
Recognition of a Clickable Abasic Site Analog by DNA Polymerases and DNA Repair Enzymes
 , ,
, ,
Abstract
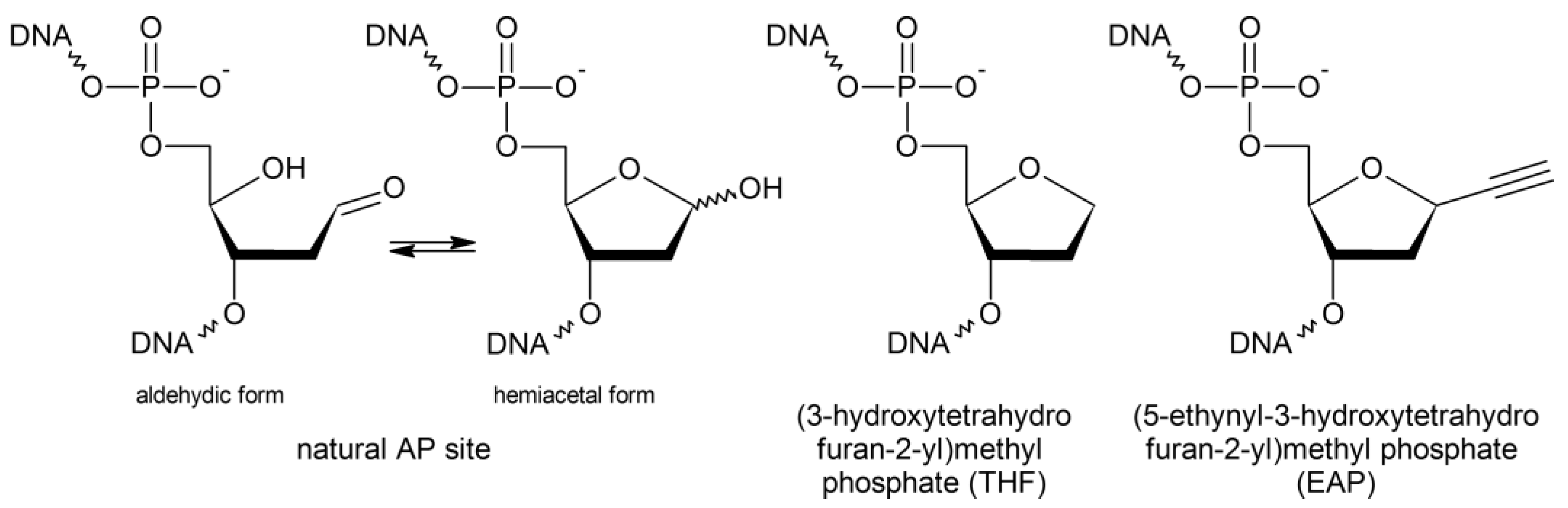
:1. Introduction
2. Results
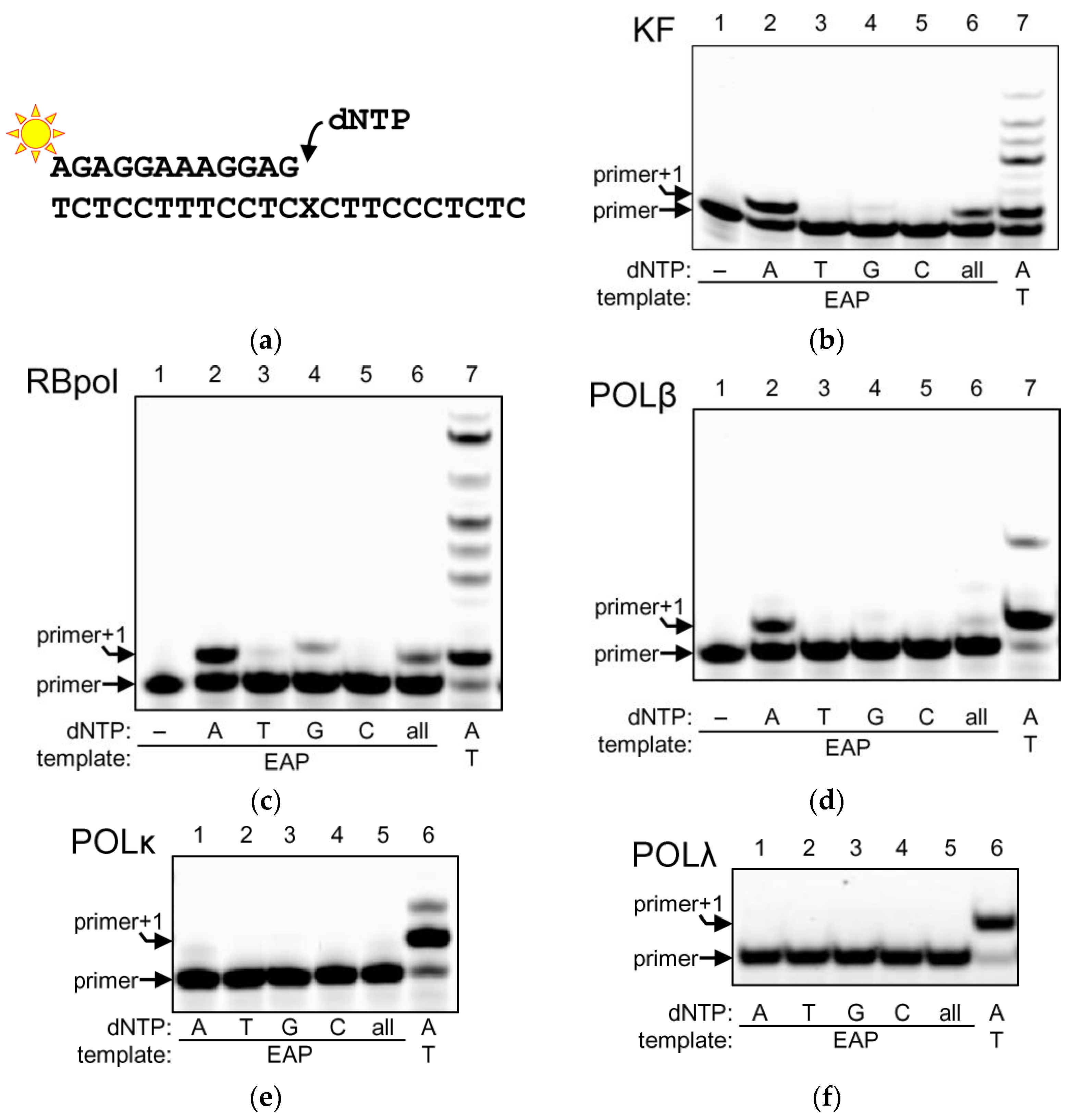
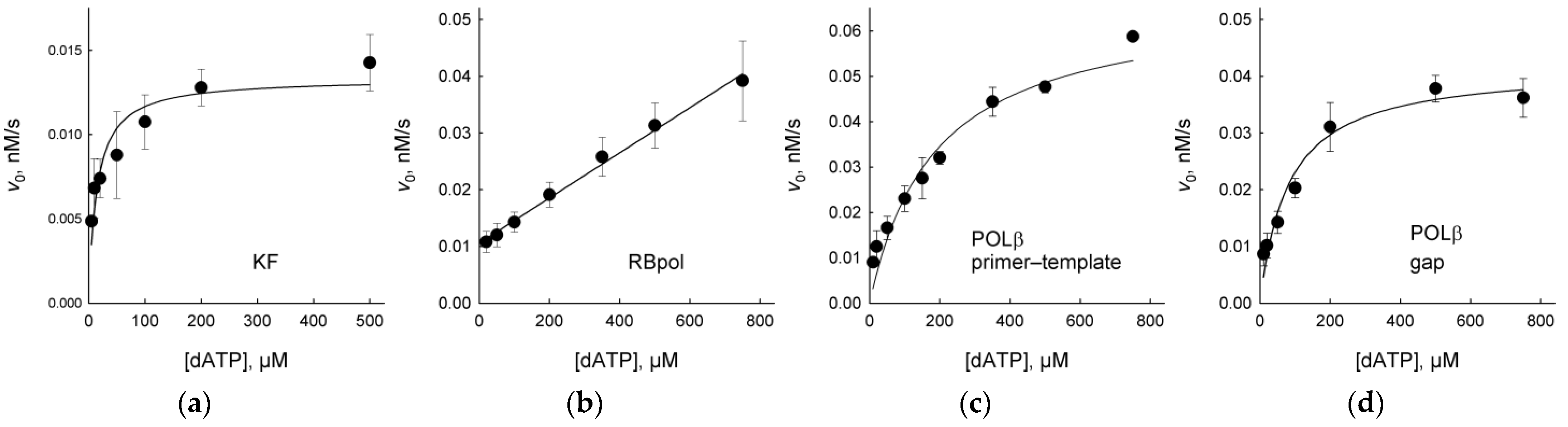
2.1. DNA Polymerases Preferentially Incorporate dAMP Opposite to EAP
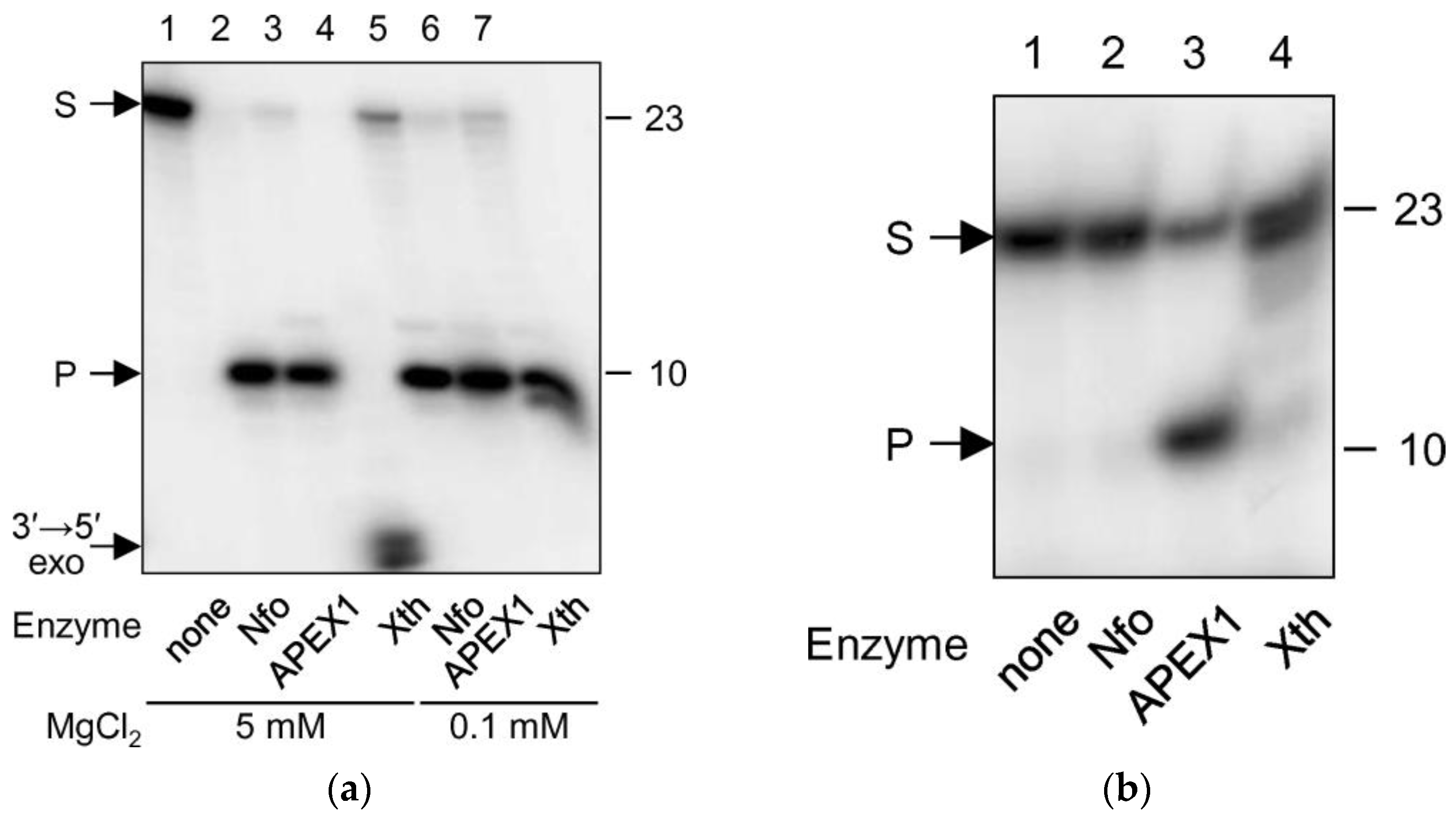
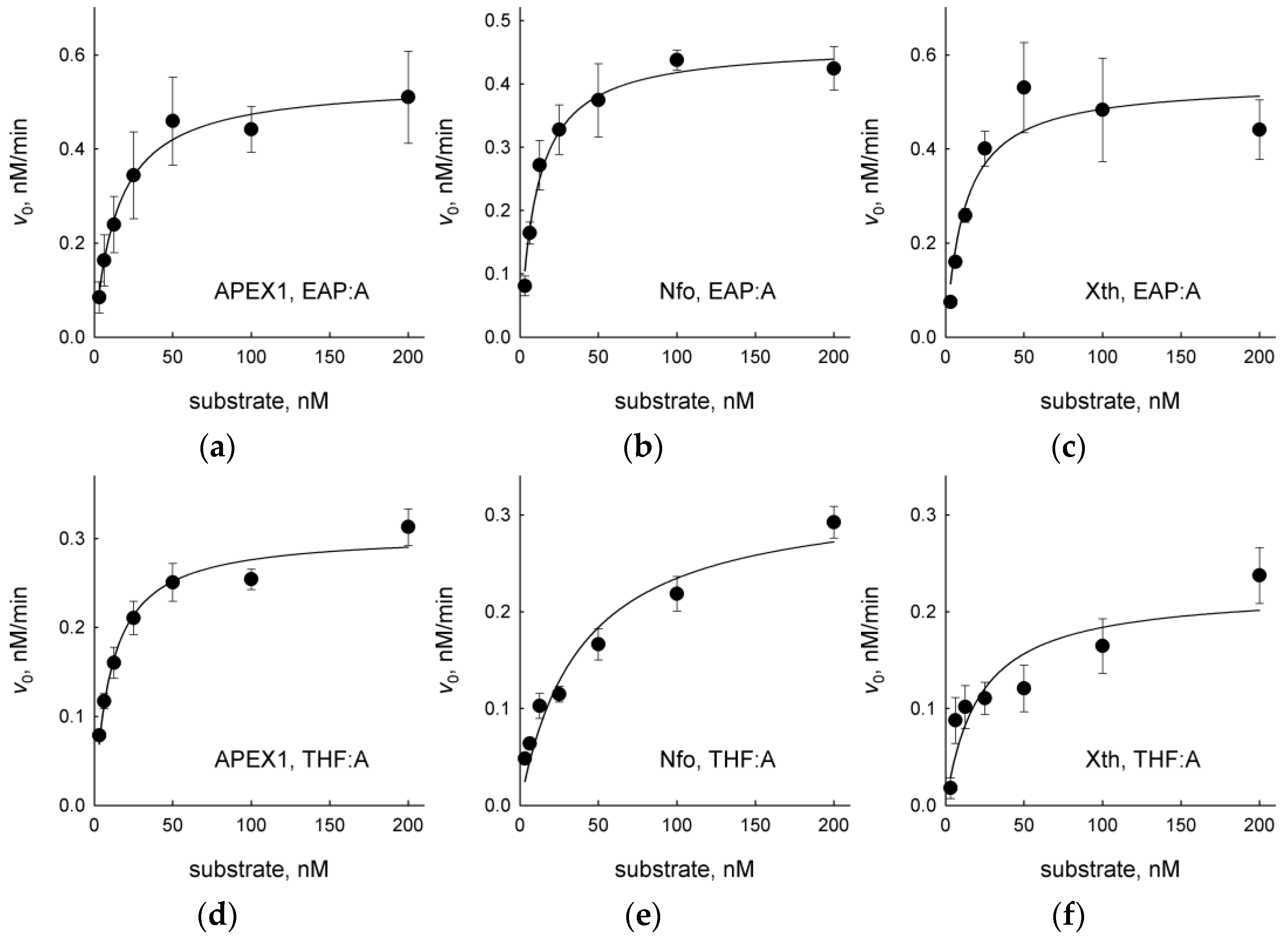
2.2. EAP Is Processed by AP Endonucleases
2.3. EAP Is Resistant to DNA Glycosylases
3. Discussion
4. Materials and Methods
4.1. Enzymes
4.2. POLβ Cloning and Purification
4.3. Oligonucleotides
4.4. Standing-Start DNA Polymerase Assay
4.5. DNA Polymerase Steady-State Kinetics
4.6. AP Endonuclease and DNA Glycosylase Assays
4.7. AP Endonuclease Steady-State Kinetics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zargar, A.; Payne, G.F.; Bentley, W.E. A ‘bioproduction breadboard’: Programming, assembling, and actuating cellular networks. Curr. Opin. Biotechnol. 2015, 36, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Elowitz, M.B. Programmable protein circuit design. Cell 2021, 184, 2284–2301. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V. Expanding the synthetic biology toolbox: Engineering orthogonal regulators of gene expression. Curr. Opin. Biotechnol. 2012, 23, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Bervoets, I.; Charlier, D. Diversity, versatility and complexity of bacterial gene regulation mechanisms: Opportunities and drawbacks for applications in synthetic biology. FEMS Microbiol. Rev. 2019, 43, 304–339. [Google Scholar] [CrossRef] [Green Version]
- Prescher, J.A.; Bertozzi, C.R. Chemistry in living systems. Nat. Chem. Biol. 2005, 1, 13–21. [Google Scholar] [CrossRef]
- Lim, R.K.V.; Lin, Q. Bioorthogonal chemistry: Recent progress and future directions. Chem. Commun. 2010, 46, 1589–1600. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Best, M.D. Click chemistry and bioorthogonal reactions: Unprecedented selectivity in the labeling of biological molecules. Biochemistry 2009, 48, 6571–6584. [Google Scholar] [CrossRef]
- Oliveira, B.L.; Guo, Z.; Bernardes, G.J.L. Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef]
- Smeenk, M.L.W.J.; Agramunt, J.; Bonger, K.M. Recent developments in bioorthogonal chemistry and the orthogonality within. Curr. Opin. Chem. Biol. 2021, 60, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Burrows, C.J.; Muller, J.G. Oxidative nucleobase modifications leading to strand scission. Chem. Rev. 1998, 98, 1109–1151. [Google Scholar] [CrossRef] [PubMed]
- Gierlich, J.; Burley, G.A.; Gramlich, P.M.E.; Hammond, D.M.; Carell, T. Click chemistry as a reliable method for the high-density postsynthetic functionalization of alkyne-modified DNA. Org. Lett. 2006, 8, 3639–3642. [Google Scholar] [CrossRef] [PubMed]
- Seela, F.; Sirivolu, V.R. DNA containing side chains with terminal triple bonds: Base-pair stability and functionalization of alkynylated pyrimidines and 7-deazapurines. Chem. Biodivers. 2006, 3, 509–514. [Google Scholar] [CrossRef]
- Nakahara, M.; Kuboyama, T.; Izawa, A.; Hari, Y.; Imanishi, T.; Obika, S. Synthesis and base-pairing properties of C-nucleotides having 1-substituted 1H-1,2,3-triazoles. Bioorg. Med. Chem. Lett. 2009, 19, 3316–3319. [Google Scholar] [CrossRef]
- Hari, Y.; Nakahara, M.; Pang, J.; Akabane, M.; Kuboyama, T.; Obika, S. Synthesis and triplex-forming ability of oligonucleotides bearing 1-substituted 1H-1,2,3-triazole nucleobases. Bioorg. Med. Chem. 2011, 19, 1162–1166. [Google Scholar] [CrossRef]
- Fujimoto, K.; Ishida, K.; Xue, L.; Nakamura, S. Effect of linker length on photo-cross-linking position mediated by click chemistry via [2+2]photocycloaddition. Photochem. Photobiol. Sci. 2020, 19, 776–782. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2006; p. 1118. [Google Scholar]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [Green Version]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef]
- Boiteux, S.; Guillet, M. Abasic sites in DNA: Repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 2004, 3, 1–12. [Google Scholar] [CrossRef]
- McCullough, A.K.; Sanchez, A.; Dodson, M.L.; Marapaka, P.; Taylor, J.-S.; Lloyd, R.S. The reaction mechanism of DNA glycosylase/AP lyases at abasic sites. Biochemistry 2001, 40, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Strauss, B.S. The “A” rule revisited: Polymerases as determinants of mutational specificity. DNA Repair 2002, 1, 125–135. [Google Scholar] [CrossRef]
- Taylor, J.-S. New structural and mechanistic insight into the A-rule and the instructional and non-instructional behavior of DNA photoproducts and other lesions. Mutat. Res. 2002, 510, 55–70. [Google Scholar] [CrossRef]
- Eun, H.-M. DNA Polymerases. In Enzymology Primer for Recombinant DNA Technology; Academic Press: San Diego, CA, USA, 1996; pp. 345–489. [Google Scholar]
- Xia, S.; Konigsberg, W.H. RB69 DNA polymerase structure, kinetics, and fidelity. Biochemistry 2014, 53, 2752–2767. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Bebenek, K. DNA replication fidelity. Annu. Rev. Biochem. 2000, 69, 497–529. [Google Scholar] [CrossRef] [Green Version]
- Belousova, E.A.; Lavrik, O.I. DNA polymerases β and λ and their roles in cell. DNA Repair 2015, 29, 112–126. [Google Scholar] [CrossRef]
- Stern, H.R.; Sefcikova, J.; Chaparro, V.E.; Beuning, P.J. Mammalian DNA polymerase kappa activity and specificity. Molecules 2019, 24, 2805. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yuan, F.; Wu, X.; Wang, M.; Rechkoblit, O.; Taylor, J.-S.; Geacintov, N.E.; Wang, Z. Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res. 2000, 28, 4138–4146. [Google Scholar] [CrossRef] [Green Version]
- Wolfle, W.T.; Washington, M.T.; Prakash, L.; Prakash, S. Human DNA polymerase κ uses template-primer misalignment as a novel means for extending mispaired termini and for generating single-base deletions. Genes Dev. 2003, 17, 2191–2199. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-Y.; Lim, S.; Kim, E.-J.; Jo, A.; Guengerich, F.P. Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases α, δ, η, ι, κ, and REV1. J. Mol. Biol. 2010, 404, 34–44. [Google Scholar] [CrossRef]
- Sherrer, S.M.; Fiala, K.A.; Fowler, J.D.; Newmister, S.A.; Pryor, J.M.; Suo, Z. Quantitative analysis of the efficiency and mutagenic spectra of abasic lesion bypass catalyzed by human Y-family DNA polymerases. Nucleic Acids Res. 2011, 39, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudkina, A.V.; Zharkov, D.O. Miscoding and DNA polymerase stalling by methoxyamine-adducted abasic sites. Chem. Res. Toxicol. 2022, 35, 303–314. [Google Scholar] [CrossRef]
- Efrati, E.; Tocco, G.; Eritja, R.; Wilson, S.H.; Goodman, M.F. Abasic translesion synthesis by DNA polymerase β violates the “A-rule”: Novel types of nucleotide incorporation by human DNA polymerase β at an abasic lesion in different sequence contexts. J. Biol. Chem. 1997, 272, 2559–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieseking, S.; Bergen, K.; Di Pasquale, F.; Diederichs, K.; Welte, W.; Marx, A. Human DNA polymerase β mutations allowing efficient abasic site bypass. J. Biol. Chem. 2011, 286, 4011–4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, R.; Beard, W.A.; Wilson, S.H. Studies of gapped DNA substrate binding by mammalian DNA polymerase β: Dependence on 5′-phosphate group. J. Biol. Chem. 1994, 269, 18096–18101. [Google Scholar] [CrossRef]
- Chagovetz, A.M.; Sweasy, J.B.; Preston, B.D. Increased activity and fidelity of DNA polymerase β on single-nucleotide gapped DNA. J. Biol. Chem. 1997, 272, 27501–27504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, W.A.; Shock, D.D.; Yang, X.-P.; DeLauder, S.F.; Wilson, S.H. Loss of DNA polymerase β stacking interactions with templating purines, but not pyrimidines, alters catalytic efficiency and fidelity. J. Biol. Chem. 2002, 277, 8235–8242. [Google Scholar] [CrossRef] [Green Version]
- Mol, C.D.; Hosfield, D.J.; Tainer, J.A. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: The 3′ ends justify the means. Mutat. Res. 2000, 460, 211–229. [Google Scholar] [CrossRef]
- Hitomi, K.; Iwai, S.; Tainer, J.A. The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal, and repair. DNA Repair 2007, 6, 410–428. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Freudenthal, B.D. APE1: A skilled nucleic acid surgeon. DNA Repair 2018, 71, 93–100. [Google Scholar] [CrossRef]
- Talpaert-Borlé, M.; Liuzzi, M. Reaction of apurinic/apyrimidinic sites with [14C]methoxyamine: A method for the quantitative assay of AP sites in DNA. Biochim. Biophys. Acta 1983, 740, 410–416. [Google Scholar] [CrossRef]
- Atamna, H.; Cheung, I.; Ames, B.N. A method for detecting abasic sites in living cells: Age-dependent changes in base excision repair. Proc. Natl Acad. Sci. USA 2000, 97, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marenstein, D.R.; Wilson, D.M., III.; Teebor, G.W. Human AP endonuclease (APE1) demonstrates endonucleolytic activity against AP sites in single-stranded DNA. DNA Repair 2004, 3, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, M.; Chang, C.-N.; Johnson, F.; Will, S.; Grollman, A.P. Oligodeoxynucleotides containing synthetic abasic sites: Model substrates for DNA polymerases and apurinic/apyrimidinic endonucleases. J. Biol. Chem. 1987, 262, 10171–10179. [Google Scholar] [CrossRef]
- Wilson, D.M., III.; Takeshita, M.; Grollman, A.P.; Demple, B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem. 1995, 270, 16002–16007. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.M., III.; Takeshita, M.; Demple, B. Abasic site binding by the human apurinic endonuclease, Ape, and determination of the DNA contact sites. Nucleic Acids Res. 1997, 25, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Erzberger, J.P.; Barsky, D.; Schärer, O.D.; Colvin, M.E.; Wilson, D.M., III. Elements in abasic site recognition by the major human and Escherichia coli apurinic/apyrimidinic endonucleases. Nucleic Acids Res. 1998, 26, 2771–2778. [Google Scholar] [CrossRef] [Green Version]
- Mundle, S.T.; Delaney, J.C.; Essigmann, J.M.; Strauss, P.R. Enzymatic mechanism of human apurinic/apyrimidinic endonuclease against a THF AP site model substrate. Biochemistry 2009, 48, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Schermerhorn, K.M.; Delaney, S. Transient-state kinetics of apurinic/apyrimidinic (AP) endonuclease 1 acting on an authentic AP site and commonly used substrate analogs: The effect of diverse metal ions and base mismatches. Biochemistry 2013, 52, 7669–7677. [Google Scholar] [CrossRef] [Green Version]
- Endutkin, A.V.; Zharkov, D.O. Substrate specificities of DNA glycosylases in vitro and in vivo. In DNA Damage, DNA Repair and Disease; Dizdaroglu, M., Lloyd, R.S., Eds.; Royal Society of Chemistry: London, UK, 2021; Volume 1, pp. 175–203. [Google Scholar]
- David, S.S.; Williams, S.D. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev. 1998, 98, 1221–1261. [Google Scholar] [CrossRef]
- Stivers, J.T.; Jiang, Y.L. A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev. 2003, 103, 2729–2760. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Michaels, M.L.; Miller, J.H.; Grollman, A.P. Function of the zinc finger in Escherichia coli Fpg protein. J. Biol. Chem. 1993, 268, 26738–26744. [Google Scholar] [CrossRef]
- Kropachev, K.Y.; Zharkov, D.O.; Grollman, A.P. Catalytic mechanism of Escherichia coli endonuclease VIII: Roles of the intercalation loop and the zinc finger. Biochemistry 2006, 45, 12039–12049. [Google Scholar] [CrossRef] [Green Version]
- Guibourt, N.; Castaing, B.; Auffret van der Kemp, P.; Boiteux, S. Catalytic and DNA binding properties of the Ogg1 protein of Saccharomyces cerevisiae: Comparison between the wild type and the K241R and K241Q active-site mutant proteins. Biochemistry 2000, 39, 1716–1724. [Google Scholar] [CrossRef]
- Fromme, J.C.; Verdine, G.L. Structure of a trapped endonuclease III–DNA covalent intermediate. EMBO J. 2003, 22, 3461–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Lu, L.; Zhang, J.; Yue, Z.; Song, J.; Zong, S.; Liu, M.; Stovicek, O.; Gao, Y.Q.; Yi, C. Tautomerization-dependent recognition and excision of oxidation damage in base-excision DNA repair. Proc. Natl Acad. Sci. USA 2016, 113, 7792–7797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaing, B.; Boiteux, S.; Zelwer, C. DNA containing a chemically reduced apurinic site is a high affinity ligand for the E. coli formamidopyrimidine-DNA glycosylase. Nucleic Acids Res. 1992, 20, 389–394. [Google Scholar] [CrossRef]
- Thomas, M.; Castaing, B.; Fourrey, J.-L.; Zelwer, C. Synthesis of an enantiomerically pure carbocyclic DNA abasic site analogue. Nucleosides Nucleotides 1999, 18, 239–243. [Google Scholar] [CrossRef]
- de los Santos, C.; El-khateeb, M.; Rege, P.; Tian, K.; Johnson, F. Impact of the C1′ configuration of abasic sites on DNA duplex structure. Biochemistry 2004, 43, 15349–15357. [Google Scholar] [CrossRef]
- Schärer, O.D.; Ortholand, J.-Y.; Ganesan, A.; Ezaz-Nikpay, K.; Verdine, G.L. Specific binding of the DNA repair enzyme AlkA to a pyrrolidine-based inhibitor. J. Am. Chem. Soc. 1995, 117, 6623–6624. [Google Scholar] [CrossRef]
- Chu, A.M.; Fettinger, J.C.; David, S.S. Profiling base excision repair glycosylases with synthesized transition state analogs. Bioorg. Med. Chem. Lett. 2011, 21, 4969–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, M.; Chevrie, K.; Chassignol, M.; Thuong, N.T.; Maurizot, J.C. Circular dichroism studies of an oligodeoxyribonucleotide containing a hairpin loop made of a hexaethylene glycol chain: Conformation and stability. Nucleic Acids Res. 1990, 18, 6353–6359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyshnaya, I.A.; Pyshnii, D.V.; Ivanova, E.M.; Zarytova, V.F.; Bonora, G.M.; Scalfi-Happ, C.; Seliger, H. Oligonucleotide conjugates designed for discriminative hybridization at physiological temperature. Nucleosides Nucleotides 1998, 17, 1289–1297. [Google Scholar] [CrossRef]
- Castaing, B.; Fourrey, J.-L.; Hervouet, N.; Thomas, M.; Boiteux, S.; Zelwer, C. AP site structural determinants for Fpg specific recognition. Nucleic Acids Res. 1999, 27, 608–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, M.H.; Sieber, S.A. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33, 681–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, D.; Harijan, D.; Wagenknecht, H.-A. Labelling of DNA and RNA in the cellular environment by means of bioorthogonal cycloaddition chemistry. RSC Chem. Biol. 2020, 1, 86–97. [Google Scholar] [CrossRef]
- Obeid, S.; Blatter, N.; Kranaster, R.; Schnur, A.; Diederichs, K.; Welte, W.; Marx, A. Replication through an abasic DNA lesion: Structural basis for adenine selectivity. EMBO J. 2010, 29, 1738–1747. [Google Scholar] [CrossRef] [Green Version]
- Obeid, S.; Welte, W.; Diederichs, K.; Marx, A. Amino acid templating mechanisms in selection of nucleotides opposite abasic sites by a family A DNA polymerase. J. Biol. Chem. 2012, 287, 14099–14108. [Google Scholar] [CrossRef] [Green Version]
- Zahn, K.E.; Belrhali, H.; Wallace, S.S.; Doublié, S. Caught bending the A-rule: Crystal structures of translesion DNA synthesis with a non-natural nucleotide. Biochemistry 2007, 46, 10551–10561. [Google Scholar] [CrossRef]
- Xia, S.; Vashishtha, A.; Bulkley, D.; Eom, S.H.; Wang, J.; Konigsberg, W.H. Contribution of partial charge interactions and base stacking to the efficiency of primer extension at and beyond abasic sites in DNA. Biochemistry 2012, 51, 4922–4931. [Google Scholar] [CrossRef]
- Beard, W.A.; Shock, D.D.; Batra, V.K.; Pedersen, L.C.; Wilson, S.H. DNA polymerase β substrate specificity: Side chain modulation of the “A-rule”. J. Biol. Chem. 2009, 284, 31680–31689. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Kim, K.; Hurwitz, J.; Gary, R.; Levin, D.S.; Tomkinson, A.E.; Park, M.S. Reconstitution of proliferating cell nuclear antigen-dependent repair of apurinic/apyrimidinic sites with purified human proteins. J. Biol. Chem. 1999, 274, 33703–33708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyamichev, V.; Brow, M.A.D.; Dahlberg, J.E. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science 1993, 260, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jinks-Robertson, S. Abasic sites in the transcribed strand of yeast DNA are removed by transcription-coupled nucleotide excision repair. Mol. Cell. Biol. 2010, 30, 3206–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitsera, N.; Rodriguez-Alvarez, M.; Emmert, S.; Carell, T.; Khobta, A. Nucleotide excision repair of abasic DNA lesions. Nucleic Acids Res. 2019, 47, 8537–8547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair 2007, 6, 317–328. [Google Scholar] [CrossRef]
- Katafuchi, A.; Nakano, T.; Masaoka, A.; Terato, H.; Iwai, S.; Hanaoka, F.; Ide, H. Differential specificity of human and Escherichia coli endonuclease III and VIII homologues for oxidative base lesions. J. Biol. Chem. 2004, 279, 14464–14471. [Google Scholar] [CrossRef] [Green Version]
- Kakhkharova, Z.I.; Zharkov, D.O.; Grin, I.R. A low-activity polymorphic variant of human NEIL2 DNA glycosylase. Int. J. Mol. Sci. 2022, 23, 2212. [Google Scholar] [CrossRef]
- Moréra, S.; Grin, I.; Vigouroux, A.; Couvé, S.; Henriot, V.; Saparbaev, M.; Ishchenko, A.A. Biochemical and structural characterization of the glycosylase domain of MBD4 bound to thymine and 5-hydroxymethyuracil-containing DNA. Nucleic Acids Res. 2012, 40, 9917–9926. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Koval, V.V.; Zharkov, D.O.; Nevinsky, G.A.; Douglas, K.T.; Fedorova, O.S. Kinetics of substrate recognition and cleavage by human 8-oxoguanine-DNA glycosylase. Nucleic Acids Res. 2005, 33, 3919–3931. [Google Scholar] [CrossRef]
- Grin, I.R.; Mechetin, G.V.; Kasymov, R.D.; Diatlova, E.A.; Yudkina, A.V.; Shchelkunov, S.N.; Gileva, I.P.; Denisova, A.A.; Stepanov, G.A.; Chilov, G.G.; et al. A new class of uracil–DNA glycosylase inhibitors active against human and vaccinia virus enzyme. Molecules 2021, 26, 6668. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, E.; Ogi, T.; Kusumoto, R.; Iwai, S.; Masutani, C.; Hanaoka, F.; Ohmori, H. Error-prone bypass of certain DNA lesions by the human DNA polymerase κ. Genes Dev. 2000, 14, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, M.; Bebenek, K.; Krahn, J.M.; Blanco, L.; Kunkel, T.A.; Pedersen, L.C. A structural solution for the DNA polymerase λ-dependent repair of DNA gaps with minimal homology. Mol. Cell 2004, 13, 561–572. [Google Scholar] [CrossRef]
- Gilboa, R.; Zharkov, D.O.; Golan, G.; Fernandes, A.S.; Gerchman, S.E.; Matz, E.; Kycia, J.H.; Grollman, A.P.; Shoham, G. Structure of formamidopyrimidine-DNA glycosylase covalently complexed to DNA. J. Biol. Chem. 2002, 277, 19811–19816. [Google Scholar] [CrossRef] [Green Version]
- Miller, H.; Grollman, A.P. Kinetics of DNA polymerase I (Klenow fragment exo–) activity on damaged DNA templates: Effect of proximal and distal template damage on DNA synthesis. Biochemistry 1997, 36, 15336–15342. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Gilboa, R.; Yagil, I.; Kycia, J.H.; Gerchman, S.E.; Shoham, G.; Grollman, A.P. Role for lysine 142 in the excision of adenine from A:G mispairs by MutY DNA glycosylase of Escherichia coli. Biochemistry 2000, 39, 14768–14778. [Google Scholar] [CrossRef]
- Rieger, R.A.; McTigue, M.M.; Kycia, J.H.; Gerchman, S.E.; Grollman, A.P.; Iden, C.R. Characterization of a cross-linked DNA-endonuclease VIII repair complex by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2000, 11, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Ishchenko, A.A.; Sanz, G.; Privezentzev, C.V.; Maksimenko, A.V.; Saparbaev, M. Characterisation of new substrate specificities of Escherichia coli and Saccharomyces cerevisiae AP endonucleases. Nucleic Acids Res. 2003, 31, 6344–6353. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, N.A.; Kladova, O.A.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.K.; Zharkov, D.O.; Fedorova, O.S. Conformational dynamics of DNA repair by Escherichia coli endonuclease III. J. Biol. Chem. 2015, 290, 14338–14349. [Google Scholar] [CrossRef] [Green Version]
- Freisinger, E.; Grollman, A.P.; Miller, H.; Kisker, C. Lesion (in)tolerance reveals insights into DNA replication fidelity. EMBO J. 2004, 23, 1494–1505. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Template Nucleotide | KM, μM | kcat, s−1 | kcat/KM, μM−1 × s−1 |
|---|---|---|---|---|
| KF | EAP | 14 ± 4 | (2.7 ± 0.2) × 10−2 | (1.9 ± 0.5) × 10−3 |
| AP 1 | 19 ± 5 | (2.1 ± 0.1) × 10−3 | (1.1 ± 0.3) × 10−4 | |
| T 1 | 0.77 ± 0.32 | (2.7 ± 0.2) × 10−2 | (3.5 ± 1.5) × 10−2 | |
| RBpol 2 | EAP | – | – | (4.0 ± 0.3) × 10−6 |
| AP 1 | 110 ± 30 | (2.7 ± 0.3) × 10−2 | (2.5 ± 0.7) × 10−4 | |
| T 1 | 0.35 ± 0.09 | (2.7 ± 0.1) × 10−2 | (7.7 ± 1.9) × 10−2 | |
| POLβ | EAP (primer-template) | 190 ± 30 | (6.7 ± 0.4) × 10−3 | (3.5 ± 0.6) × 10−5 |
| EAP (gap) | 81 ± 14 | (4.2 ± 0.2) × 10−3 | (5.2 ± 0.9) × 10−5 | |
| THF or AP (primer-template) | negligible 3 | |||
| AP (gap) 1 | 2.9 ± 1.5 | (4.4 ± 0.2) × 10−4 | (1.5 ± 0.8) × 10−4 | |
| T (primer-template) 1 | 4.4 ± 1.7 | (9.7 ± 0.7) × 10−2 | (2.2 ± 0.9) × 10−2 | |
| T (gap) 4 | 0.9 ± 0.2 | 0.69 ± 0.01 | 0.77 ± 0.17 | |
| Enzyme | KM, nM | kcat, min−1 | kcat/KM, nM−1 × min−1 | |||
|---|---|---|---|---|---|---|
| THF:A | EAP:A | THF:A | EAP:A | THF:A | EAP:A | |
| APEX1 | 11 ± 2 | 15 ± 6 | 25 ± 1 | 43 ± 5 | 2.3 ± 0.4 | 2.9 ± 1.2 |
| Nfo | 38 ± 8 | 11 ± 2 | 1.8 ± 0.1 | 2.6 ± 0.2 | 0.05 ± 0.01 | 0.24 ± 0.05 |
| Xth | 21 ± 8 | 12 ± 4 | 73 ± 7 | 180 ± 20 | 3 ± 1 | 15 ± 5 |
| Oligonucleotide | Sequence, 5′→3′ | Modification |
|---|---|---|
| modified | CTCTCCCTTCXCTCCTTTCCTCT | X = EAP, THF or T |
| complementary | AGAGGAAAGGAGNGAAGGGAGAG | N = A, C, G or T |
| primer | [Fluo]AGAGGAAAGGAG | fluorescein |
| downstream | GAAGGGAGAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endutkin, A.V.; Yudkina, A.V.; Zharkov, T.D.; Kim, D.V.; Zharkov, D.O. Recognition of a Clickable Abasic Site Analog by DNA Polymerases and DNA Repair Enzymes. Int. J. Mol. Sci. 2022, 23, 13353. https://doi.org/10.3390/ijms232113353
Endutkin AV, Yudkina AV, Zharkov TD, Kim DV, Zharkov DO. Recognition of a Clickable Abasic Site Analog by DNA Polymerases and DNA Repair Enzymes. International Journal of Molecular Sciences. 2022; 23(21):13353. https://doi.org/10.3390/ijms232113353
Chicago/Turabian StyleEndutkin, Anton V., Anna V. Yudkina, Timofey D. Zharkov, Daria V. Kim, and Dmitry O. Zharkov. 2022. "Recognition of a Clickable Abasic Site Analog by DNA Polymerases and DNA Repair Enzymes" International Journal of Molecular Sciences 23, no. 21: 13353. https://doi.org/10.3390/ijms232113353