

The Autism Spectrum Disorder-Associated Bacterial Metabolite p-Cresol Derails the Neuroimmune Response of Microglial Cells Partially via Reduction of ADAM17 and ADAM10
 , ,
, ,
Abstract
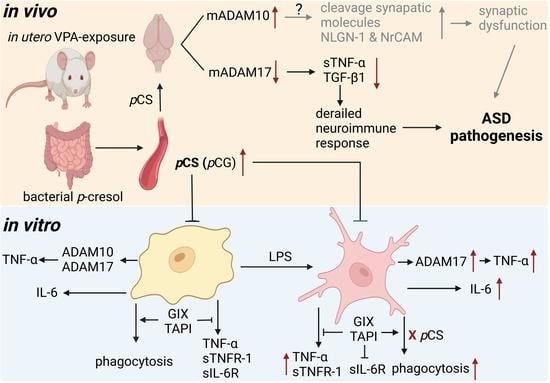
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effect of pCS on the Expression of ADAM10 and ADAM17 of Microglial Cells Constitutively and during LPS-Induced Inflammation
2.2. ADAM10 and ADAM17 Cleave TNF-α, TNFR-1 and IL-6R from BV2 Microglia
2.3. The Effect of ADAM10 and ADAM17 Inhibition on Constitutive and LPS-Induced Phagocytosis by BV2 Cells
2.4. Effect of pCS Exposure on Innate Immune Response of Microglial Cells
2.5. Effect of pCS on Phagocytosis Response of Microglial Cells
3. Discussion
4. Materials and Methods
4.1. BV2 Microglial Cell Culture and Treatments
4.2. BV2 Microglial Cell Viability
4.3. BV2 Microglial ADAM10 and ADAM17 Expression and Inflammatory Response
4.4. Phagocytosis Activity Assay
4.5. Cytokine and Cytokine Receptor ELISAs
4.6. Immunoblotting Brain Tissue and BV2 Microglial Cells
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Roussin, L.; Prince, N.; Perez-Pardo, P.; Kraneveld, A.D.; Rabot, S.; Naudon, L. Role of the gut microbiota in the pathophysiology of autism spectrum disorder: Clinical and preclinical evidence. Microorganisms 2020, 8, 1369. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Marzal, L.N.; Prince, N.; Bajic, D.; Roussin, L.; Naudon, L.; Rabot, S.; Garssen, J.; Kraneveld, A.D.; Perez-Pardo, P. The impact of gut microbiota-derived metabolites in autism spectrum disorders. Int. J. Mol. Sci. 2021, 22, 10052. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, E.; MacFarlane, H.; Salem, A.C. Epidemiological surveys of ASD: Advances and remaining challenges. J. Autism Dev. Disord. 2021, 51, 4271–4290. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Bourson, L.; Prevost, C. Characteristics of restricted interests in girls with ASD compared to boys: A systematic review of the literature. Eur. Child Adolesc. Psychiatry 2022. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Reichow, B.; Hume, K.; Barton, E.E.; Boyd, B.A. Early intensive behavioral intervention (EIBI) for young children with autism spectrum disorders (ASD). Cochrane Database Syst. Rev. 2018, 5, Cd009260. [Google Scholar] [CrossRef] [PubMed]
- Walsh, P.; Elsabbagh, M.; Bolton, P.; Singh, I. In search of biomarkers for autism: Scientific, social and ethical challenges. Nat. Rev. Neurosci. 2011, 12, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Caporale, N.; Testa, G. Autism spectrum disorder at the crossroad between genes and environment: Contributions, convergences, and interactions in ASD developmental pathophysiology. Mol. Autism 2020, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Chaste, P.; Leboyer, M. Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, T.; Colamartino, M.; Fiori, E.; Sacco, R.; Coviello, A.; Ventura, R.; Puglisi-Allegra, S.; Turriziani, L.; Persico, A.M. P-cresol alters brain dopamine metabolism and exacerbates autism-like behaviors in the BTBR mouse. Brain Sci. 2020, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Hertz-Picciotto, I.; Schmidt, R.J.; Krakowiak, P. Understanding environmental contributions to autism: Causal concepts and the state of science. Autism Res. 2018, 11, 554–586. [Google Scholar] [CrossRef] [PubMed]
- de Theije, C.G.; Koelink, P.J.; Korte-Bouws, G.A.; da Silva, S.L.; Korte, S.M.; Olivier, B.; Garssen, J.; Kraneveld, A.D. Intestinal inflammation in a murine model of autism spectrum disorders. Brain Behav. Immun. 2014, 37, 240–247. [Google Scholar] [CrossRef] [PubMed]
- de Theije, C.G.; Wopereis, H.; Ramadan, M.; van Eijndthoven, T.; Lambert, J.; Knol, J.; Garssen, J.; Kraneveld, A.D.; Oozeer, R. Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav. Immun. 2014, 37, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Chaliha, D.; Albrecht, M.; Vaccarezza, M.; Takechi, R.; Lam, V.; Al-Salami, H.; Mamo, J. A systematic review of the valproic-acid-induced rodent model of autism. Dev. Neurosci. 2020, 42, 12–48. [Google Scholar] [CrossRef] [PubMed]
- Roullet, F.I.; Wollaston, L.; Decatanzaro, D.; Foster, J.A. Behavioral and molecular changes in the mouse in response to prenatal exposure to the anti-epileptic drug valproic acid. Neuroscience 2010, 170, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, N.; Seiffe, A.; Campolongo, M.; Zappala, C.; Depino, A.M. Sex-specific effects of prenatal valproic acid exposure on sociability and neuroinflammation: Relevance for susceptibility and resilience in autism. Psychoneuroendocrinology 2019, 110, 104441. [Google Scholar] [CrossRef] [PubMed]
- Davoli-Ferreira, M.; Thomson, C.A.; McCoy, K.D. Microbiota and microglia interactions in ASD. Front. Immunol. 2021, 12, 676255. [Google Scholar] [CrossRef] [PubMed]
- Koyama, R.; Ikegaya, Y. Microglia in the pathogenesis of autism spectrum disorders. Neurosci. Res. 2015, 100, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Azmitia, E.C.; Whitaker-Azmitia, P.M. Developmental microglial priming in postmortem autism spectrum disorder temporal cortex. Brain Behav. Immun. 2017, 62, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.J.; Deshpande, P.; Mehta, P.; Wagner, G.C.; Kusnecov, A.W. The effect of valproic acid exposure throughout development on microglia number in the prefrontal cortex, hippocampus and cerebellum. Neuroscience 2022, 481, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Gąssowska-Dobrowolska, M.; Cieślik, M.; Czapski, G.A.; Jęśko, H.; Frontczak-Baniewicz, M.; Gewartowska, M.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Babiec, L.; et al. Prenatal exposure to valproic acid affects microglia and synaptic ultrastructure in a brain-region-specific manner in young-adult male rats: Relevance to Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3576. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, X.; Jiang, R.; Cai, J.; Lin, Q.; Fan, Z.; Zhao, P.; Wang, S.; Zou, C.; Du, W.; et al. CQMUH-011 inhibits LPS-induced microglia activation and ameliorates brain ischemic injury in mice. Inflammation 2021, 44, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; de Angelis, A.L.H.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Bek, M.K.; Prince, N.Z.; Peralta Marzal, L.N.; Garssen, J.; Perez Pardo, P.; Kraneveld, A.D. The role of bacterial-derived aromatic amino acids metabolites relevant in autism spectrum disorders: A comprehensive review. Front. Neurosci. 2021, 15, 738220. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Duncan, S.H.; Scobbie, L.; Duncan, G.; Cantlay, L.; Calder, A.G.; Anderson, S.E.; Flint, H.J. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol. Nutr. Food Res. 2013, 57, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Sato, T.; Nomoto, K.; Tsuji, H. Identification of phenol- and p-cresol-producing intestinal bacteria by using media supplemented with tyrosine and its metabolites. FEMS Microbiol. Ecol. 2018, 94, fiy125. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Kiang, T.K.L. Characterization of human sulfotransferases catalyzing the formation of p-cresol sulfate and identification of mefenamic acid as a potent metabolism inhibitor and potential therapeutic agent for detoxification. Toxicol. Appl. Pharmacol. 2021, 425, 115553. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, B.S.; Roberts-Thomson, I.C.; Pannall, P.R.; Roediger, W.E. Impaired sulphation of phenol by the colonic mucosa in quiescent and active ulcerative colitis. Gut 1991, 32, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Persico, A.M.; Napolioni, V. Urinary p-cresol in autism spectrum disorder. Neurotoxicol. Teratol. 2013, 36, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Adams, J.B.; Vargason, T.; Santiago, M.; Hahn, J.; Krajmalnik-Brown, R. Distinct fecal and plasma metabolites in children with Autism Spectrum Disorders and their modulation after microbiota transfer therapy. MSphere 2020, 5, e00314-20. [Google Scholar] [CrossRef]
- Gabriele, S.; Sacco, R.; Cerullo, S.; Neri, C.; Urbani, A.; Tripi, G.; Malvy, J.; Barthelemy, C.; Bonnet-Brihault, F.; Persico, A.M. Urinary p-cresol is elevated in young French children with autism spectrum disorder: A replication study. Biomark. Biochem. Indic. Expo. Response Susceptibil. Chem. 2014, 19, 463–470. [Google Scholar]
- Sun, C.Y.; Li, J.R.; Wang, Y.Y.; Lin, S.Y.; Ou, Y.C.; Lin, C.J.; Wang, J.D.; Liao, S.L.; Chen, C.J. p-Cresol sulfate caused behavior disorders and neurodegeneration in mice with unilateral nephrectomy involving oxidative stress and neuroinflammation. Int. J. Mol. Sci. 2020, 21, 6687. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, B.; Księżarczyk, K.; Raćkowska, E.; Szlufik, S.; Koziorowski, D.; Giebułtowicz, J. Higher cerebrospinal fluid to plasma ratio of p-cresol sulfate and indoxyl sulfate in patients with Parkinson’s disease. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 501, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Altieri, L.; Neri, C.; Sacco, R.; Curatolo, P.; Benvenuto, A.; Muratori, F.; Santocchi, E.; Bravaccio, C.; Lenti, C.; Saccani, M.; et al. Urinary p-cresol is elevated in small children with severe autism spectrum disorder. Biomark. Biochem. Indic. Expo. Response Susceptibil. Chem. 2011, 16, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Martin, P.; Becker, J.A.J.; Caramello, N.; Fernandez, S.P.; Costa-Campos, R.; Canaguier, J.; Barbosa, S.; Martinez-Gili, L.; Myridakis, A.; Dumas, M.E.; et al. The microbial metabolite p-Cresol induces autistic-like behaviors in mice by remodeling the gut microbiota. Microbiome 2021, 9, 157. [Google Scholar] [CrossRef]
- Zheng, Y.; Verhoeff, T.A.; Perez Pardo, P.; Garssen, J.; Kraneveld, A.D. The gut-brain axis in Autism Spectrum Disorder: A focus on the metalloproteases ADAM10 and ADAM17. Int. J. Mol. Sci. 2020, 22, 118. [Google Scholar] [CrossRef] [PubMed]
- Malinverno, M.; Carta, M.; Epis, R.; Marcello, E.; Verpelli, C.; Cattabeni, F.; Sala, C.; Mulle, C.; Di Luca, M.; Gardoni, F. Synaptic localization and activity of ADAM10 regulate excitatory synapses through N-cadherin cleavage. J. Neurosci. 2010, 30, 16343–16355. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Ahmed, T.; Wahle, T.; Gardoni, F.; D’Andrea, L.; Pacini, L.; Jacquemont, S.; Tassone, F.; Balschun, D.; Dotti, C.G.; et al. Dysregulated ADAM10-mediated processing of APP during a critical time window leads to synaptic deficits in fragile X syndrome. Neuron 2015, 87, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Sommer, D.; Corstjens, I.; Sanchez, S.; Dooley, D.; Lemmens, S.; Van Broeckhoven, J.; Bogie, J.; Vanmierlo, T.; Vidal, P.M.; Rose-John, S.; et al. ADAM17-deficiency on microglia but not on macrophages promotes phagocytosis and functional recovery after spinal cord injury. Brain Behav. Immun. 2019, 80, 129–145. [Google Scholar] [CrossRef]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef]
- Ray, B.; Sokol, D.K.; Maloney, B.; Lahiri, D.K. Finding novel distinctions between the sAPPα-mediated anabolic biochemical pathways in Autism Spectrum Disorder and fragile X syndrome plasma and brain tissue. Sci. Rep. 2016, 6, 26052. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Makino, I.; Kawakami, K.; Kato, I.; Kobayashi, T.; Kaneko, K. p-Cresyl sulfate suppresses lipopolysaccharide-induced anti-bacterial immune responses in murine macrophages in vitro. Toxicol. Lett. 2016, 245, 24–30. [Google Scholar] [CrossRef]
- Shiba, T.; Kawakami, K.; Sasaki, T.; Makino, I.; Kato, I.; Kobayashi, T.; Uchida, K.; Kaneko, K. Effects of intestinal bacteria-derived p-cresyl sulfate on Th1-type immune response in vivo and in vitro. Toxicol. Appl. Pharmacol. 2014, 274, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Kawata, A.; Ito, S.; Katayama, T.; Fujisawa, S. Inhibitory effects of p-cresol and p-hydroxy anisole dimers on expression of the cyclooxygenase-2 gene and lipopolysaccharide-stimulated activation of nuclear factor-κB in RAW264.7 cells. In Vivo 2014, 28, 719–725. [Google Scholar] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Freimuth, J.; Meyer, D.S.; Lee, M.M.; Tochimoto-Okamoto, A.; Benzinou, M.; Clermont, F.F.; Wu, G.; Roy, R.; Letteboer, T.G.; et al. Genetic variants of Adam17 differentially regulate TGFβ signaling to modify vascular pathology in mice and humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7723–7728. [Google Scholar] [CrossRef]
- Malapeira, J.; Esselens, C.; Bech-Serra, J.J.; Canals, F.; Arribas, J. ADAM17 (TACE) regulates TGFβ signaling through the cleavage of vasorin. Oncogene 2011, 30, 1912–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hikita, A.; Tanaka, N.; Yamane, S.; Ikeda, Y.; Furukawa, H.; Tohma, S.; Suzuki, R.; Tanaka, S.; Mitomi, H.; Fukui, N. Involvement of a disintegrin and metalloproteinase 10 and 17 in shedding of tumor necrosis factor-alpha. Biochem. Cell Biol.—Biochim. Biol. Cell. 2009, 87, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Yan, I.; Schwarz, J.; Lücke, K.; Schumacher, N.; Schumacher, V.; Schmidt, S.; Rabe, B.; Saftig, P.; Donners, M.; Rose-John, S.; et al. ADAM17 controls IL-6 signaling by cleavage of the murine IL-6Rα from the cell surface of leukocytes during inflammatory responses. J. Leukoc. Biol. 2016, 99, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Shahid, S.; Blumberg, B.R.; Suzuki, M.; Bartlett, J.D. ADAM10 is expressed by ameloblasts, cleaves the RELT TNF receptor extracellular domain and facilitates enamel development. Sci. Rep. 2019, 9, 14086. [Google Scholar] [CrossRef] [PubMed]
- Vezzoli, E.; Caron, I.; Talpo, F.; Besusso, D.; Conforti, P.; Battaglia, E.; Sogne, E.; Falqui, A.; Petricca, L.; Verani, M.; et al. Inhibiting pathologically active ADAM10 rescues synaptic and cognitive decline in Huntington’s disease. J. Clin. Investig. 2019, 129, 2390–2403. [Google Scholar] [CrossRef]
- Kwon, J.; Jeong, S.M.; Choi, I.; Kim, N.H. ADAM10 is involved in cell junction assembly in early porcine embryo development. PLoS ONE 2016, 11, e0152921. [Google Scholar] [CrossRef]
- Muliyil, S.; Levet, C.; Düsterhöft, S.; Dulloo, I.; Cowley, S.A.; Freeman, M. ADAM17-triggered TNF signalling protects the ageing Drosophila retina from lipid droplet-mediated degeneration. EMBO J. 2020, 39, e104415. [Google Scholar] [CrossRef]
- Pruessmeyer, J.; Hess, F.M.; Alert, H.; Groth, E.; Pasqualon, T.; Schwarz, N.; Nyamoya, S.; Kollert, J.; van der Vorst, E.; Donners, M.; et al. Leukocytes require ADAM10 but not ADAM17 for their migration and inflammatory recruitment into the alveolar space. Blood 2014, 123, 4077–4088. [Google Scholar] [CrossRef]
- Ardestani, S.; Deskins, D.L.; Young, P.P. Membrane TNF-alpha-activated programmed necrosis is mediated by Ceramide-induced reactive oxygen species. J. Mol. Signal. 2013, 8, 12. [Google Scholar] [CrossRef]
- Richter, C.; Messerschmidt, S.; Holeiter, G.; Tepperink, J.; Osswald, S.; Zappe, A.; Branschädel, M.; Boschert, V.; Mann, D.A.; Scheurich, P.; et al. The tumor necrosis factor receptor stalk regions define responsiveness to soluble versus membrane-bound ligand. Mol. Cell. Biol. 2012, 32, 2515–2529. [Google Scholar] [CrossRef]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeh, H.; Rudolph, N.; Billing, U.; Christen, H.; Streif, S.; Bullinger, E.; Schliemann-Bullinger, M.; Findeisen, R.; Schaper, F.; Huber, H.J.; et al. Response to IL-6 trans- and IL-6 classic signalling is determined by the ratio of the IL-6 receptor α to gp130 expression: Fusing experimental insights and dynamic modelling. Cell Commun. Signal. CCS 2019, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Müllberg, J.; Durie, F.H.; Otten-Evans, C.; Alderson, M.R.; Rose-John, S.; Cosman, D.; Black, R.A.; Mohler, K.M. A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. J. Immunol. 1995, 155, 5198–5205. [Google Scholar]
- Li, Y.Y.; Hsieh, L.L.; Tang, R.P.; Liao, S.K.; Yeh, K.Y. Interleukin-6 (IL-6) released by macrophages induces IL-6 secretion in the human colon cancer HT-29 cell line. Hum. Immunol. 2009, 70, 151–158. [Google Scholar] [CrossRef]
- Seifert, A.; Düsterhöft, S.; Wozniak, J.; Koo, C.Z.; Tomlinson, M.G.; Nuti, E.; Rossello, A.; Cuffaro, D.; Yildiz, D.; Ludwig, A. The metalloproteinase ADAM10 requires its activity to sustain surface expression. Cell. Mol. Life Sci. CMLS 2021, 78, 715–732. [Google Scholar] [CrossRef]
- Folgosa, L.; Zellner, H.B.; El Shikh, M.E.; Conrad, D.H. Disturbed follicular architecture in B cell A disintegrin and metalloproteinase (ADAM)10 knockouts is mediated by compensatory increases in ADAM17 and TNF-α shedding. J. Immunol. 2013, 191, 5951–5958. [Google Scholar] [CrossRef]
- Mezyk-Kopeć, R.; Bzowska, M.; Stalińska, K.; Chełmicki, T.; Podkalicki, M.; Jucha, J.; Kowalczyk, K.; Mak, P.; Bereta, J. Identification of ADAM10 as a major TNF sheddase in ADAM17-deficient fibroblasts. Cytokine 2009, 46, 309–315. [Google Scholar] [CrossRef]
- Azevedo, M.L.; Bonan, N.B.; Dias, G.; Brehm, F.; Steiner, T.M.; Souza, W.M.; Stinghen, A.E.; Barreto, F.C.; Elifio-Esposito, S.; Pecoits-Filho, R.; et al. p-Cresyl sulfate affects the oxidative burst, phagocytosis process, and antigen presentation of monocyte-derived macrophages. Toxicol. Lett. 2016, 263, 1–5. [Google Scholar] [CrossRef]
- Zhan, Y.; Paolicelli, R.C.; Sforazzini, F.; Weinhard, L.; Bolasco, G.; Pagani, F.; Vyssotski, A.L.; Bifone, A.; Gozzi, A.; Ragozzino, D.; et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 2014, 17, 400–406. [Google Scholar] [CrossRef]
- Xu, Z.X.; Kim, G.H.; Tan, J.W.; Riso, A.E.; Sun, Y.; Xu, E.Y.; Liao, G.Y.; Xu, H.; Lee, S.H.; Do, N.Y.; et al. Elevated protein synthesis in microglia causes autism-like synaptic and behavioral aberrations. Nat. Commun. 2020, 11, 1797. [Google Scholar] [CrossRef]
- Elsden, S.R.; Hilton, M.G.; Waller, J.M. The end products of the metabolism of aromatic amino acids by Clostridia. Arch. Microbiol. 1976, 107, 283–288. [Google Scholar] [CrossRef]
- Selmer, T.; Andrei, P.I. p-Hydroxyphenylacetate decarboxylase from Clostridium difficile. A novel glycyl radical enzyme catalysing the formation of p-cresol. Eur. J. Biochem. 2001, 268, 1363–1372. [Google Scholar] [CrossRef]
- Bone, E.; Tamm, A.; Hill, M. The production of urinary phenols by gut bacteria and their possible role in the causation of large bowel cancer. Am. J. Clin. Nutr. 1976, 29, 1448–1454. [Google Scholar] [CrossRef]
- Smith, E.A.; Macfarlane, G.T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: Effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 1996, 81, 288–302. [Google Scholar] [CrossRef]
- Kandeel, W.A.; Meguid, N.A.; Bjørklund, G.; Eid, E.M.; Farid, M.; Mohamed, S.K.; Wakeel, K.E.; Chirumbolo, S.; Elsaeid, A.; Hammad, D.Y. Impact of clostridium bacteria in children with Autism Spectrum Disorder and their anthropometric measurements. J. Mol. Neurosci. MN 2020, 70, 897–907. [Google Scholar] [CrossRef]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef]
- Daneberga, Z.; Nakazawa-Miklasevica, M.; Berga-Svitina, E.; Murmane, D.; Isarova, D.; Cupane, L.; Masinska, M.; Nartisa, I.; Lazdane, A.; Miklasevics, E. Urinary organic acids spectra in children with altered gut microbiota composition and autistic spectrum disorder. Nord. J. Psychiatry 2021, 1–7, Online ahead of print. [Google Scholar] [CrossRef]
- Gevi, F.; Belardo, A.; Zolla, L. A metabolomics approach to investigate urine levels of neurotransmitters and related metabolites in autistic children. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165859. [Google Scholar] [CrossRef]
- Southan, C.; DeWolf, W.E., Jr.; Kruse, L.I. Inactivation of dopamine beta-hydroxylase by p-cresol: Evidence for a second, minor site of covalent modification at tyrosine 357. Biochim. Biophys. Acta 1990, 1037, 256–258. [Google Scholar] [CrossRef]
- Prinzen, C.; Trümbach, D.; Wurst, W.; Endres, K.; Postina, R.; Fahrenholz, F. Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice. BMC Genom. 2009, 10, 66. [Google Scholar] [CrossRef]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia 2020, 68, 1421–1434. [Google Scholar] [CrossRef]
- Arenas, Y.M.; Cabrera-Pastor, A.; Juciute, N.; Mora-Navarro, E.; Felipo, V. Blocking glycine receptors reduces neuroinflammation and restores neurotransmission in cerebellum through ADAM17-TNFR1-NF-κβ pathway. J. Neuroinflamm. 2020, 17, 269. [Google Scholar] [CrossRef]
- Takaki, J.; Fujimori, K.; Miura, M.; Suzuki, T.; Sekino, Y.; Sato, K. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: The ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J. Neuroinflamm. 2012, 9, 275. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Colombo, A.V.; Schusser, B.; Dreymueller, D.; Wetzel, S.; Schepers, U.; Herber, J.; Ludwig, A.; Kremmer, E.; Montag, D.; et al. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. ELife 2016, 5, e12748. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Hick, M.; Herrmann, U.; Weyer, S.W.; Mallm, J.P.; Tschäpe, J.A.; Borgers, M.; Mercken, M.; Roth, F.C.; Draguhn, A.; Slomianka, L.; et al. Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol. 2015, 129, 21–37. [Google Scholar] [CrossRef]
- Tyan, S.H.; Shih, A.Y.; Walsh, J.J.; Maruyama, H.; Sarsoza, F.; Ku, L.; Eggert, S.; Hof, P.R.; Koo, E.H.; Dickstein, D.L. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell. Neurosci. 2012, 51, 43–52. [Google Scholar] [CrossRef]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef]
- Brummer, T.; Müller, S.A.; Pan-Montojo, F.; Yoshida, F.; Fellgiebel, A.; Tomita, T.; Endres, K.; Lichtenthaler, S.F. NrCAM is a marker for substrate-selective activation of ADAM10 in Alzheimer’s disease. EMBO Mol. Med. 2019, 11, e9695. [Google Scholar] [CrossRef]
- Borcel, E.; Palczynska, M.; Krzisch, M.; Dimitrov, M.; Ulrich, G.; Toni, N.; Fraering, P.C. Shedding of neurexin 3β ectodomain by ADAM10 releases a soluble fragment that affects the development of newborn neurons. Sci. Rep. 2016, 6, 39310. [Google Scholar] [CrossRef]
- Groth, E.; Pruessmeyer, J.; Babendreyer, A.; Schumacher, J.; Pasqualon, T.; Dreymueller, D.; Higashiyama, S.; Lorenzen, I.; Grötzinger, J.; Cataldo, D.; et al. Stimulated release and functional activity of surface expressed metalloproteinase ADAM17 in exosomes. Biochim. Biophys. Acta 2016, 1863, 2795–2808. [Google Scholar] [CrossRef]
- Scharfenberg, F.; Helbig, A.; Sammel, M.; Benzel, J.; Schlomann, U.; Peters, F.; Wichert, R.; Bettendorff, M.; Schmidt-Arras, D.; Rose-John, S.; et al. Degradome of soluble ADAM10 and ADAM17 metalloproteases. Cell. Mol. Life Sci. CMLS 2020, 77, 331–350. [Google Scholar] [CrossRef]
- Bertram, A.; Lovric, S.; Engel, A.; Beese, M.; Wyss, K.; Hertel, B.; Park, J.K.; Becker, J.U.; Kegel, J.; Haller, H.; et al. Circulating ADAM17 level reflects disease activity in proteinase-3 ANCA-associated vasculitis. J. Am. Soc. Nephrol. JASN 2015, 26, 2860–2870. [Google Scholar] [CrossRef]
- Sun, Q.; Hampel, H.; Blennow, K.; Lista, S.; Levey, A.; Tang, B.; Li, R.; Shen, Y. Increased plasma TACE activity in subjects with mild cognitive impairment and patients with Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2014, 41, 877–886. [Google Scholar] [CrossRef]
- de Queiroz, T.M.; Lakkappa, N.; Lazartigues, E. ADAM17-mediated shedding of inflammatory cytokines in hypertension. Front. Pharmacol. 2020, 11, 1154. [Google Scholar] [CrossRef]
- Ashwood, P.; Enstrom, A.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.L.; Croen, L.A.; Ozonoff, S.; Pessah, I.N.; Van de Water, J. Decreased transforming growth factor beta1 in autism: A potential link between immune dysregulation and impairment in clinical behavioral outcomes. J. Neuroimmunol. 2008, 204, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Hashimoto, K.; Iwata, Y.; Nakamura, K.; Tsujii, M.; Tsuchiya, K.J.; Sekine, Y.; Suda, S.; Suzuki, K.; Sugihara, G.; et al. Decreased serum levels of transforming growth factor-beta1 in patients with autism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Lucchina, L.; Depino, A.M. Altered peripheral and central inflammatory responses in a mouse model of autism. Autism Res. 2014, 7, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef]
- Zhuang, J.; Wei, Q.; Lin, Z.; Zhou, C. Effects of ADAM10 deletion on Notch-1 signaling pathway and neuronal maintenance in adult mouse brain. Gene 2015, 555, 150–158. [Google Scholar] [CrossRef]
- Bocchini, V.; Mazzolla, R.; Barluzzi, R.; Blasi, E.; Sick, P.; Kettenmann, H. An immortalized cell line expresses properties of activated microglial cells. J. Neurosci. Res. 1992, 31, 616–621. [Google Scholar] [CrossRef]
- Avallone, R.; Lucchi, C.; Puja, G.; Codeluppi, A.; Filaferro, M.; Vitale, G.; Rustichelli, C.; Biagini, G. BV-2 microglial cells respond to rotenone toxic insult by modifying pregnenolone, 5α-dihydroprogesterone and pregnanolone levels. Cells 2020, 9, 2091. [Google Scholar] [CrossRef]
- Günaydin, C.; Çelik, Z.B.; Bilge, S.S.; Avci, B.; Kara, N. SAHA attenuates rotenone-induced toxicity in primary microglia and HT-22 cells. Toxicol. Ind. Health 2021, 37, 23–33. [Google Scholar] [CrossRef]
- Yarmohammadi, F.; Hayes, A.W.; Najafi, N.; Karimi, G. The protective effect of natural compounds against rotenone-induced neurotoxicity. J. Biochem. Mol. Toxicol. 2020, 34, e22605. [Google Scholar] [CrossRef] [PubMed]
- Brummer, T.; Pigoni, M.; Rossello, A.; Wang, H.; Noy, P.J.; Tomlinson, M.G.; Blobel, C.P.; Lichtenthaler, S.F. The metalloprotease ADAM10 (a disintegrin and metalloprotease 10) undergoes rapid, postlysis autocatalytic degradation. FASEB J. 2018, 32, 3560–3573. [Google Scholar] [CrossRef] [PubMed]
- Herzog, C.; Haun, R.S.; Ludwig, A.; Shah, S.V.; Kaushal, G.P. ADAM10 is the major sheddase responsible for the release of membrane-associated meprin A. J. Biol. Chem. 2014, 289, 13308–13322. [Google Scholar] [CrossRef] [PubMed]
- Pigoni, M.; Hsia, H.E.; Hartmann, J.; Rudan Njavro, J.; Shmueli, M.D.; Müller, S.A.; Güner, G.; Tüshaus, J.; Kuhn, P.H.; Kumar, R.; et al. Seizure protein 6 controls glycosylation and trafficking of kainate receptor subunits GluK2 and GluK3. EMBO J. 2020, 39, e103457. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Prince, N.Z.; Peralta Marzal, L.N.; Ahmed, S.; Garssen, J.; Perez Pardo, P.; Kraneveld, A.D. The Autism Spectrum Disorder-Associated Bacterial Metabolite p-Cresol Derails the Neuroimmune Response of Microglial Cells Partially via Reduction of ADAM17 and ADAM10. Int. J. Mol. Sci. 2022, 23, 11013. https://doi.org/10.3390/ijms231911013
Zheng Y, Prince NZ, Peralta Marzal LN, Ahmed S, Garssen J, Perez Pardo P, Kraneveld AD. The Autism Spectrum Disorder-Associated Bacterial Metabolite p-Cresol Derails the Neuroimmune Response of Microglial Cells Partially via Reduction of ADAM17 and ADAM10. International Journal of Molecular Sciences. 2022; 23(19):11013. https://doi.org/10.3390/ijms231911013
Chicago/Turabian StyleZheng, Yuanpeng, Naika Z. Prince, Lucia N. Peralta Marzal, Sabbir Ahmed, Johan Garssen, Paula Perez Pardo, and Aletta D. Kraneveld. 2022. "The Autism Spectrum Disorder-Associated Bacterial Metabolite p-Cresol Derails the Neuroimmune Response of Microglial Cells Partially via Reduction of ADAM17 and ADAM10" International Journal of Molecular Sciences 23, no. 19: 11013. https://doi.org/10.3390/ijms231911013