
Fenbendazole Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice via Suppression of Fibroblast-to-Myofibroblast Differentiation
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
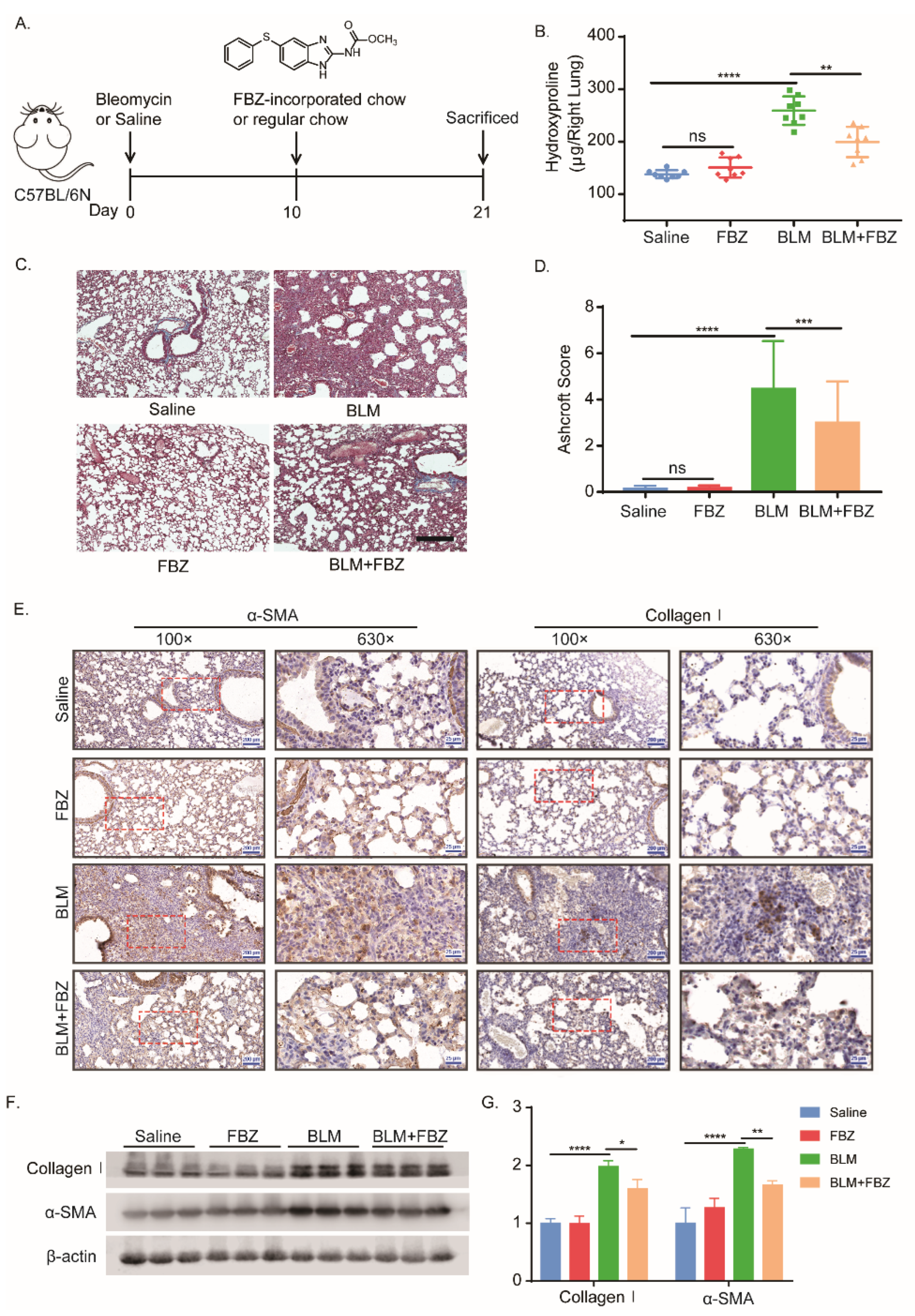
2.1. Fenbendazole Attenuated Bleomycin-Induced Lung Fibrosis in Mice
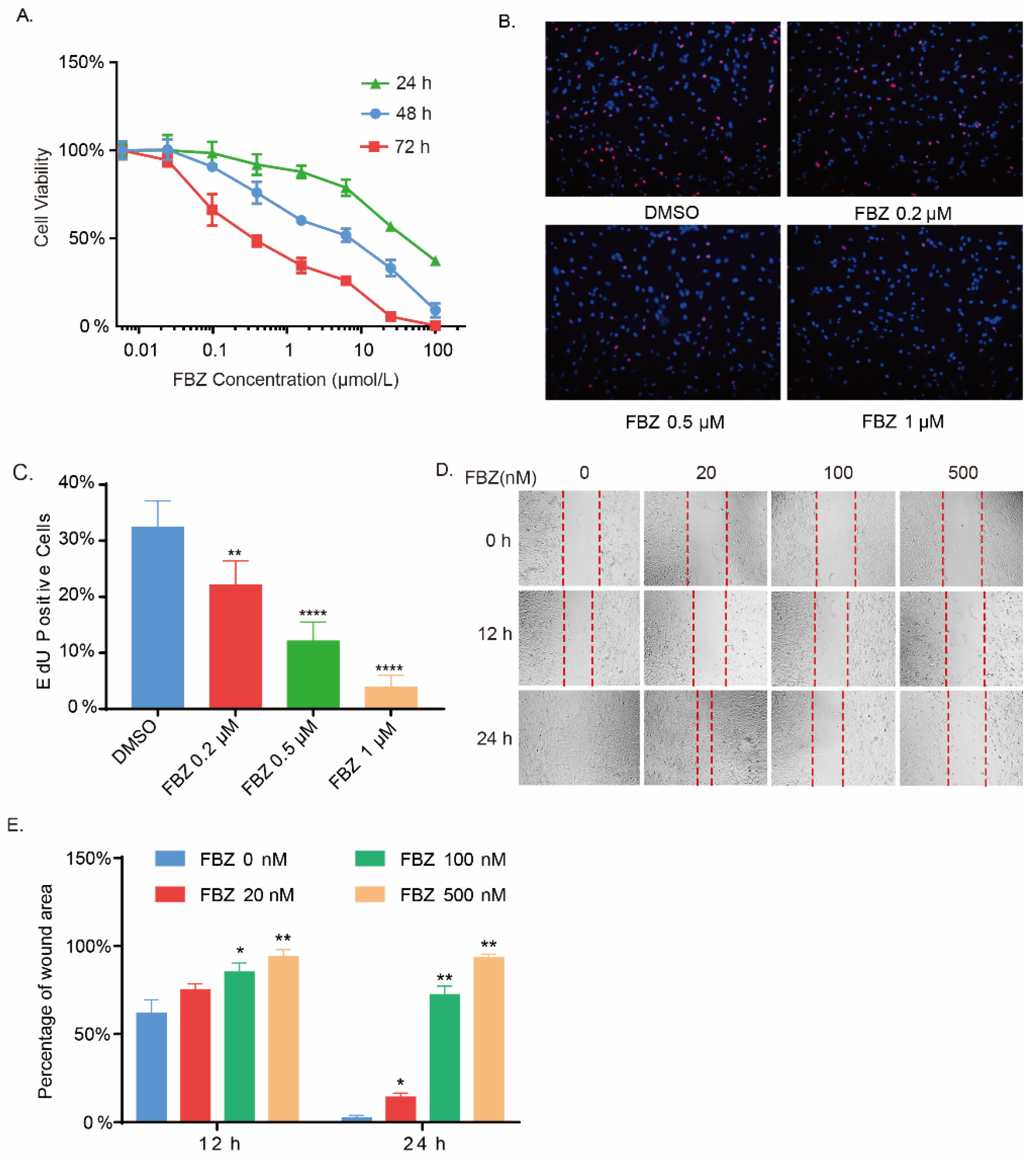
2.2. Fenbendazole Inhibited Fibroblasts Growth and Mobility
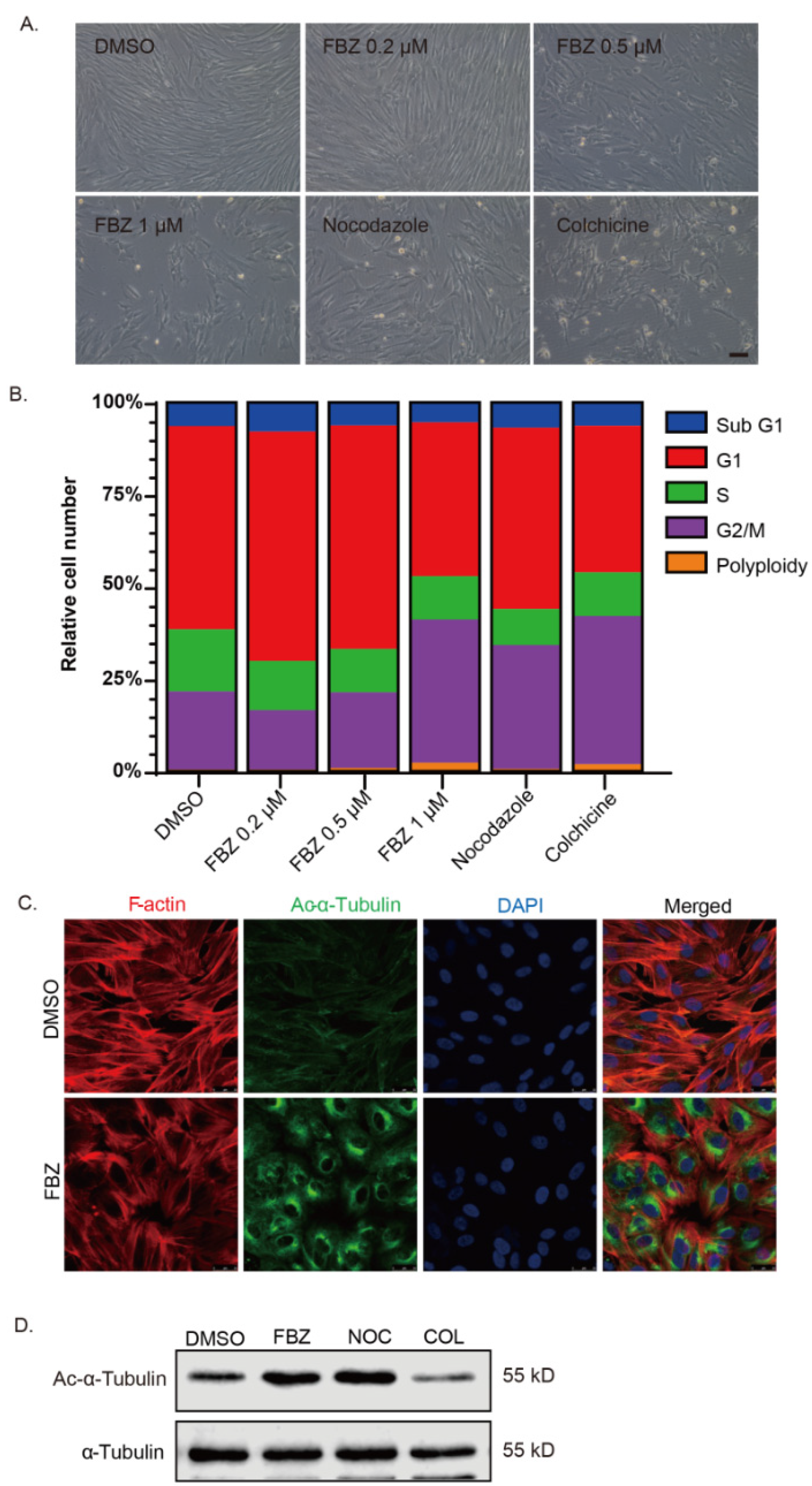
2.3. Fenbendazole Disrupted Microtubule Cytoskeleton and Caused Cell Cycle Arrest
2.4. Fenbendazole Inhibited Glycolytic Metabolism in Fibroblasts
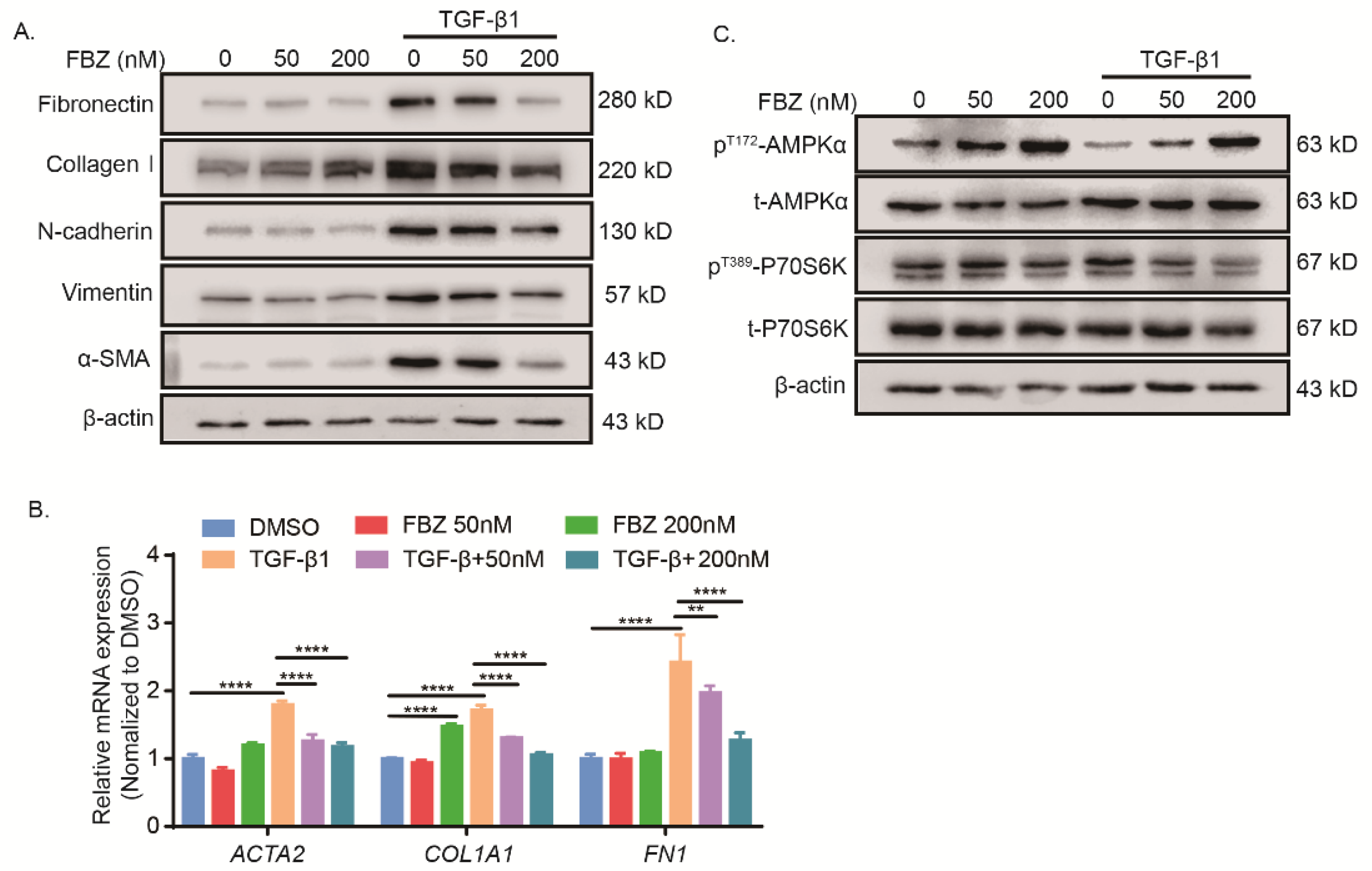
2.5. Fenbendazole Inhibited TGF-β1-Induced Fibroblast-to-Myofibroblast Differentiation and Collagen Formation through AMPK Activation and mTOR Suppression
3. Discussion
4. Material and Methods
4.1. Reagents and Antibodies
4.2. Mice and the Mouse Model of Bleomycin-Induced Lung Fibrosis
4.3. Lung Histological Analysis and Immunohistochemical Staining
4.4. Hydroxyproline
4.5. Cell Culture
4.6. CCK-8 and EdU Incorporation Assay
4.7. Determination of Glucose Concentration in Culture Medium
4.8. Cell Cycle Analysis
4.9. Immunofluorescence Staining
4.10. Glycolytic Function Assay
4.11. Western Blot Analysis
4.12. Quantitative RT-PCR Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lederer, D.J.; Longo, D.L.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [PubMed]
- Misharin, A.V.; Budinger, G.R.S. Targeting the Myofibroblast in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 198, 834–835. [Google Scholar]
- Cui, H.; Xie, N.; Banerjee, S.; Dey, T.; Liu, R.-M.; Antony, V.B.; Sanders, Y.Y.; Adams, T.S.; Gomez, J.L.; Thannickal, V.J.; et al. CD38 Mediates Lung Fibrosis by Promoting Alveolar Epithelial Cell Aging. Am. J. Respir. Crit. Care Med. 2022, 206, 459–475. [Google Scholar] [PubMed]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 515–546. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar]
- Ruaro, B.; Pozzan, R.; Confalonieri, P.; Tavano, S.; Hughes, M.; Matucci Cerinic, M.; Baratella, E.; Zanatta, E.; Lerda, S.; Geri, P.; et al. Gastroesophageal Reflux Disease in Idiopathic Pulmonary Fibrosis: Viewer or Actor? To Treat or Not to Treat? Pharmaceuticals 2022, 15, 1033. [Google Scholar] [CrossRef]
- Borie, R.; Cardwell, J.; Konigsberg, I.R.; Moore, C.M.; Zhang, W.; Sasse, S.K.; Gally, F.; Dobrinskikh, E.; Walts, A.; Powers, J.; et al. Colocalization of Gene Expression and DNA Methylation with Genetic Risk Variants Supports Functional Roles of MUC5B and DSP in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022. [Google Scholar] [CrossRef]
- Somogyi, V.; Chaudhuri, N.; Torrisi, S.E.; Kahn, N.; Müller, V.; Kreuter, M. The therapy of idiopathic pulmonary fibrosis: What is next? Eur. Respir. Rev. 2019, 28, 190021. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.-F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2019, 19, 57–75. [Google Scholar] [PubMed]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [PubMed] [Green Version]
- Hamanaka, R.B.; Mutlu, G.M. Metabolic requirements of pulmonary fibrosis: Role of fibroblast metabolism. FEBS J. 2021, 288, 6331–6352. [Google Scholar] [PubMed]
- Hamanaka, R.B.; O’Leary, E.M.; Witt, L.J.; Tian, Y.; Gökalp, G.A.; Meliton, A.Y.; Dulin, N.O.; Mutlu, G.M. Glutamine Metabolism Is Required for Collagen Protein Synthesis in Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 597–606. [Google Scholar]
- Chang, L.; Zhu, L. Dewormer drug fenbendazole has antiviral effects on BoHV-1 productive infection in cell cultures. J. Vet. Sci. 2020, 21, e72. [Google Scholar] [CrossRef]
- Duan, Q.W.; Liu, Y.F.; Rockwell, S. Fenbendazole as a Potential Anticancer Drug. Anticancer Res. 2013, 33, 355–362. [Google Scholar]
- Cray, C.; Altman, N.H. An Update on the Biologic Effects of Fenbendazole. Comp. Med. 2022, 72, 215–219. [Google Scholar]
- Park, H.; Lim, W.; You, S.; Song, G. Fenbendazole induces apoptosis of porcine uterine luminal epithelial and trophoblast cells during early pregnancy. Sci. Total Environ. 2019, 681, 28–38. [Google Scholar] [CrossRef]
- Dogra, N.; Kumar, A.; Mukhopadhyay, T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci. Rep. 2018, 8, 11926. [Google Scholar]
- Jasra, N.; Sanyal, S.N.; Khera, S. Effect of thiabendazole and fenbendazole on glucose uptake and carbohydrate metabolism in trichuris globulosa. Vet. Parasitol. 1990, 35, 201–209. [Google Scholar] [CrossRef]
- Sultana, T.; Jan, U.; Lee, H.; Lee, H.; Lee, J.I. Exceptional Repositioning of Dog Dewormer: Fenbendazole Fever. Curr. Issues Mol. Biol. 2022, 44, 4977–4986. [Google Scholar] [PubMed]
- Yu, C.G.; Singh, R.; Crowdus, C.; Raza, K.; Kincer, J.; Geddes, J.W. Fenbendazole improves pathological and functional recovery following traumatic spinal cord injury. Neuroscience 2014, 256, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, H.; Gigant, B.; Yu, Y.; Wu, Y.; Chen, X.; Lai, Q.; Yang, Z.; Chen, Q.; Yang, J. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283, 102–111. [Google Scholar]
- You, E.; Ko, P.; Jeong, J.; Keum, S.; Kim, J.-W.; Seo, Y.-J.; Song, W.K.; Rhee, S. Dynein-mediated nuclear translocation of yes-associated protein through microtubule acetylation controls fibroblast activation. Cell. Mol. Life Sci. 2020, 77, 4143–4161. [Google Scholar]
- Sandbo, N.; Ngam, C.; Torr, E.; Kregel, S.; Kach, J.; Dulin, N. Control of myofibroblast differentiation by microtubule dynamics through a regulated localization of mDia2. J. Biol. Chem. 2013, 288, 15466–15473. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Yang, J.-J.; Shi, K.-H.; Li, J. Epigenetic factors MeCP2 and HDAC6 control α-tubulin acetylation in cardiac fibroblast proliferation and fibrosis. Inflamm. Res. 2016, 65, 415–426. [Google Scholar] [CrossRef]
- Xiaojun, W.; Yan, L.; Hong, X.; Xianghong, Z.; Shifeng, L.; Dingjie, X.; Xuemin, G.; Lijuan, Z.; Bonan, Z.; Zhongqiu, W.; et al. Acetylated α-Tubulin Regulated by N-Acetyl-Seryl-Aspartyl-Lysyl-Proline(Ac-SDKP) Exerts the Anti-fibrotic Effect in Rat Lung Fibrosis Induced by Silica. Sci. Rep. 2016, 6, 32257. [Google Scholar]
- Para, R.; Romero, F.; George, G.; Summer, R. Metabolic Reprogramming as a Driver of Fibroblast Activation in Pulmonary Fibrosis. Am. J. Med. Sci. 2019, 357, 394–398. [Google Scholar] [CrossRef]
- Cho, S.J.; Moon, J.-S.; Lee, C.-M.; Choi, A.M.K.; Stout-Delgado, H.W. Glucose Transporter 1–Dependent Glycolysis Is Increased during Aging-Related Lung Fibrosis, and Phloretin Inhibits Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 521–531. [Google Scholar] [CrossRef]
- Cho, S.J.; Moon, J.-S.; Nikahira, K.; Yun, H.S.; Harris, R.; Hong, K.S.; Huang, H.; Choi, A.M.K.; Stout-Delgado, H. GLUT1-dependent glycolysis regulates exacerbation of fibrosis via AIM2 inflammasome activation. Thorax 2020, 75, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Sun, H.; Zhang, B.; Liu, S.; Deng, S.; Weng, Z.; Zuo, B.; Yang, J.; He, Y. 18F-FDG PET imaging for monitoring the early anti-tumor effect of albendazole on triple-negative breast cancer. Breast Cancer 2019, 27, 372–380. [Google Scholar] [PubMed]
- Son, D.-S.; Lee, E.-S.; Adunyah, S.E. The Antitumor Potentials of Benzimidazole Anthelmintics as Repurposing Drugs. Immune Netw. 2020, 20, e29. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Pan, J.; Ou, F.; Wang, W.; Hu, H.; Chen, L.; Zeng, S.; Zeng, K.; Yu, L. Fenbendazole and its synthetic analog interfere with HeLa cells’ proliferation and energy metabolism via inducing oxidative stress and modulating MEK3/6-p38-MAPK pathway. Chem.-Biol. Interact. 2022, 361, 109983. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef]
- Tang, C.-J.; Xu, J.; Ye, H.-Y.; Wang, X.-B. Metformin prevents PFKFB3-related aerobic glycolysis from enhancing collagen synthesis in lung fibroblasts by regulating AMPK/mTOR pathway. Exp. Ther. Med. 2021, 21, 581. [Google Scholar] [PubMed]
- González, A.; Hall, M.N.; Lin, S.-C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Romero, Y.; Bueno, M.; Ramirez, R.; Álvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef]
- Chung, E.J.; Sowers, A.; Thetford, A.; McKay-Corkum, G.; Chung, S.I.; Mitchell, J.B.; Citrin, D.E. Mammalian Target of Rapamycin Inhibition With Rapamycin Mitigates Radiation-Induced Pulmonary Fibrosis in a Murine Model. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 857–866. [Google Scholar]
- Lawrence, J.; Nho, R. The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 778. [Google Scholar] [CrossRef]
- Liu, M.-W.; Su, M.-X.; Tang, D.-Y.; Hao, L.; Xun, X.-H.; Huang, Y.-Q. Ligustrazin increases lung cell autophagy and ameliorates paraquat-induced pulmonary fibrosis by inhibiting PI3K/Akt/mTOR and hedgehog signalling via increasing miR-193a expression. BMC Pulm. Med. 2019, 19, 35. [Google Scholar] [CrossRef]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal v-ATPase-Ragulator Complex Is a Common Activator for AMPK and mTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [PubMed] [Green Version]
- Li, M.; Zhang, C.-S.; Feng, J.-W.; Wei, X.; Zhang, C.; Xie, C.; Wu, Y.; Hawley, S.A.; Atrih, A.; Lamont, D.J.; et al. Aldolase is a sensor for both low and high glucose, linking to AMPK and mTORC1. Cell Res. 2020, 31, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; de Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2017, 24, 39–49. [Google Scholar] [PubMed]
- Hübner, R.-H.; Gitter, W.; Eddine El Mokhtari, N.; Mathiak, M.; Both, M.; Bolte, H.; Freitag-Wolf, S.; Bewig, B. Standardized quantification of pulmonary fibrosis in histological samples. BioTechniques 2008, 44, 507–517. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Xu, K.; Wang, N.; Ding, L.; Zhao, W.; Wan, R.; Zhao, W.; Guo, X.; Pan, X.; Yang, J.; et al. Fenbendazole Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice via Suppression of Fibroblast-to-Myofibroblast Differentiation. Int. J. Mol. Sci. 2022, 23, 14088. https://doi.org/10.3390/ijms232214088
Wang L, Xu K, Wang N, Ding L, Zhao W, Wan R, Zhao W, Guo X, Pan X, Yang J, et al. Fenbendazole Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice via Suppression of Fibroblast-to-Myofibroblast Differentiation. International Journal of Molecular Sciences. 2022; 23(22):14088. https://doi.org/10.3390/ijms232214088
Chicago/Turabian StyleWang, Lan, Kai Xu, Ningdan Wang, Linke Ding, Wenyu Zhao, Ruyan Wan, Weiming Zhao, Xiaoshu Guo, Xin Pan, Juntang Yang, and et al. 2022. "Fenbendazole Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice via Suppression of Fibroblast-to-Myofibroblast Differentiation" International Journal of Molecular Sciences 23, no. 22: 14088. https://doi.org/10.3390/ijms232214088