
Residual Interactions of LL-37 with POPC and POPE:POPG Bilayer Model Studied by All-Atom Molecular Dynamics Simulation
 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Results
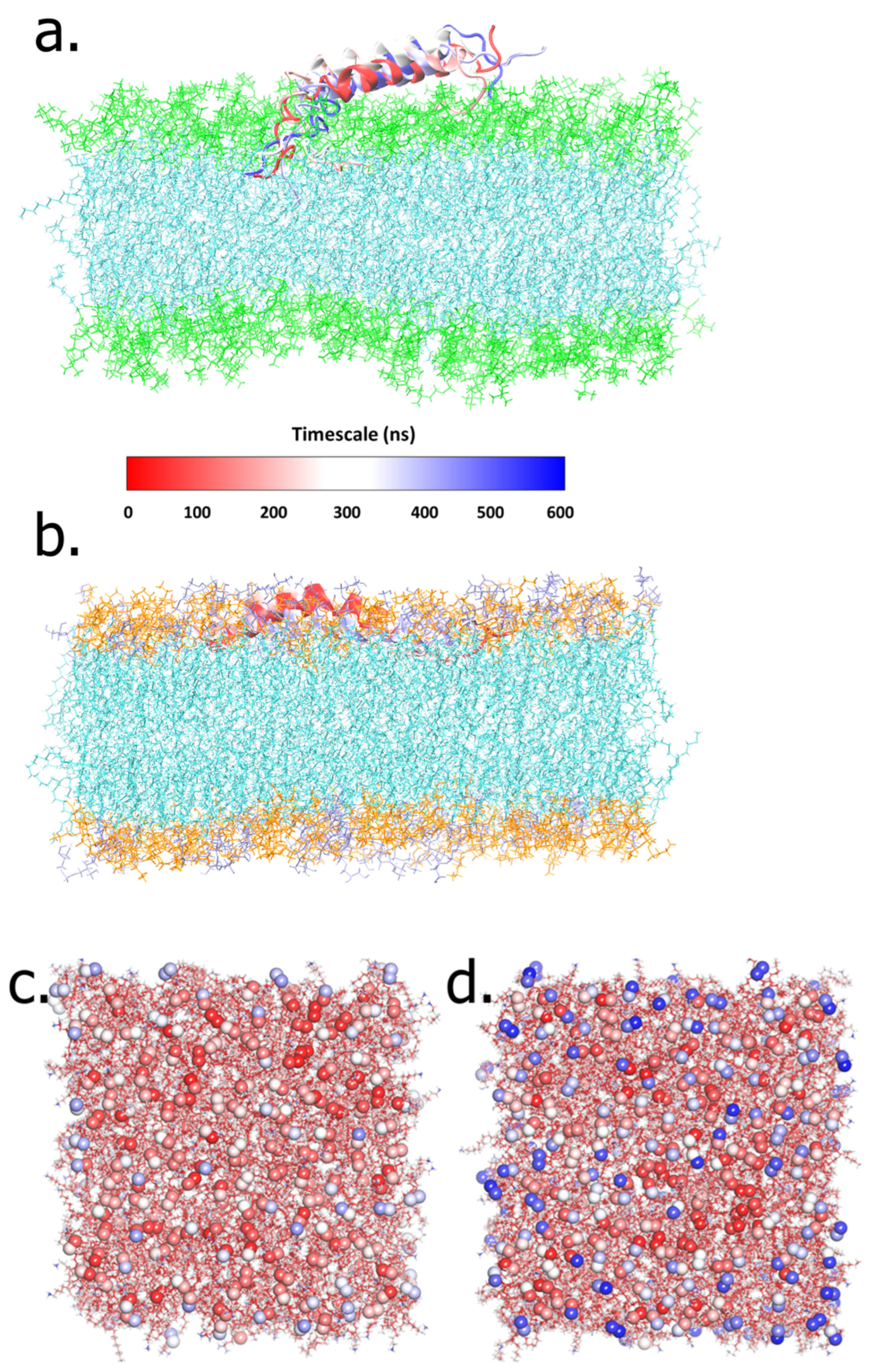
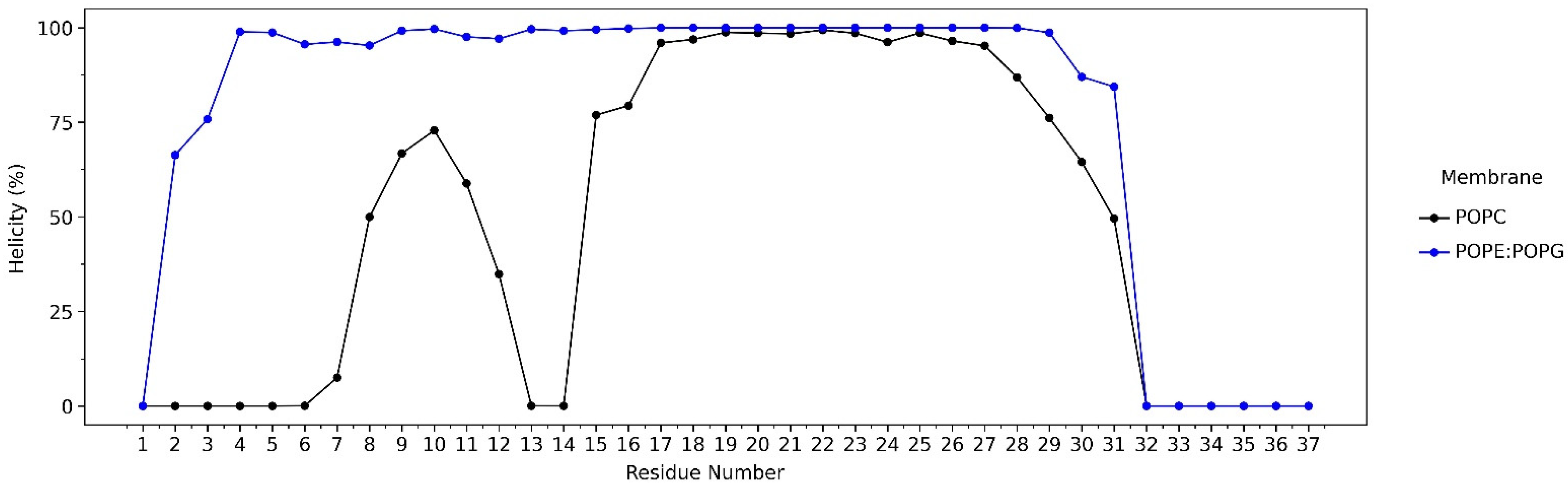
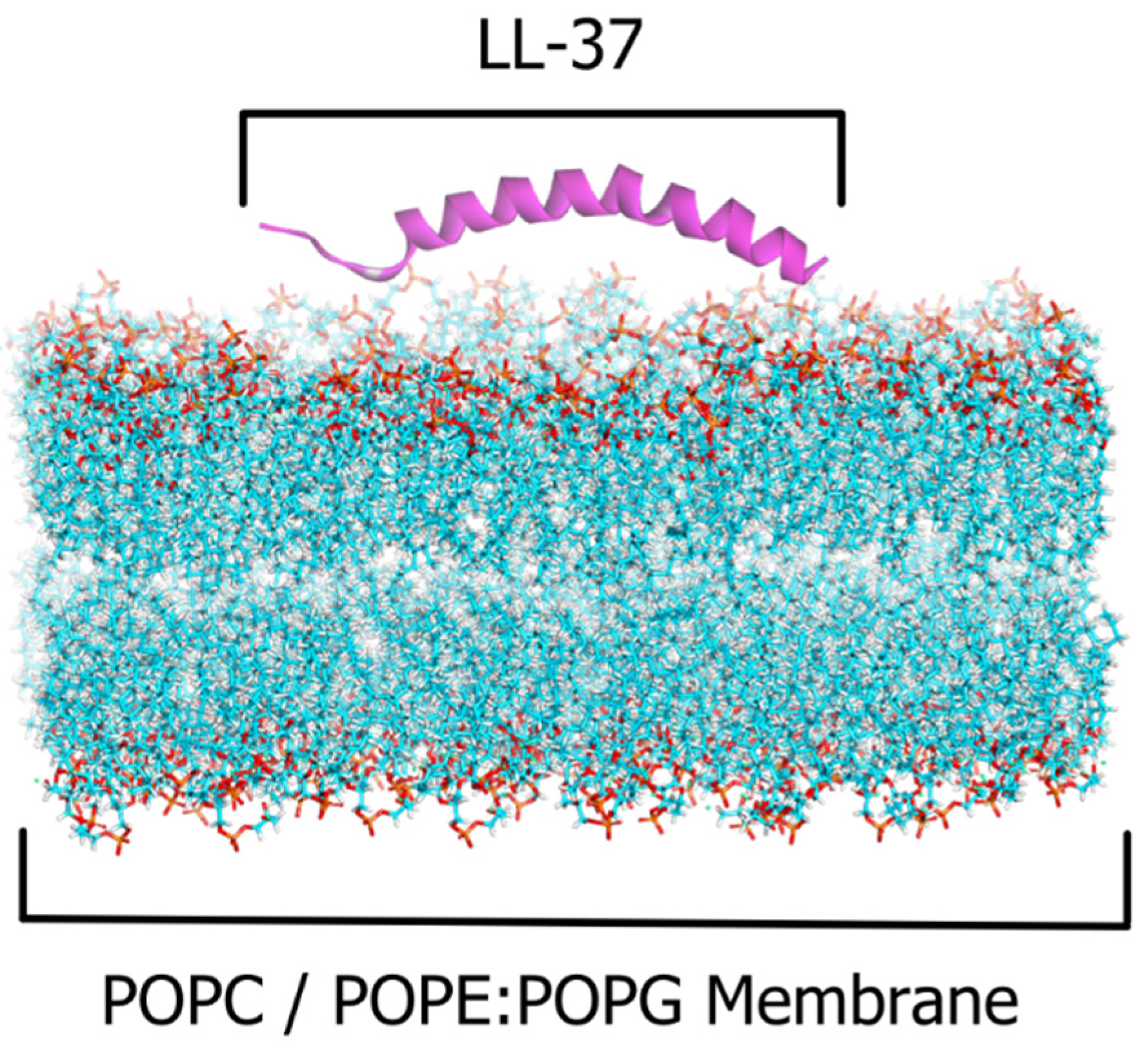
2.1. The LL-37–POPC/POPE:POPG Membrane Systems
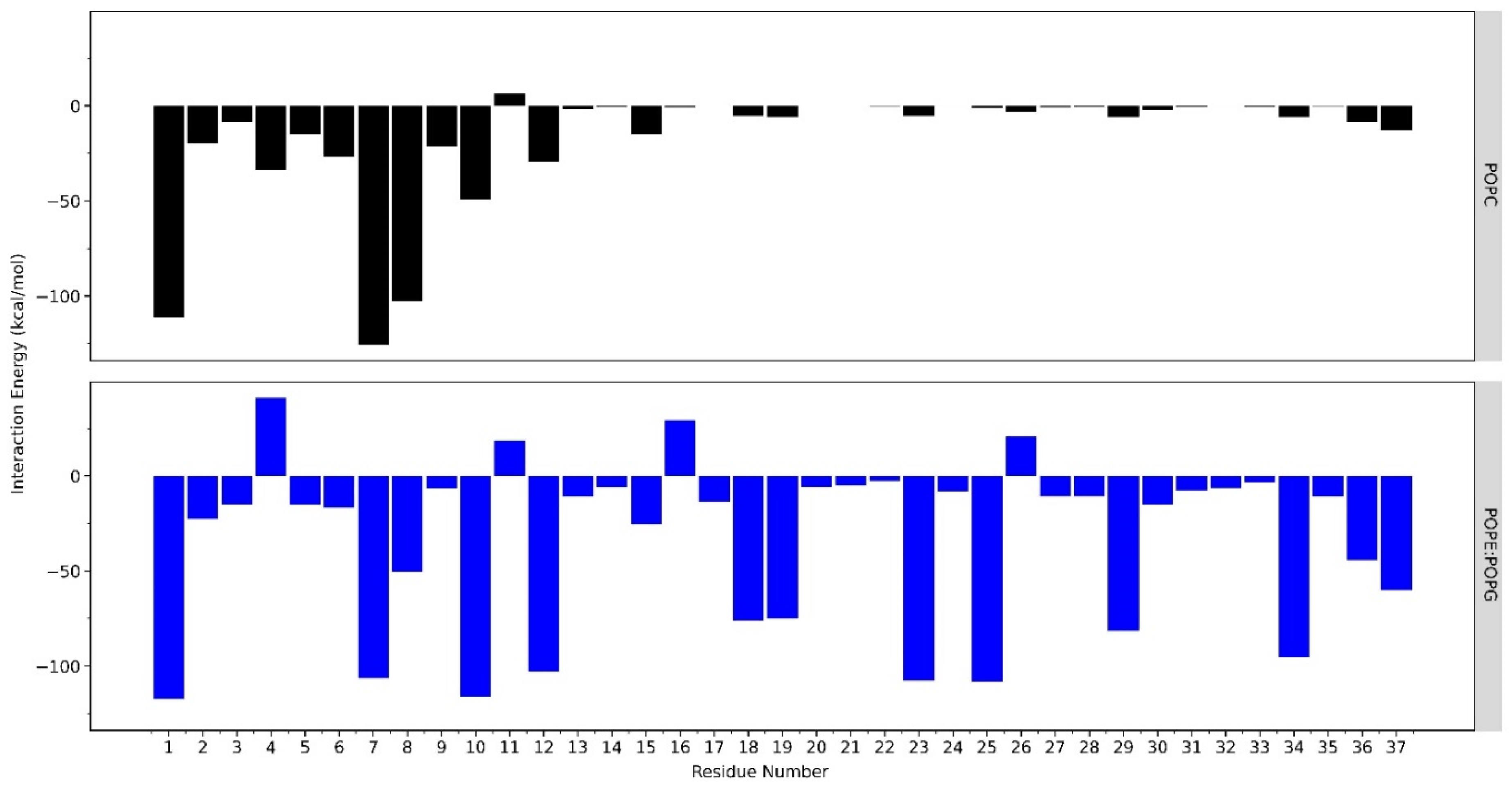
2.2. Interactions of LL-37 with POPC and POPE:POPG Membrane
2.3. Alteration of POPC and POPG Membranes
3. Discussion
4. Materials and Methods
4.1. LL-37 and Membrane System Construction
4.2. Molecular Dynamics Simulations
4.3. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van ’T Hof, W.; Veerman, E.C.I.; Heimerhorst, E.J.; Nieuw Amerongen, A.V. Antimicrobial Peptides: Properties and Applicability. Biol. Chem. 2001, 382, 597–619. [Google Scholar] [PubMed]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The Expanding Scope of Antimicrobial Peptide Structures and Their Modes of Action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Szliter-Berger, E.A.; Hazlett, L.D. Corneal Epithelium: Response to Infection. In Encyclopedia of the Eye; Elsevier: Amsterdam, The Netherlands, 2010; pp. 442–448. ISBN 9780123742032. [Google Scholar]
- Currie, S.M.; Findlay, E.G.; McHugh, B.J.; Mackellar, A.; Man, T.; Macmillan, D.; Wang, H.; Fitch, P.M.; Schwarze, J.; Davidson, D.J. The Human Cathelicidin LL-37 Has Antiviral Activity against Respiratory Syncytial Virus. PLoS ONE 2013, 8, 73659. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, S.R.; Amarullah, I.H.; Wubbolts, R.W.; Veldhuizen, E.J.A.; Haagsman, H.P. Fungicidal Mechanisms of Cathelicidins LL-37 and CATH-2 Revealed by Live-Cell Imaging. Antimicrob. Agents Chemother. 2014, 58, 2240–2248. [Google Scholar] [CrossRef] [Green Version]
- Crauwels, P.; Bank, E.; Walber, B.; Wenzel, U.A.; Agerberth, B.; Chanyalew, M.; Abebe, M.; König, R.; Ritter, U.; Reiling, N.; et al. Cathelicidin Contributes to the Restriction of Leishmania in Human Host Macrophages. Front. Immunol. 2019, 10, 2697. [Google Scholar] [CrossRef]
- Henzler Wildman, K.A.; Lee, D.K.; Ramamoorthy, A. Mechanism of Lipid Bilayer Disruption by the Human Antimicrobial Peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Kaplan, M.J. Little Peptide, Big Effects: The Role of LL-37 in Inflammation and Autoimmune Disease. J. Immunol. 2013, 191, 4895–4901. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.; Epand, R.F.; Epand, R.M.; Wang, G. Structural Location Determines Functional Roles of the Basic Amino Acids of KR-12, the Smallest Antimicrobial Peptide from Human Cathelicidin LL-37. RSC Adv. 2013, 3, 19560–19571. [Google Scholar] [CrossRef] [Green Version]
- Jacob, B.; Park, I.S.; Bang, J.K.; Shin, S.Y. Short KR-12 Analogs Designed from Human Cathelicidin LL-37 Possessing Both Antimicrobial and Antiendotoxic Activities without Mammalian Cell Toxicity. J. Pept. Sci. 2013, 19, 700–707. [Google Scholar] [CrossRef]
- Li, H.; Zhang, S.; Nie, B.; Du, Z.; Long, T.; Yue, B. The Antimicrobial Peptide KR-12 Promotes the Osteogenic Differentiation of Human Bone Marrow Stem Cells by Stimulating BMP/SMAD Signaling. RSC Adv. 2018, 8, 15547–15557. [Google Scholar] [CrossRef]
- Wang, G.; Elliott, M.; Cogen, A.L.; Ezell, E.L.; Gallo, R.L.; Hancock, R.E.W. Structure, Dynamics, and Antimicrobial and Immune Modulatory Activities of Human LL-23 and Its Single-Residue Variants Mutated on the Basis of Homologous Primate Cathelicidins. Biochemistry 2012, 51, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Epand, R.F.; Mishra, B.; Lushnikova, T.; Thomas, V.C.; Bayles, K.W.; Epand, R.M. Decoding the Functional Roles of Cationic Side Chains of the Major Antimicrobial Region of Human Cathelicidin LL-37. Antimicrob. Agents Chemother. 2012, 56, 845–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xhindoli, D.; Pacor, S.; Benincasa, M.; Scocchi, M.; Gennaro, R.; Tossi, A. The Human Cathelicidin LL-37-A Pore-Forming Antibacterial Peptide and Host-Cell Modulator. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 546–566. [Google Scholar] [CrossRef]
- Zeth, K.; Sancho-Vaello, E. The Human Antimicrobial Peptides Dermcidin and LL-37 Show Novel Distinct Pathways in Membrane Interactions. Front. Chem. 2017, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Verjans, E.T.; Zels, S.; Luyten, W.; Landuyt, B.; Schoofs, L. Molecular Mechanisms of LL-37-Induced Receptor Activation: An Overview. Peptides 2016, 85, 16–26. [Google Scholar] [CrossRef]
- Murzyn, K.; Róg, T.; Pasenkiewicz-Gierula, M. Phosphatidylethanolamine-Phosphatidylglycerol Bilayer as a Model of the Inner Bacterial Membrane. Biophys. J. 2005, 88, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Shahane, G.; Ding, W.; Palaiokostas, M.; Orsi, M. Physical Properties of Model Biological Lipid Bilayers: Insights from All-Atom Molecular Dynamics Simulations. J. Mol. Model. 2019, 25, 76. [Google Scholar] [CrossRef] [Green Version]
- Marrink, S.J.; Corradi, V.; Souza, P.C.T.; Ingólfsson, H.I.; Tieleman, D.P.; Sansom, M.S.P. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, T.; Wei, D.; Strandberg, E.; Ulrich, A.S.; Ulmschneider, J.P. How Reliable Are Molecular Dynamics Simulations of Membrane Active Antimicrobial Peptides? Biochim. Biophys. Acta-Biomembr. 2014, 1838, 2280–2288. [Google Scholar] [CrossRef] [Green Version]
- Ingolfsson, H.I.; Arnarez, C.; Periole, X.; Marrink, S.J. Computational ’microscopy of Cellular Membranes. J. Cell Sci. 2016, 129, 257–268. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Z.; Bian, Y.; Hu, G.; Wang, J.; Zhou, Y. Molecular Dynamics Simulations of Human Antimicrobial Peptide LL-37 in Model POPC and POPG Lipid Bilayers. Int. J. Mol. Sci. 2018, 19, 1186. [Google Scholar] [CrossRef] [Green Version]
- Hills, R.D.; McGlinchey, N. Model Parameters for Simulation of Physiological Lipids. J. Comput. Chem. 2016, 37, 1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, G.C.Q.; Silva, G.M.; Tavares, F.W.; Fleming, F.P.; Horta, B.A.C. Are All-Atom Any Better than United-Atom Force Fields for the Description of Liquid Properties of Alkanes? J. Mol. Model. 2020, 26, 296. [Google Scholar] [CrossRef]
- Chothia, C. Hydrophobic Bonding and Accessible Surface Area in Proteins. Nature 1974, 248, 338–339. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Lesk, A.M.; Janin, J.; Chothia, C. The Accessible Surface Area and Stability of Oligomeric Proteins. Nature 1987, 328, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Dürr, U.H.N.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the Only Human Member of the Cathelicidin Family of Antimicrobial Peptides. Biochim. Biophys. Acta-Biomembr. 2006, 1758, 1408–1425. [Google Scholar] [CrossRef] [Green Version]
- Ciornei, C.D.; Sigurdardóttir, T.; Schmidtchen, A.; Bodelsson, M. Antimicrobial and Chemoattractant Activity, Lipopolysaccharide Neutralization, Cytotoxicity, and Inhibition by Serum of Analogs of Human Cathelicidin LL-37. Antimicrob. Agents Chemother. 2005, 49, 2845–2850. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.F.; Wang, G.; Berno, B.; Epand, R.M. Lipid Segregation Explains Selective Toxicity of a Series of Fragments Derived from the Human Cathelicidin LL-37. Antimicrob. Agents Chemother. 2009, 53, 3705–3714. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Hanke, M.L.; Mishra, B.; Lushnikova, T.; Heim, C.E.; Thomas, V.C.; Bayles, K.W.; Kielian, T. Transformation of Human Cathelicidin LL-37 into Selective, Stable, and Potent Antimicrobial Compounds. ACS Chem. Biol. 2014, 9, 1997–2002. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Sun, L.C.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q.Y. The Antimicrobial Peptides and Their Potential Clinical Applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Gunasekera, S.; Muhammad, T.; Strömstedt, A.A.; Rosengren, K.J.; Göransson, U. Alanine and Lysine Scans of the LL-37-Derived Peptide Fragment KR-12 Reveal Key Residues for Antimicrobial Activity. ChemBioChem 2018, 19, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Structures of Human Host Defense Cathelicidin LL-37 and Its Smallest Antimicrobial Peptide KR-12 in Lipid Micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamala, G.; Vutukuru, S.S.; Pasha, C. In Silico Modelling of Engineered LL-37. Int. J. Biotechnol. Biochem. 2016, 12, 197–205. [Google Scholar]
- Marimuthu, S.K.; Nagarajan, K.; Perumal, S.K.; Palanisamy, S.; Subbiah, L. Structural Stability of Antimicrobial Peptides Rich in Tryptophan, Proline and Arginine: A Computational Study. J. Biomol. Struct. Dyn. 2020, 40, 3551–3559. [Google Scholar] [CrossRef]
- Malaspina, T.; Colherinhas, G.; De Oliveira Outi, F.; Fileti, E.E. Assessing the Interaction between Surfactant-like Peptides and Lipid Membranes. RSC Adv. 2017, 7, 35973–35981. [Google Scholar] [CrossRef] [Green Version]
- Petkov, P.; Lilkova, E.; Ilieva, N.; Litov, L. Self-Association of Antimicrobial Peptides: A Molecular Dynamics Simulation Study on Bombinin. Int. J. Mol. Sci. 2019, 20, 5450. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Sun, Y.; Qian, S.; Huang, H.W. Transmembrane Pores Formed by Human Antimicrobial Peptide LL-37. Biophys. J. 2011, 100, 1688–1696. [Google Scholar] [CrossRef] [Green Version]
- Guterstam, P.; Madani, F.; Hirose, H.; Takeuchi, T.; Futaki, S.; EL Andaloussi, S.; Gräslund, A.; Langel, Ü. Elucidating Cell-Penetrating Peptide Mechanisms of Action for Membrane Interaction, Cellular Uptake, and Translocation Utilizing the Hydrophobic Counter-Anion Pyrenebutyrate. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 2509–2517. [Google Scholar] [CrossRef] [Green Version]
- Pushpanathan, M.; Gunasekaran, P.; Rajendhran, J. Antimicrobial Peptides: Versatile Biological Properties. Int. J. Pept. 2013, 2013, 675391. [Google Scholar] [CrossRef] [Green Version]
- Sancho-Vaello, E.; Gil-Carton, D.; François, P.; Bonetti, E.J.; Kreir, M.; Pothula, K.R.; Kleinekathöfer, U.; Zeth, K. The Structure of the Antimicrobial Human Cathelicidin LL-37 Shows Oligomerization and Channel Formation in the Presence of Membrane Mimics. Sci. Rep. 2020, 10, 17356. [Google Scholar] [CrossRef]
- Engelberg, Y.; Landau, M. The Human LL-37(17-29) Antimicrobial Peptide Reveals a Functional Supramolecular Structure. Nat. Commun. 2020, 11, 3894. [Google Scholar] [CrossRef] [PubMed]
- Horn, J.N.; Romo, T.D.; Grossfield, A. Simulating the Mechanism of Antimicrobial Lipopeptides with All- Atom Molecular Dynamics. Biochem. J. 2014, 52, 5604–5610. [Google Scholar] [CrossRef]
- Shabala, L.; Bowman, J.; Brown, J.; Ross, T.; McMeekin, T.; Shabala, S. Ion Transport and Osmotic Adjustment in Escherichia coli in Response to Ionic and Non-Ionic Osmotica. Environ. Microbiol. 2009, 11, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Florova, P.; Sklenovsky, P.; Bana, P. Explicit Water Models Affect the Specific Solvation and Dynamics of Unfolded Peptides While the Conformational Behavior and Flexibility of Folded Peptides Remain Intact. J. Chem. Theory Comput. 2010, 6, 3569–3579. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2008, 26, 1781–1802. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Calculations | POPE (kcal/mol) | POPE:POPG (kcal/mol) |
|---|---|---|
| E Van der Waals | −95.4395 | −242.2387 |
| E Electrostatic | −627.1922 | −6199.7519 |
| E PB | 641.2761 | 6208.2515 |
| E Polar | −86.3656 | −199.5350 |
| ΔG Gas | −722.6318 | −6441.9906 |
| ΔG Solv | 703.3229 | 6356.4950 |
| ΔG Total | −19.3089 | −85.4955 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yusuf, M.; Destiarani, W.; Firdaus, A.R.R.; Rohmatulloh, F.G.; Novianti, M.T.; Pradini, G.W.; Dwiyana, R.F. Residual Interactions of LL-37 with POPC and POPE:POPG Bilayer Model Studied by All-Atom Molecular Dynamics Simulation. Int. J. Mol. Sci. 2022, 23, 13413. https://doi.org/10.3390/ijms232113413
Yusuf M, Destiarani W, Firdaus ARR, Rohmatulloh FG, Novianti MT, Pradini GW, Dwiyana RF. Residual Interactions of LL-37 with POPC and POPE:POPG Bilayer Model Studied by All-Atom Molecular Dynamics Simulation. International Journal of Molecular Sciences. 2022; 23(21):13413. https://doi.org/10.3390/ijms232113413
Chicago/Turabian StyleYusuf, Muhammad, Wanda Destiarani, Ade Rizqi Ridwan Firdaus, Fauzian Giansyah Rohmatulloh, Mia Tria Novianti, Gita Widya Pradini, and Reiva Farah Dwiyana. 2022. "Residual Interactions of LL-37 with POPC and POPE:POPG Bilayer Model Studied by All-Atom Molecular Dynamics Simulation" International Journal of Molecular Sciences 23, no. 21: 13413. https://doi.org/10.3390/ijms232113413