
ATP8B1 Deficiency Results in Elevated Mitochondrial Phosphatidylethanolamine Levels and Increased Mitochondrial Oxidative Phosphorylation in Human Hepatoma Cells
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
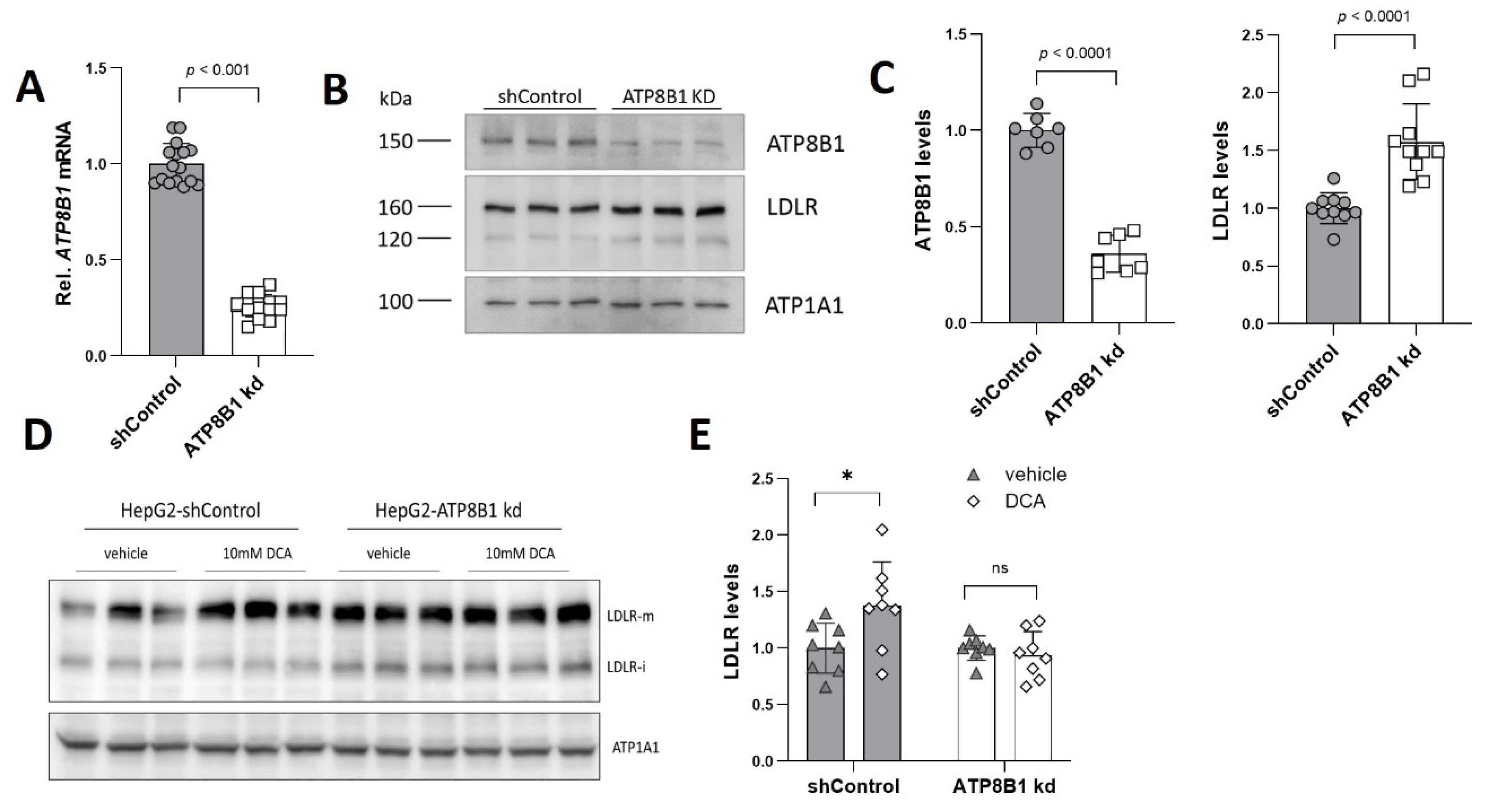
2.1. ATP8B1 Knockdown in HepG2 Cells Leads to Elevated Levels of LDLR
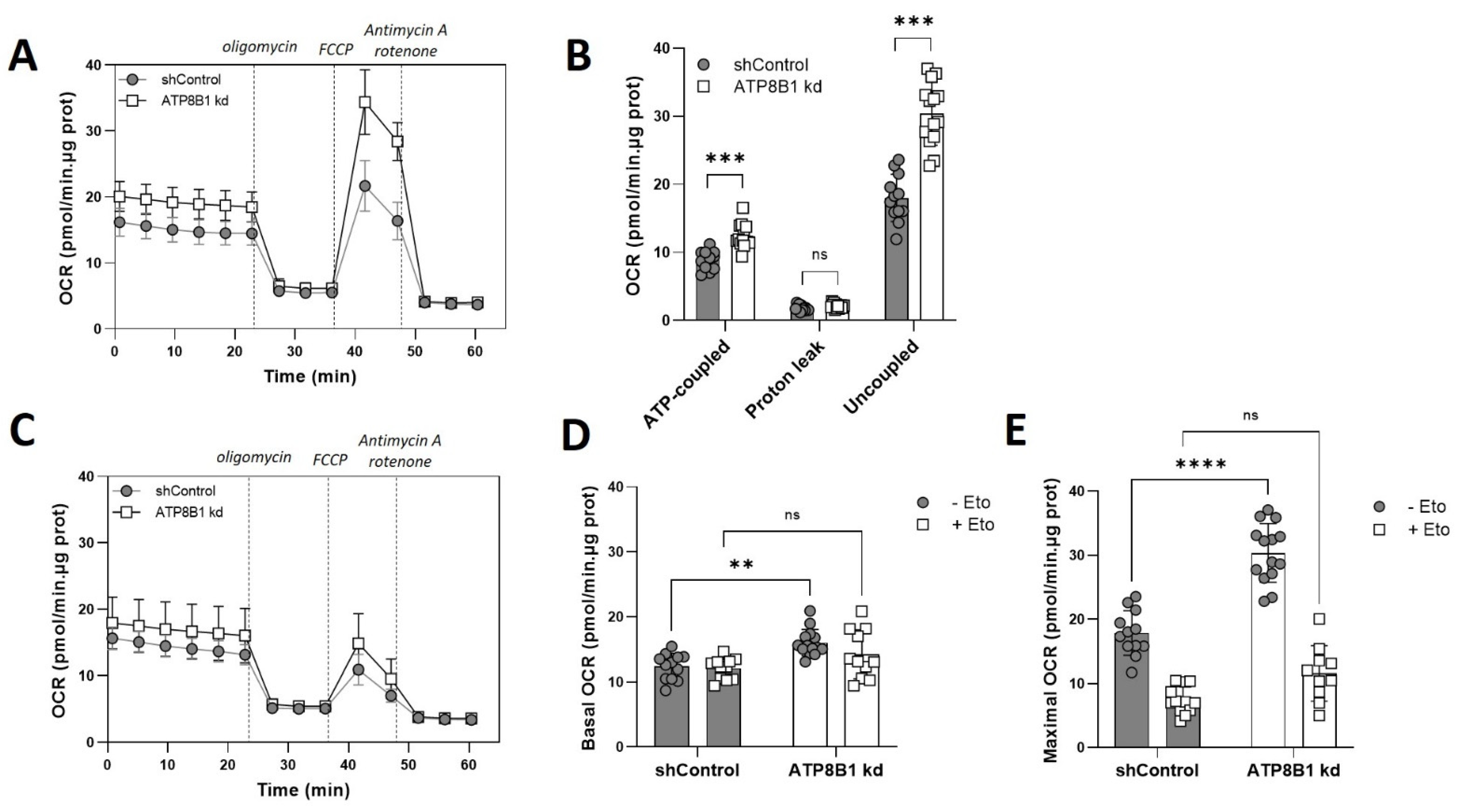
2.2. ATP8B1 Knockdown HepG2 Cells Show Increased Mitochondrial OXPHOS
2.3. ATP8B1 Knockdown Coincides with a Shift towards Mitochondrial ß-Oxidation
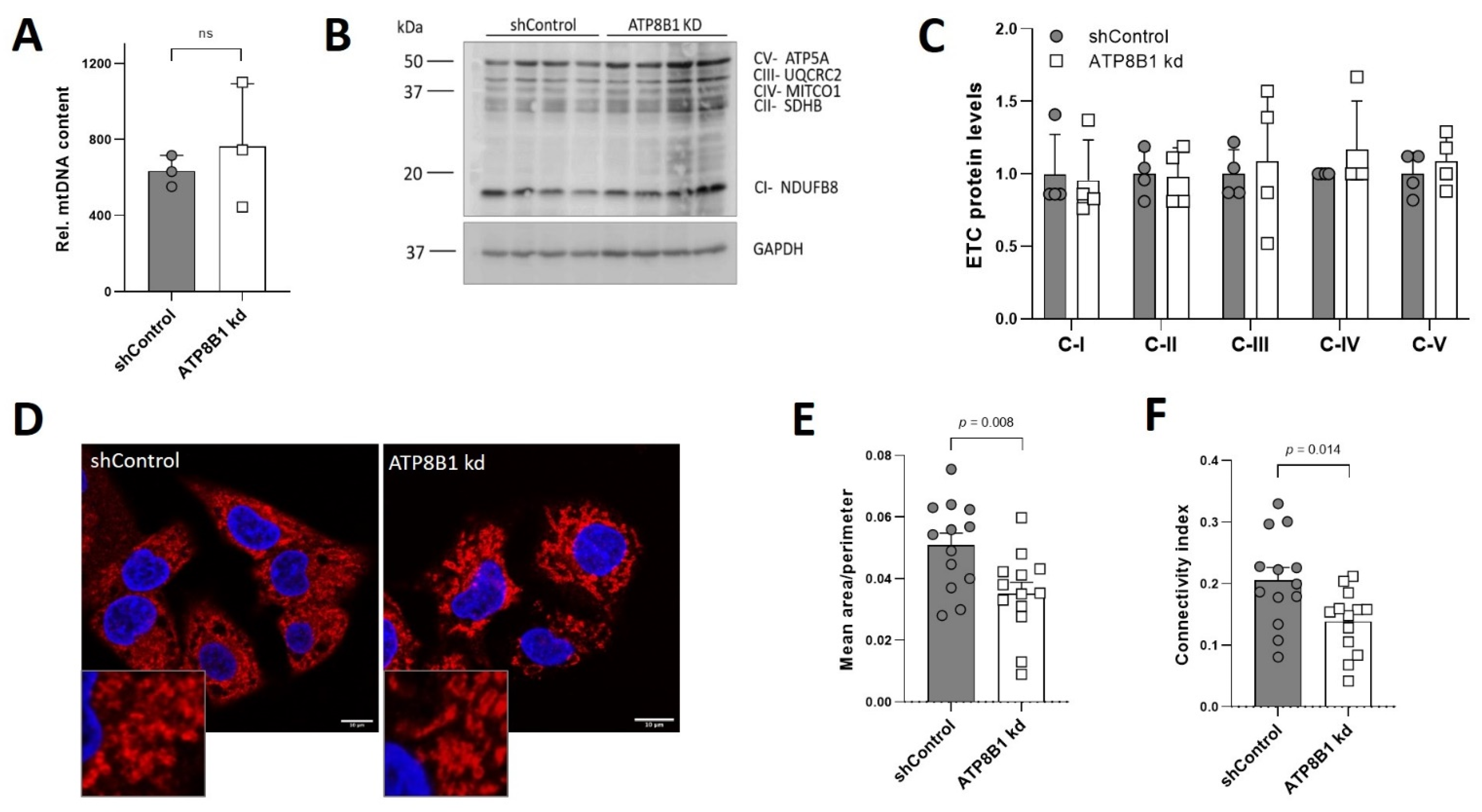
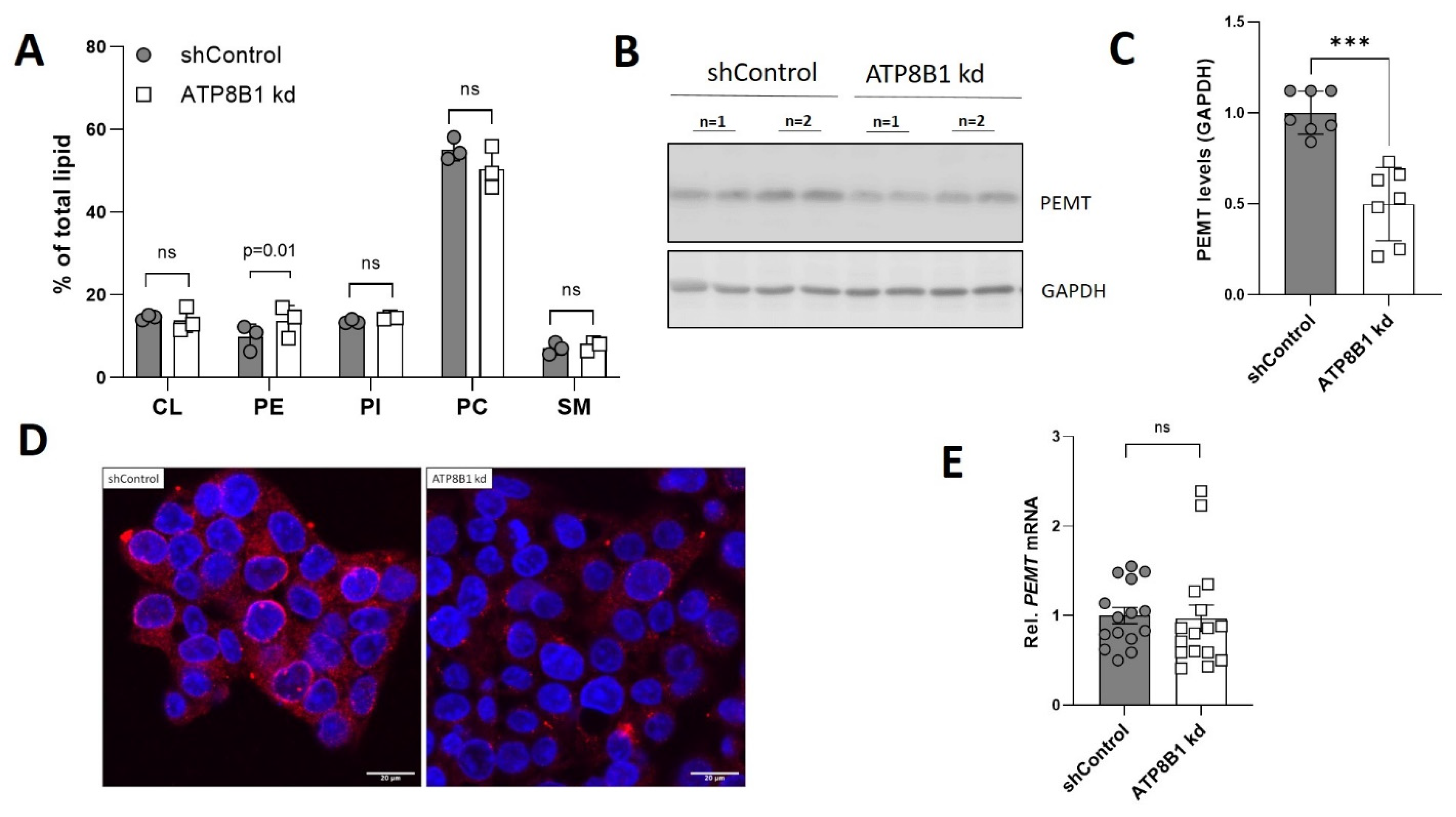
2.4. ATP8B1 Knockdown Cells Have Increased Mitochondrial Fragmentation and Elevated Mitochondrial PE Levels
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Quantitative RT-PCR
4.3. SDS Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blotting
4.4. Indirect Immunofluorescence and Mitochondrial Network Analysis
4.5. Extracellular Flux Analysis
4.6. Isolation of Mitochondria and High-Performance Thin-Layer Chromatography
4.7. Data Presentation and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Best, J.T.; Xu, P.; Graham, T.R. Phospholipid flippases in membrane remodeling and transport carrier biogenesis. Curr. Opin. Cell Biol. 2019, 59, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Panatala, R.; Hennrich, H.; Holthuis, J.C. Inner workings and biological impact of phospholipid flippases. J. Cell Sci. 2015, 128, 2021–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Mark, V.A.; Oude Elferink, R.P.J.; Paulusma, C.C. P4 ATPases: Flippases in health and disease. Int. J. Mol. Sci. 2013, 14, 7897–7922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, L.N.; van Eijk, M.J.; Pawlikowska, L.; DeYoung, J.A.; Juijn, J.A.; Liao, M.; Klomp, L.W.; Lomri, N.; Berger, R.; Scharschmidt, B.F.; et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet. 1998, 18, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Van der Woerd, W.L.; van Mil, S.W.; Stapelbroek, J.M.; Klomp, L.W.; van de Graaf, S.F.; Houwen, R.H. Familial cholestasis: Progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis and intrahepatic cholestasis of pregnancy. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Paulusma, C.C.; Elferink, R.P.; Jansen, P.L. Progressive familial intrahepatic cholestasis type 1. Semin Liver Dis 2010, 30, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Pawlikowska, L.; Strautnieks, S.; Jankowska, I.; Czubkowski, P.; Emerick, K.; Antoniou, A.; Wanty, C.; Fischler, B.; Jacquemin, E.; Wali, S.; et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J. Hepatol. 2010, 53, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapelbroek, J.M.; Peters, T.A.; van Beurden, D.H.; Curfs, J.H.; Joosten, A.; Beynon, A.J.; van Leeuwen, B.M.; van der Velden, L.M.; Bull, L.; Oude Elferink, R.P.; et al. ATP8B1 is essential for maintaining normal hearing. Proc. Natl. Acad. Sci. USA 2009, 106, 9709–9714. [Google Scholar] [CrossRef] [Green Version]
- Whitington, P.F.; Freese, D.K.; Alonso, E.M.; Schwarzenberg, S.J.; Sharp, H.L. Clinical and biochemical findings in progressive familial intrahepatic cholestasis. J. Pediatr. Gastroenterol. Nutr. 1994, 18, 134–141. [Google Scholar] [CrossRef]
- Verhulst, P.M.; van der Velden, L.M.; Oorschot, V.; van Faassen, E.E.; Klumperman, J.; Houwen, R.H.; Pomorski, T.G.; Holthuis, J.C.; Klomp, L.W. A flippase-independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatology 2010, 51, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Van der Mark, V.A.; de Jonge, H.R.; Chang, J.C.; Ho-Mok, K.S.; Duijst, S.; Vidovic, D.; Carlon, M.S.; Oude Elferink, R.P.; Paulusma, C.C. The phospholipid flippase ATP8B1 mediates apical localization of the cystic fibrosis transmembrane regulator. Biochim. Biophys. Acta 2016, 1863, 2280–2288. [Google Scholar] [CrossRef]
- Van der Mark, V.A.; de Waart, D.R.; Ho-Mok, K.S.; Tabbers, M.M.; Voogt, H.W.; Oude Elferink, R.P.; Knisely, A.S.; Paulusma, C.C. The lipid flippase heterodimer ATP8B1-CDC50A is essential for surface expression of the apical sodium-dependent bile acid transporter (SLC10A2/ASBT) in intestinal Caco-2 cells. Biochim. Biophys. Acta 2014, 1842, 2378–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasaka, H.; Yorifuji, T.; Egawa, H.; Yanai, H.; Fujisawa, T.; Kosugiyama, K.; Matsui, A.; Hasegawa, M.; Okada, T.; Takayanagi, M.; et al. Evaluation of risk for atherosclerosis in Alagille syndrome and progressive familial intrahepatic cholestasis: Two congenital cholestatic diseases with different lipoprotein metabolisms. J. Pediatr. 2005, 146, 329–335. [Google Scholar] [CrossRef]
- Nagasaka, H.; Yorifuji, T.; Hirano, K.; Ota, A.; Toyama-Nakagawa, Y.; Takatani, T.; Tsukahara, H.; Kobayashi, K.; Takayanagi, M.; Inomata, Y.; et al. Effects of bezafibrate on dyslipidemia with cholestasis in children with familial intrahepatic cholestasis-1 deficiency manifesting progressive familial intrahepatic cholestasis. Metabolism 2009, 58, 48–54. [Google Scholar] [CrossRef]
- Nagasaka, H.; Chiba, H.; Hui, S.P.; Takikawa, H.; Miida, T.; Takayanagi, M.; Yorifuji, T.; Hasegawa, M.; Ota, A.; Hirano, K.; et al. Depletion of high-density lipoprotein and appearance of triglyceride-rich low-density lipoprotein in a Japanese patient with FIC1 deficiency manifesting benign recurrent intrahepatic cholestasis. J. Pediatr. Gastroenterol. Nutr. 2007, 45, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Toyota, Y.; Yamamura, T.; Miyake, Y.; Yamamoto, A. Low density lipoprotein (LDL) binding affinity for the LDL receptor in hyperlipoproteinemia. Atherosclerosis 1999, 147, 77–86. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Gerbal-Chaloin, S.; Gondeau, C.; Vo, D.N.; Belkahla, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; et al. The PDK1 Inhibitor Dichloroacetate Controls Cholesterol Homeostasis Through the ERK5/MEF2 Pathway. Sci. Rep. 2017, 7, 10654. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Greene, Y.J. Dichloroacetate. Diabetes Care 1992, 15, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.W.; Swift, L.L.; Rabinowitz, D.; Crofford, O.B.; Oates, J.A.; Stacpoole, P.W. Reduction of serum cholesterol in two patients with homozygous familial hypercholesterolemia by dichloroacetate. Atherosclerosis 1979, 33, 285–293. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Moore, G.W.; Kornhauser, D.M. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N. Engl. J. Med. 1978, 298, 526–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.R., Jr.; Ballinger, S.; Dell’Italia, L.; Jianhua, Z.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.C.; Go, S.; Gilglioni, E.H.; Duijst, S.; Panneman, D.M.; Rodenburg, R.J.; Li, H.L.; Huang, H.L.; Levin, L.R.; Buck, J.; et al. Soluble adenylyl cyclase regulates the cytosolic NADH/NAD(+) redox state and the bioenergetic switch between glycolysis and oxidative phosphorylation. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148367. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mookerjee, S.A.; Goncalves, R.L.S.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta 2015, 1847, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Schreurs, M.; Kuipers, F.; van der Leij, F.R. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes. Rev. 2010, 11, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, N.D.; Vance, D.E. Purification of phosphatidylethanolamine N-methyltransferase from rat liver. J. Biol. Chem. 1987, 262, 17231–17239. [Google Scholar] [CrossRef]
- Van der Veen, J.N.; Lingrell, S.; Da Silva, R.P.; Jacobs, R.L.; Vance, D.E. The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice. Diabetes 2014, 63, 2620–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Langer, T. Intramitochondrial phospholipid trafficking. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Acoba, M.G.; Senoo, N.; Claypool, S.M. Phospholipid ebb and flow makes mitochondria go. J. Cell Biol. 2020, 219, e202003131. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Heden, T.D.; Johnson, J.M.; Ferrara, P.J.; Eshima, H.; Verkerke, A.R.P.; Wentzler, E.J.; Siripoksup, P.; Narowski, T.M.; Coleman, C.B.; Lin, C.T.; et al. Mitochondrial PE potentiates respiratory enzymes to amplify skeletal muscle aerobic capacity. Sci. Adv. 2019, 5, eaax8352. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.C.; Luiken, J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Dieudonne, T.; Herrera, S.A.; Laursen, M.J.; Lejeune, M.; Stock, C.; Slimani, K.; Jaxel, C.; Lyons, J.A.; Montigny, C.; Pomorski, T.G.; et al. Autoinhibition and regulation by phosphoinositides of ATP8B1, a human lipid flippase associated with intrahepatic cholestatic disorders. Elife 2022, 11, e75272. [Google Scholar] [CrossRef]
- Paulusma, C.C.; Folmer, D.E.; Ho-Mok, K.S.; de Waart, D.R.; Hilarius, P.M.; Verhoeven, A.J.; Oude Elferink, R.P. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology 2008, 47, 268–278. [Google Scholar] [CrossRef]
- Takatsu, H.; Tanaka, G.; Segawa, K.; Suzuki, J.; Nagata, S.; Nakayama, K.; Shin, H.W. Phospholipid flippase activities and substrate specificities of human type IV P-type ATPases localized to the plasma membrane. J. Biol. Chem. 2014, 289, 33543–33556. [Google Scholar] [CrossRef] [Green Version]
- Shyu, P., Jr.; Ng, B.S.H.; Ho, N.; Chaw, R.; Seah, Y.L.; Marvalim, C.; Thibault, G. Membrane phospholipid alteration causes chronic ER stress through early degradation of homeostatic ER-resident proteins. Sci. Rep. 2019, 9, 8637. [Google Scholar] [CrossRef]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Noyon, C.; Ruysschaert, J.M.; Van Antwerpen, P.; Govaerts, C. Phosphatidylethanolamine Is a Key Regulator of Membrane Fluidity in Eukaryotic Cells. J. Biol. Chem. 2016, 291, 3658–3667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eppens, E.F.; van Mil, S.W.; de Vree, J.M.; Mok, K.S.; Juijn, J.A.; Oude Elferink, R.P.; Berger, R.; Houwen, R.H.; Klomp, L.W. FIC1, the protein affected in two forms of hereditary cholestasis, is localized in the cholangiocyte and the canalicular membrane of the hepatocyte. J. Hepatol. 2001, 35, 436–443. [Google Scholar] [CrossRef]
- Koenderink, J.B.; Geibel, S.; Grabsch, E.; De Pont, J.J.; Bamberg, E.; Friedrich, T. Electrophysiological analysis of the mutated Na,K-ATPase cation binding pocket. J. Biol. Chem. 2003, 278, 51213–51222. [Google Scholar] [CrossRef] [Green Version]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, C.D.; Wong, D.S.; Bozidis, P.; Zhang, A.; Colberg-Poley, A.M. Isolation of Endoplasmic Reticulum, Mitochondria, and Mitochondria-Associated Membrane and Detergent Resistant Membrane Fractions from Transfected Cells and from Human Cytomegalovirus-Infected Primary Fibroblasts. Curr. Protoc. Cell Biol. 2015, 68, 3.27.1–3.27.33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Paulusma, C.C.; Groen, A.; Kunne, C.; Ho-Mok, K.S.; Spijkerboer, A.L.; Rudi de Waart, D.; Hoek, F.J.; Vreeling, H.; Hoeben, K.A.; van Marle, J.; et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology 2006, 44, 195–204. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Mellado, V.E.; Chang, J.-C.; Ho-Mok, K.S.; Bernardino Morcillo, C.; Kersten, R.H.J.; Oude Elferink, R.P.J.; Verhoeven, A.J.; Paulusma, C.C. ATP8B1 Deficiency Results in Elevated Mitochondrial Phosphatidylethanolamine Levels and Increased Mitochondrial Oxidative Phosphorylation in Human Hepatoma Cells. Int. J. Mol. Sci. 2022, 23, 12344. https://doi.org/10.3390/ijms232012344
Gómez-Mellado VE, Chang J-C, Ho-Mok KS, Bernardino Morcillo C, Kersten RHJ, Oude Elferink RPJ, Verhoeven AJ, Paulusma CC. ATP8B1 Deficiency Results in Elevated Mitochondrial Phosphatidylethanolamine Levels and Increased Mitochondrial Oxidative Phosphorylation in Human Hepatoma Cells. International Journal of Molecular Sciences. 2022; 23(20):12344. https://doi.org/10.3390/ijms232012344
Chicago/Turabian StyleGómez-Mellado, Valentina E., Jung-Chin Chang, Kam S. Ho-Mok, Carmen Bernardino Morcillo, Remco H. J. Kersten, Ronald P. J. Oude Elferink, Arthur J. Verhoeven, and Coen C. Paulusma. 2022. "ATP8B1 Deficiency Results in Elevated Mitochondrial Phosphatidylethanolamine Levels and Increased Mitochondrial Oxidative Phosphorylation in Human Hepatoma Cells" International Journal of Molecular Sciences 23, no. 20: 12344. https://doi.org/10.3390/ijms232012344