) | Q13671, Q8WYP3, Q8TB24 |
| SH2B | APS, LNK, SH2B | Aps, Lnk, Sh2b | 1 | Adaptor protein | O14492, Q9UQQ2, Q9NRF2 |
| SH2D1 | SH2D1A, SH2D1B | Sh2d1A (Sap), Sh2d1B | 1 | Adaptor protein | O60880, O14796 |
| SH2D2 | SH2D2A, HSH2D, SH2D7 | Sh2d2a, Hsh2d, Sh2d7 | 1 | Adaptor protein | Q9NP31, Q96JZ2, A6NKC9 |
| SH2D3 | SH2D3A, SH2D3C, BCAR3 | Sh2d3a, Sh2d3c, Bcar3 | 1 | Adaptor protein | Q9BRG2, Q9QZS8, Q9QZK2 |
| SH2D4 | SH2D4A, SH2D4B | Sh2d4a, Sh2d4b | 1 | Adaptor protein | Q9H788, Q5SQS7 |
| SH2D5 | SH2D5 | Sh2d5 | 1 | Adaptor protein | Q6ZV89 |
| SH3BP2 | SH3BP2 | Sh3bp2 (3bp-2) | 1 | Docking protein | P78314 |
| SHB | SHB, SHD, SHE, SHF | Shb, Shd, She, Shf | 1 | Docking protein | Q15464, Q96IW2, Q5VZ18, Q7M4L6 |
| SHC | SHC1, SHC2, SHC3, SHC4 | Shc1, Shc2, Shc3, Shc4 | 1 | Docking protein | P29353, P98077, Q92529, Q6S5L8 |
| SHIP | SHIP1, SHIP2 | Ship-1, Ship-2 | 1 | Enzyme (Phosphatidylinositol (PtdIns) phosphatase) | P97573, O15357 |
| SLAP | SLAP, SLAP2 | Slap-1, Slap-2 | 1 | Adaptor protein | Q13239, Q9H6Q3 |
| SLP76 | BLNK, LCP2, CLNK, SCIMP | Slnk, Blnk, Slp76, Mist, Slp65/Slp76 | 1 | Adaptor protein | Q8WV28, Q13094, Q7Z7G1, Q6UWF3 |
| SOCS | SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, SOCS7, CISH | Socs1, Socs2, Socs3, Socs4, Socs5, Socs6, Socs7, Cish | 1 | Enzyme (protein ubiquitination) | O15524, O35717, O14543, Q8WXH5, O75159, O14544, O14512, Q9NSE2 |
| SRC | SRC, FYN, LCK, FGR, YES, LYN, HCK, BLK | Src, Fyn, Lck, Fgr, Yes, Lyn, Hck, Blk | 1 | Enzyme (Tyrosine-kinase) | P12931, P06241, P06239, P09769, P07947, P07948, P08631, P51451 |
| STAP | BRDG1, BKS | Brdg1 (Stap-1), Bks (Stap-2) | 1 | Docking protein | Q9ULZ2, Q9UGK3 |
| STAT | STAT1, STAT2, STAT3, STAT4, STAT5, STAT5B, STAT6 | Stat1, Stat2, Stat3, Stat4, Stat5, Stat5b, Stat6 | 1 | Transcription factor | P42224, P52630, P40763, Q14765, P42229, P51692, P42226 |
| SPT6 | SUPT6H | Supt6h (Spt6) | 1 | Transcription factor | P42226 |
| SYK | ZAP70, SYK | Zap-70, p72-syk | 2 | Enzyme (Tyrosine-kinase) | P43403, P43405 |
| TEC | BMX, TEC, BTK, ITK, TXK | Bmx, Tec, Btk, Itk, Txk | 1 | Enzyme (Tyrosine-kinase) | P51813, P42680, Q06187, Q08881, P42681 |
| TNS | TNS1, TENS2, TNS3, TNS4 | Tensin-1, Tensin-2, Tensin-3, Tensin-4 | 1 | Cytoskeletal protein | Q63HR2, Q63HR2, Q68CZ2, Q8IZW8 |
| VAV | VAV1, VAV2, VAV3 | Vav1, Vav2, Vav3 | 1 | Regulator (Guanine-nucleotide releasing factor) | P15498, P52735, Q9UKW4 |
| Group | SH2 Domain | βD5 | Motif | Peptide Specificity Residues | |
|---|---|---|---|---|---|
| I | IA | SRC, FYN, FRK, LCK, HCK, BLK, ABL1, ABL2, ITK, BTK, TXK, ZAP70_N & C, SYK_N &C, NCK1, NCK2, BRK, YES, LYN | Y/F | pY-σ-σ-ψ | +3 |
| IB | SAP, EAT2, SHIP1, SHIP2, CRK, CRKL, RasGAP_C, CHK, BRK | Y/F | pY-x-x-ψ | +3 | |
| IC | GRB2, GADS, GRB7, GRB10, GRB14, ALX, FES, BMX, CSK, SH3BP2, HSH2D, TENC1, FER, SRM | Y/F | pY-x-N | +2 | |
| IE | α-Chimaerin, β-Chimaerin | Y/F | n/a | ? | |
| II | IIA | VAV, VAV2, PI3K-p85α_N & C, PLC-γ1_C, PLC-γ2_N & C, SHP-1_N & C, SHP2_N & C, SOCS2 & 3 & 4 & 6 | I/C/L/V/A/T | pY-ψ-x-ψ | +3 |
| IIB | APS, SHB2, SHC1, BLNK | L/I | pY-[E/D/x]-x-ψ | +3 | |
| IIC | BRDG1, BKS, CBL | Y/T | pY-x-x-x-ψ | +4 | |
| IID | SPT6 | V | pY-[P/I]-P-[K/R]-M | ? | |
| III | III | STAT1 & 3 & 5A | V/L | pY-x-x-Q | +3 |
| Inhibitors Classes | Active Moiety or Binding Site | Inhibitor Name | Targeted Protein | KD° or IC50# | References |
|---|---|---|---|---|---|
| Peptidomimetis | |||||
| pTyr-isosteres | Phosphonomethyl-Phe | Pmp | PI 3-kinase p85 | 0.01–0.02 μM° | [120] |
| Hydroxy benzyl phosphinate | Grb2 | 0.53 μM ° | [121,122] | ||
| 4-phosphonodifluoromethylcinnamate | Stat3 | 0.16 μM # | [124,125] | ||
| Sulfonamides | C188-9 | Stat3 | [133,134] | ||
| (TTI-101) | |||||
| Photo-switchable linkers | 4-aminomethyl phenylazobenzoic acid | Zap-70 | 0.065–0.8 μM ° | [126,127,128,129] | |
| Bicyclic peptides | Carboxylate group | BC1 | Grb2 | 0.35 μM # | [130,131,132] |
| Allosteric inhibitors | |||||
| Pyrazines | N-SH2/C-SH2/PTP interface | SHP099 * | Shp2 | 0.071 μM # | [137,138] |
| N-SH2/C-SH2/PTP interface | TNO155 * | Shp2 | 0.011 μM # | [139,140] | |
| C-SH2/PTP interface | LY6 | Shp2 | 9 μM # | [141] | |
| Triazolo-quinazolinones | “latch” N-SH2/PTP binding site | SHP244 | Shp2 | 60 μM # | [142] |
| “latch” N-SH2/PTP binding site | SHP504 | Shp2 | 21 μM # | [142] | |
| “latch” N-SH2/PTP binding site | SHP844 | Shp2 | 18.9 μM # | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diop, A.; Santorelli, D.; Malagrinò, F.; Nardella, C.; Pennacchietti, V.; Pagano, L.; Marcocci, L.; Pietrangeli, P.; Gianni, S.; Toto, A. SH2 Domains: Folding, Binding and Therapeutical Approaches. Int. J. Mol. Sci. 2022, 23, 15944. https://doi.org/10.3390/ijms232415944
Diop A, Santorelli D, Malagrinò F, Nardella C, Pennacchietti V, Pagano L, Marcocci L, Pietrangeli P, Gianni S, Toto A. SH2 Domains: Folding, Binding and Therapeutical Approaches. International Journal of Molecular Sciences. 2022; 23(24):15944. https://doi.org/10.3390/ijms232415944
Chicago/Turabian StyleDiop, Awa, Daniele Santorelli, Francesca Malagrinò, Caterina Nardella, Valeria Pennacchietti, Livia Pagano, Lucia Marcocci, Paola Pietrangeli, Stefano Gianni, and Angelo Toto. 2022. "SH2 Domains: Folding, Binding and Therapeutical Approaches" International Journal of Molecular Sciences 23, no. 24: 15944. https://doi.org/10.3390/ijms232415944