
Substrate Specificity of GSDA Revealed by Cocrystal Structures and Binding Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
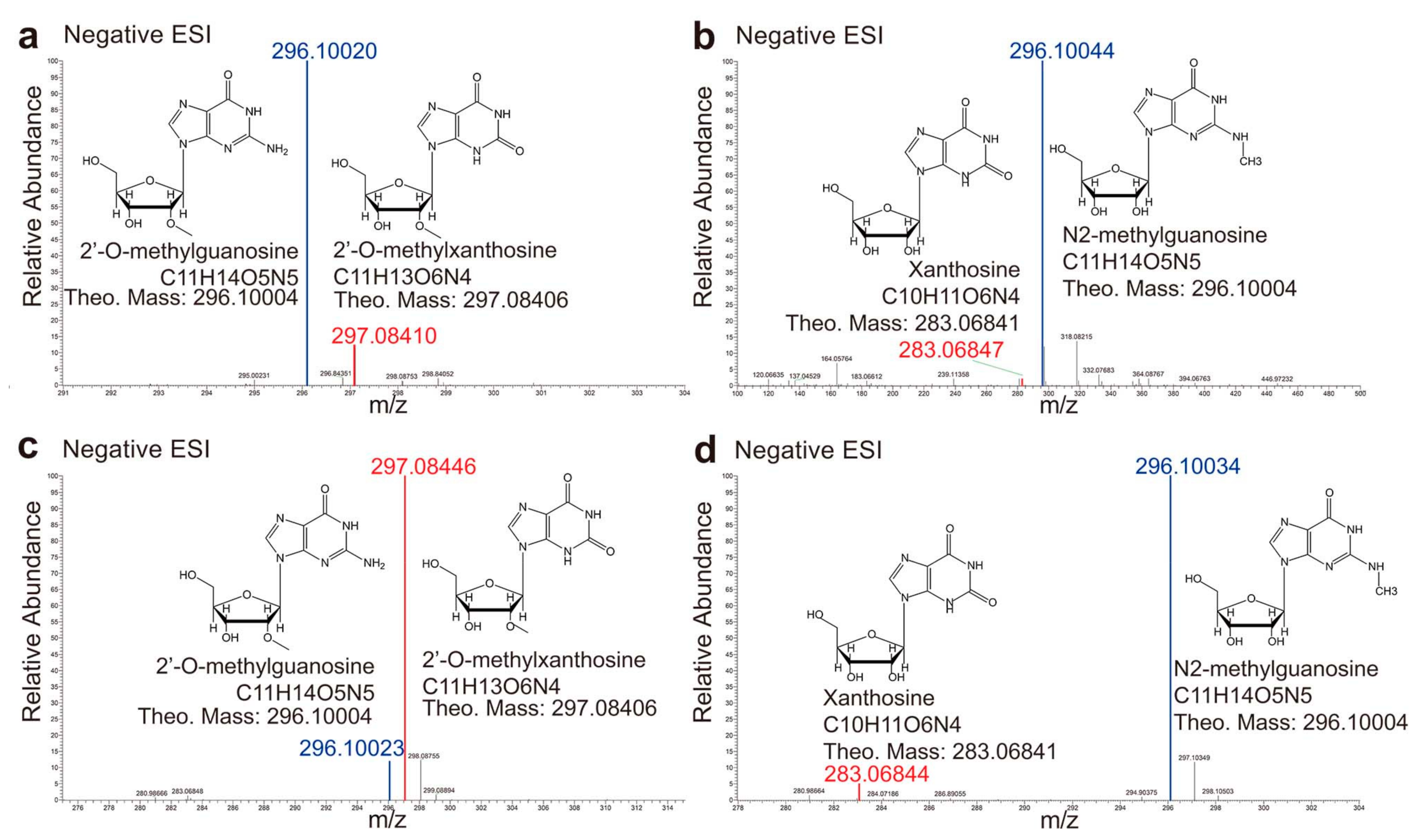
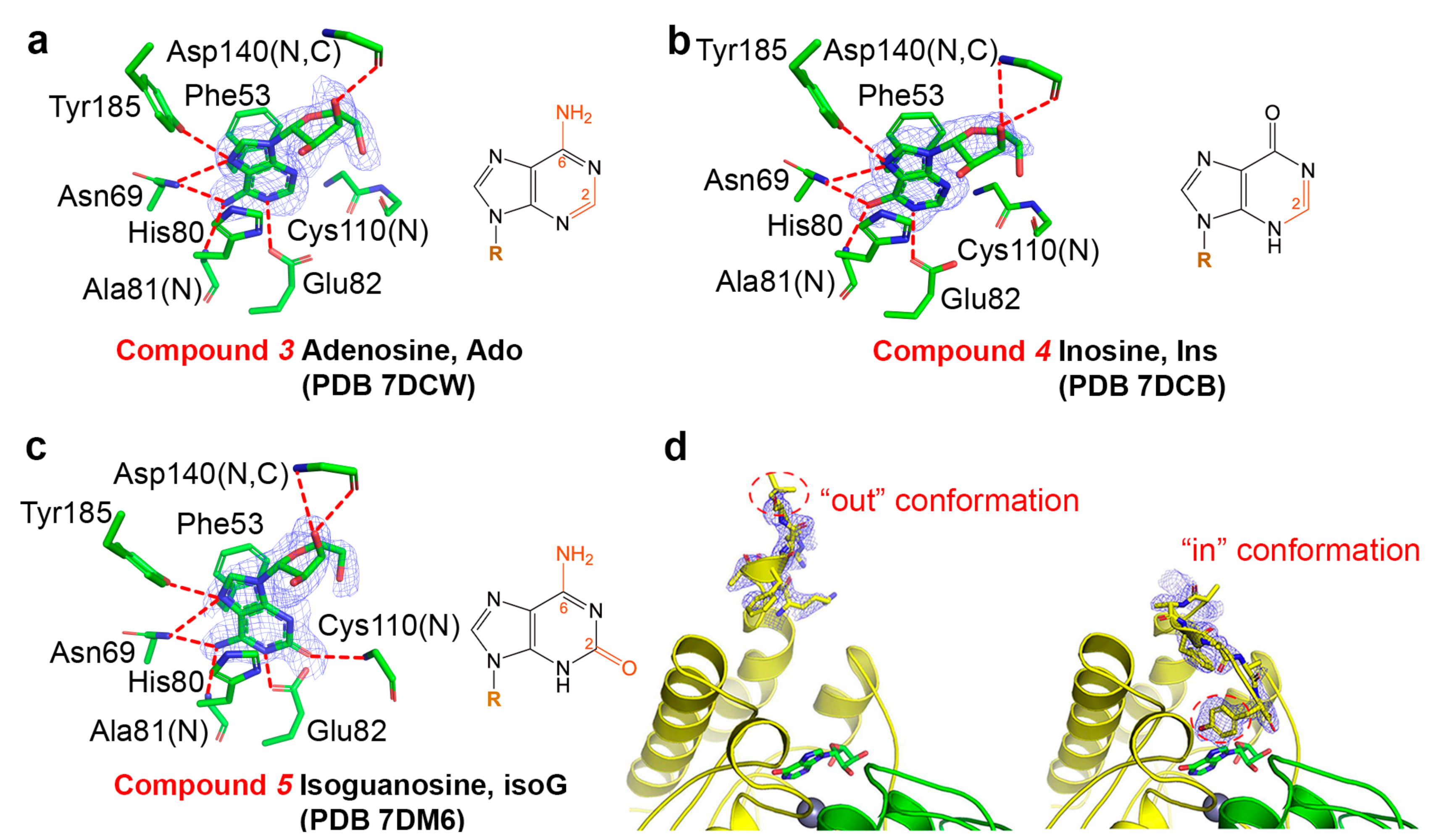
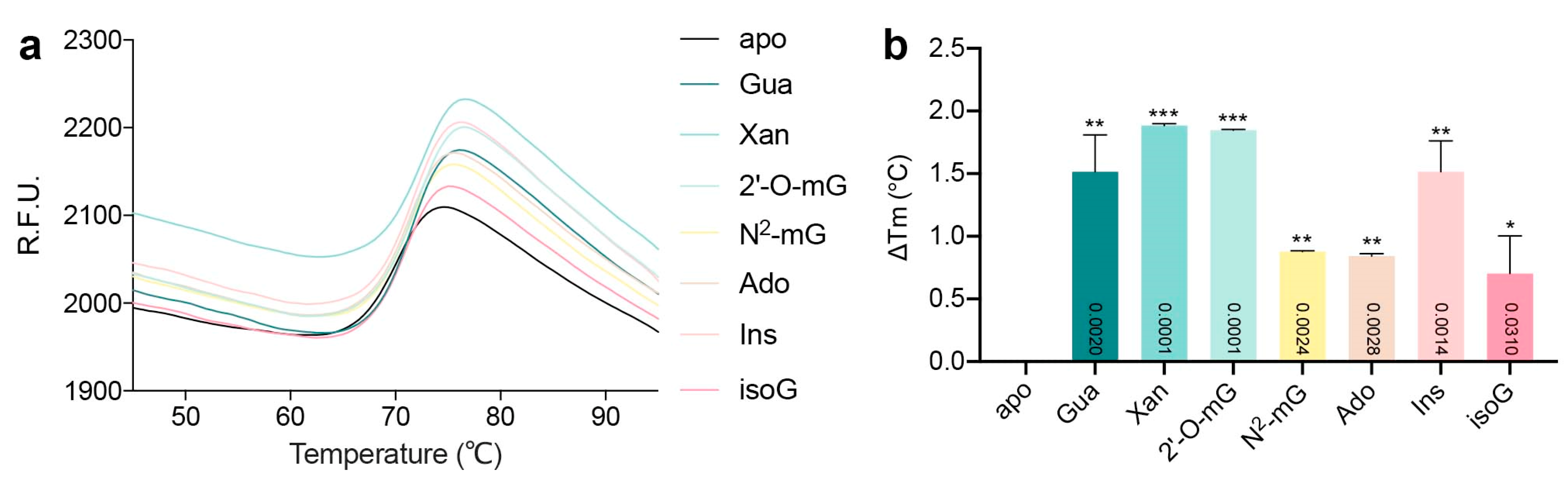
2.1. Binding Modes of Different Substrates
2.2. Binding Modes of Different Inhibitors
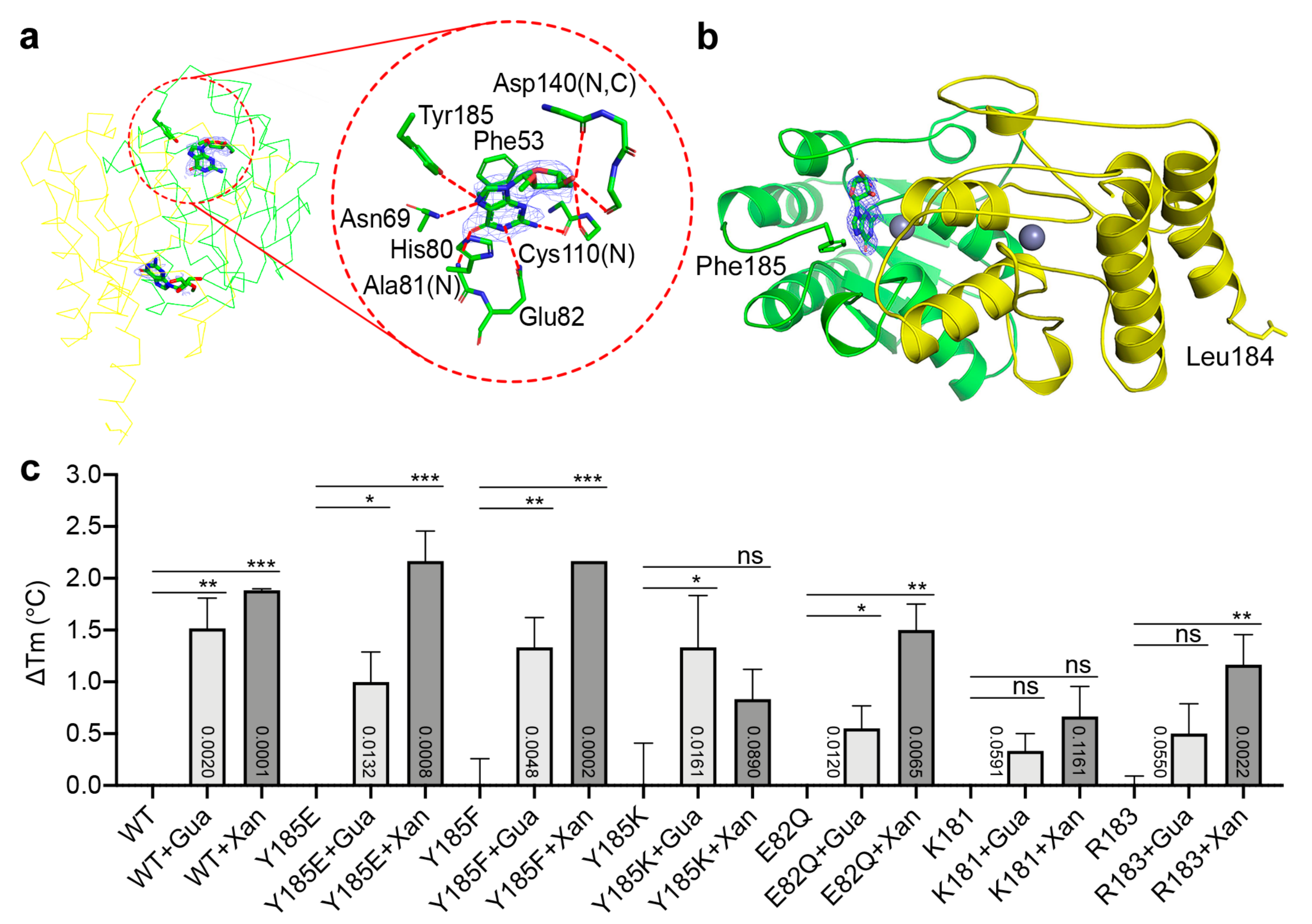
2.3. Binding Modes of Different Substrates by the Catalytically Impaired Mutants
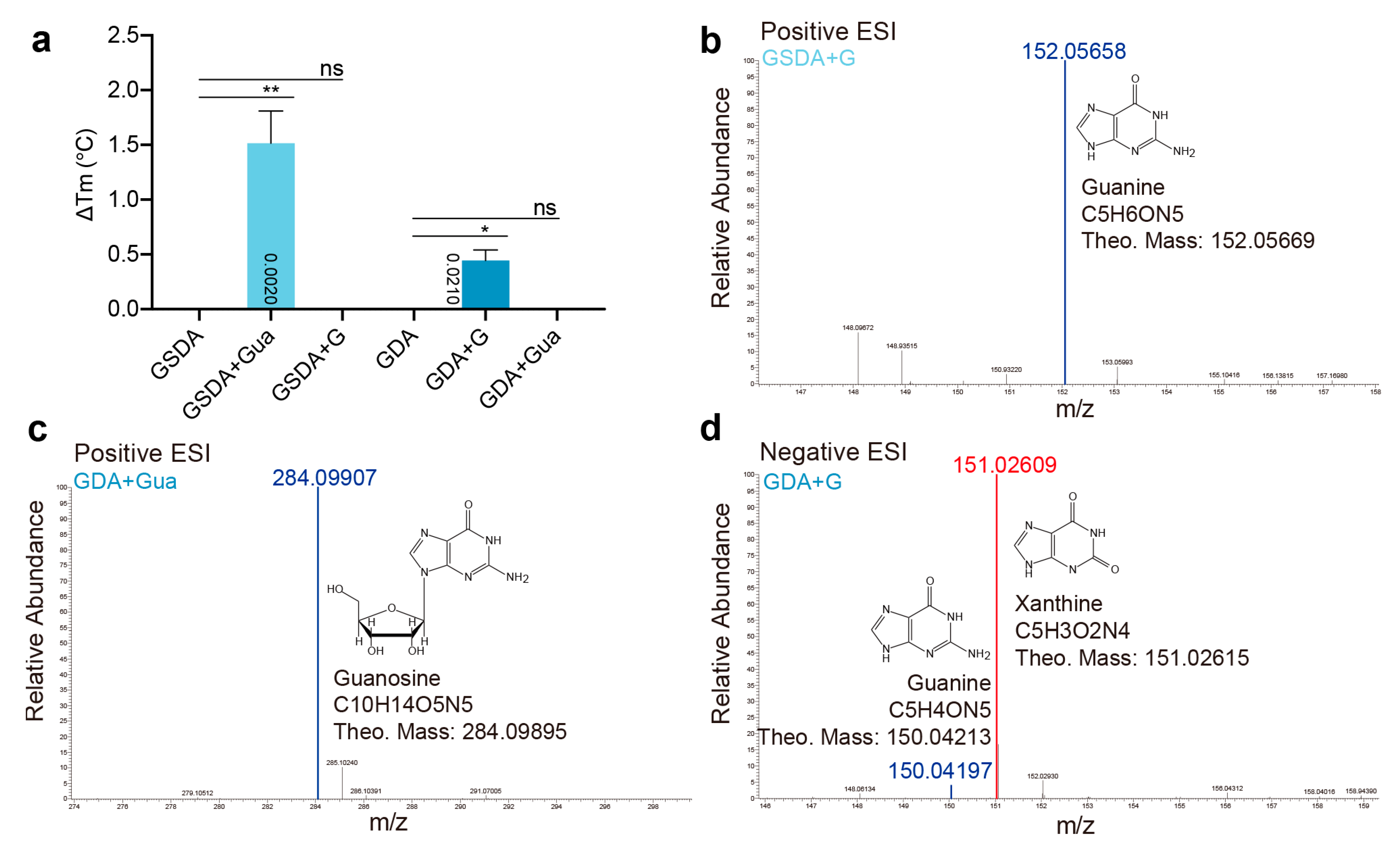
2.4. Cross Activities between GSDA and GDA
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression and Protein Purification
4.2. Crystallization and Structure Determination
4.3. Thermal Shift Analysis
4.4. Mass Spectrometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Werner, A.K.; Witte, C.P. The biochemistry of nitrogen mobilization: Purine ring catabolism. Trends Plant Sci. 2011, 6, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Stasolla, C.; Katahira, R.; Thorpe, T.A.; Ashihara, H. Purine and pyrimidine nucleotide metabolism in higher plants. J. Plant Physiol. 2003, 160, 1271–1295. [Google Scholar] [CrossRef] [PubMed]
- Zrenner, R.; Stitt, M.; Sonnewald, U.; Boldt, R. Pyrimidine and purine biosynthesis and degradation in plants. Annu. Rev. Plant Biol. 2006, 57, 805–836. [Google Scholar] [CrossRef] [PubMed]
- Ashihara, H.; Takasawa, Y.; Suzuki, T. Metabolic fate of guanosine in higher plants. Physiol. Plant. 2010, 100, 909–916. [Google Scholar] [CrossRef]
- Dahncke, K.; Witte, C.P. Plant purine nucleoside catabolism employs a guanosine deaminase required for the generation of xanthosine in Arabidopsis. Plant Cell 2013, 25, 4101–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, P.; Bested, S.M.; Andersen, K.A.K.; Saxild, H.H. Bacillus subtilis guanine deaminase is encoded by the yknA gene and is induced during growth with purines as the nitrogen source. Microbiology 2000, 146, 3061–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straube, H.; Niehaus, M.; Zwittian, S.; Witte, C.P.; Herde, M. Enhanced nucleotide analysis enables the quantification of deoxynucleotides in plants and algae revealing connections between nucleoside and deoxynucleoside metabolism. Plant Cell 2021, 33, 270–289. [Google Scholar] [CrossRef] [PubMed]
- Baccolini, C.; Witte, C.P. AMP and GMP catabolism in Arabidopsis converge on xanthosine, which is degraded by a nucleoside hydrolase heterocomplex. Plant Cell 2019, 31, 734–751. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, R.Y.; Zhu, A.; Eubel, H.; Dahncke, K.; Witte, C.P. The ribokinases of Arabidopsis thaliana and Saccharomyces cerevisiae are required for ribose recycling from nucleotide catabolism, which in plants is not essential to survive prolonged dark stress. New Phytol. 2018, 217, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Zeng, H.; Li, H.; Xiao, N.; Tang, J.; Gao, S.; Zhang, J.; Xie, W. The C-terminal loop of Arabidopsis thaliana guanosine deaminase is essential to catalysis. Chem. Commun. 2021, 57, 9748–9751. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Gaded, V.; Bitra, A.; Anand, R. Structure guided mutagenesis reveals the substrate determinants of guanine deaminase. J. Struct. Biol. 2021, 213, 107747. [Google Scholar] [CrossRef] [PubMed]
- Alunni, S.; Orrù, M.; Ottavi, L. A study on the inhibition of adenosine deaminase. J. Enzym. Inhib. Med. Chem. 2008, 23, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Xie, W. Alternative conformation induced by substrate binding for Arabidopsis thaliana N6-methyl-AMP deaminase. Nucleic Acids Res. 2019, 47, 3233–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, E.; Bao, H.; Mosley, R.T.; Du, J.; Sofia, M.J.; Furman, P.A. Adenosine deaminase-like protein 1 (ADAL1): Characterization and substrate specificity in the hydrolysis of N6- or O6-substituted purine or 2-aminopurine nucleoside monophosphates. J. Med. Chem. 2011, 54, 5902–5914. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.A.; Galellis, J.R.; McDermid, H.E. Phylogenetic analysis reveals a novel protein family closely related to adenosine deaminase. J. Mol. Evol. 2005, 61, 776–794. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M. Analyzing the topology of active sites: On the prediction of pockets and subpockets. J. Chem. Inf. Model 2010, 50, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model 2012, 52, 360–372. [Google Scholar] [CrossRef]
- Shek, R.; Hilaire, T.; Sim, J.; French, J.B. Structural determinants for substrate selectivity in guanine deaminase enzymes of the amidohydrolase superfamily. Biochemistry 2019, 58, 3280–3292. [Google Scholar] [CrossRef]
- Mccoy, A.J.; Grosse-Kunstleve, A.R.W.; Adams, B.P.D.; Winn, B.M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Q.; Zhang, J.; Zeng, H.; Tang, J.; Xiao, N.; Gao, S.; Li, H.; Xie, W. Substrate Specificity of GSDA Revealed by Cocrystal Structures and Binding Studies. Int. J. Mol. Sci. 2022, 23, 14976. https://doi.org/10.3390/ijms232314976
Jia Q, Zhang J, Zeng H, Tang J, Xiao N, Gao S, Li H, Xie W. Substrate Specificity of GSDA Revealed by Cocrystal Structures and Binding Studies. International Journal of Molecular Sciences. 2022; 23(23):14976. https://doi.org/10.3390/ijms232314976
Chicago/Turabian StyleJia, Qian, Jinbing Zhang, Hui Zeng, Jing Tang, Nan Xiao, Shangfang Gao, Huanxi Li, and Wei Xie. 2022. "Substrate Specificity of GSDA Revealed by Cocrystal Structures and Binding Studies" International Journal of Molecular Sciences 23, no. 23: 14976. https://doi.org/10.3390/ijms232314976