
Effect of Chronic Treatment with Uridine on Cardiac Mitochondrial Dysfunction in the C57BL/6 Mouse Model of High-Fat Diet–Streptozotocin-Induced Diabetes

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
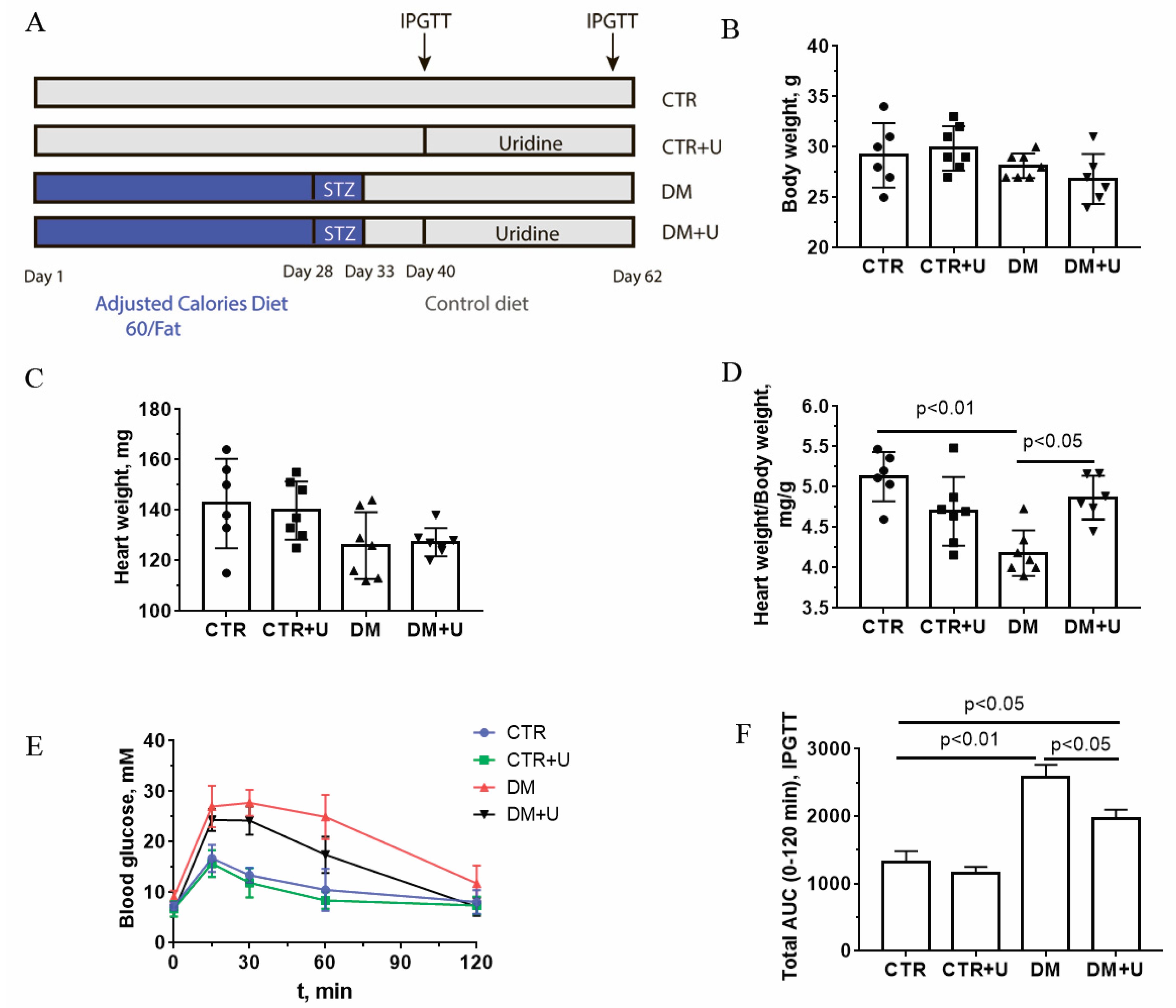
2.1. Effect of Uridine on Somatic and Biochemical Indices of C57BL/6 Mice
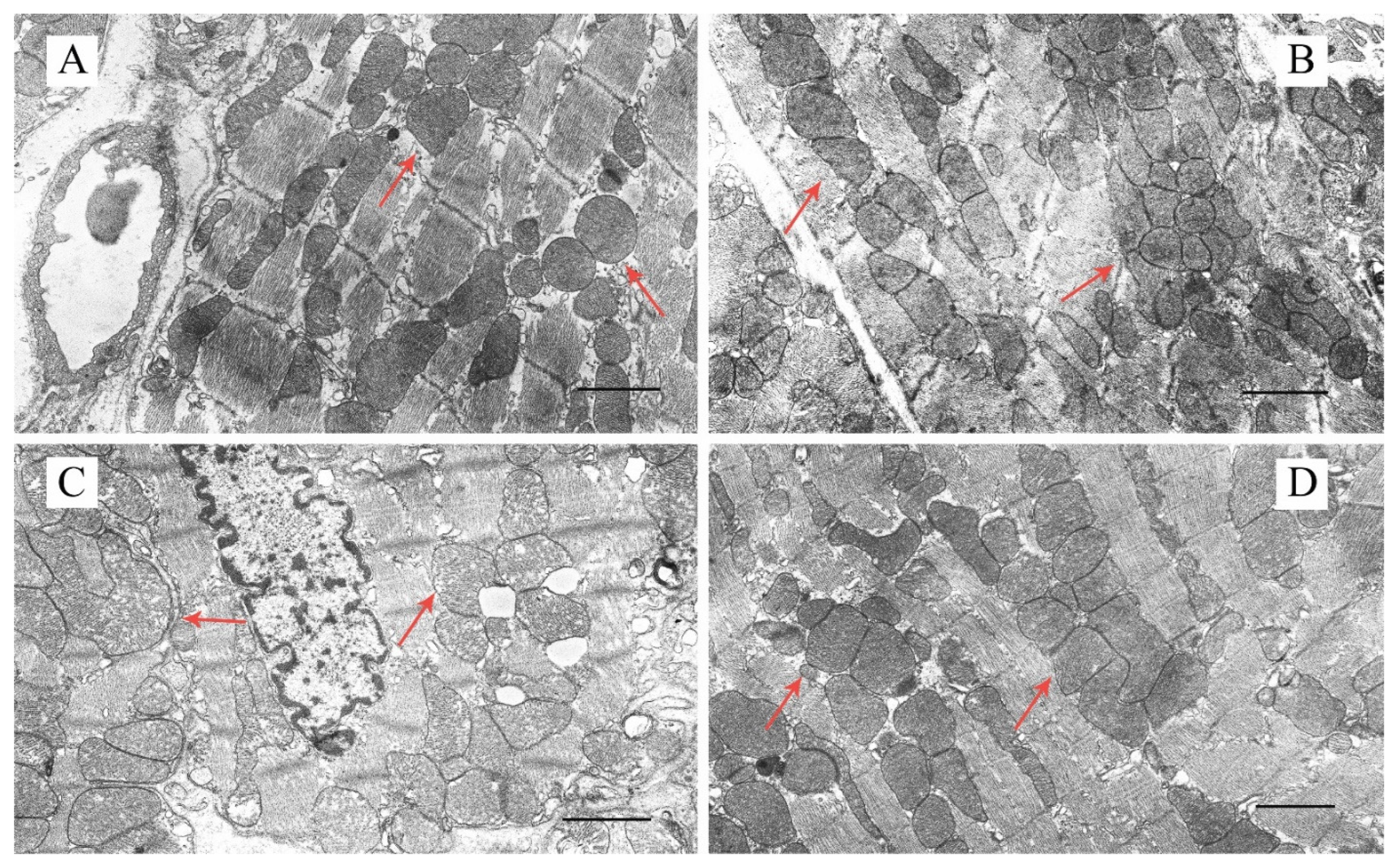
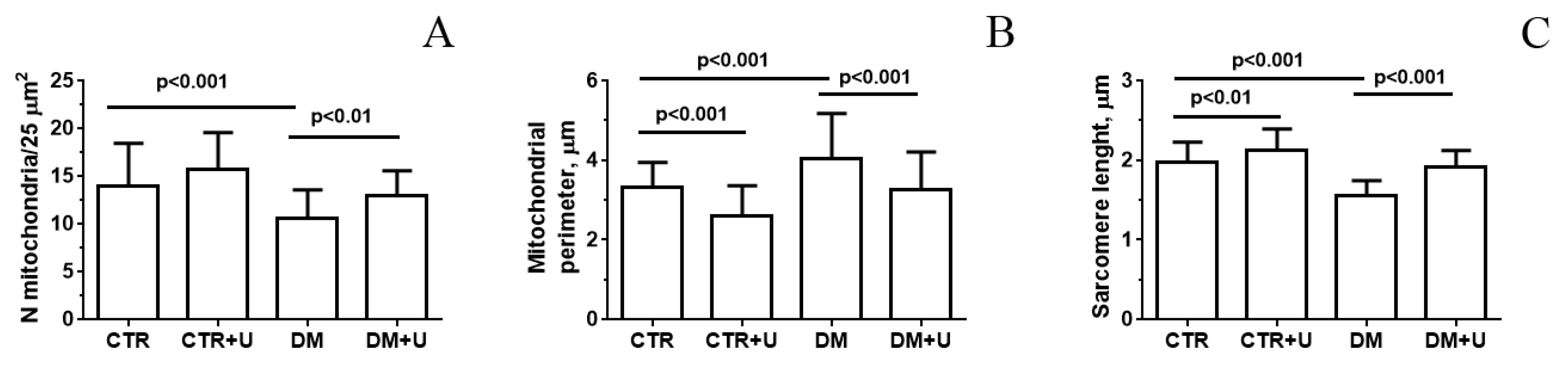
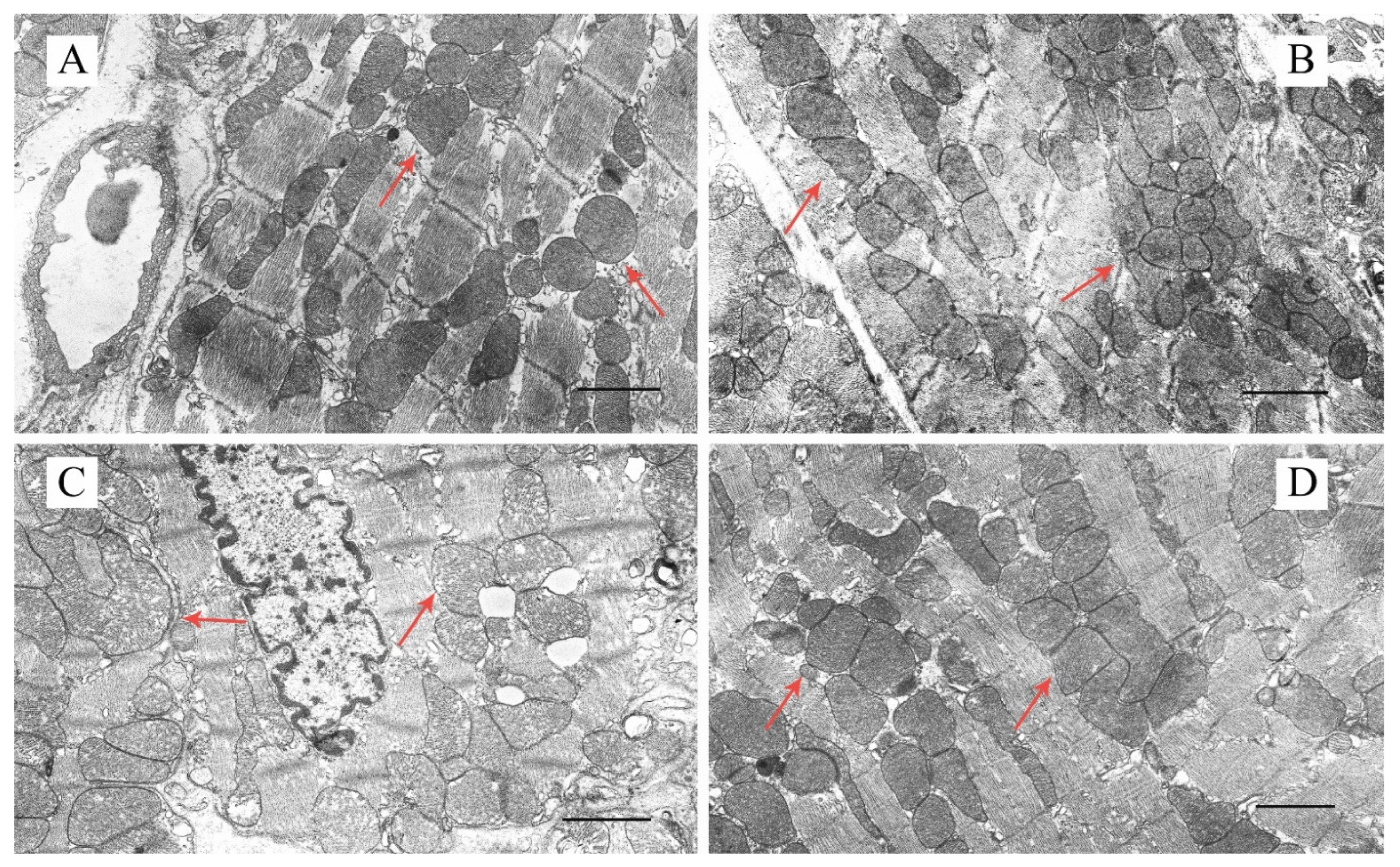
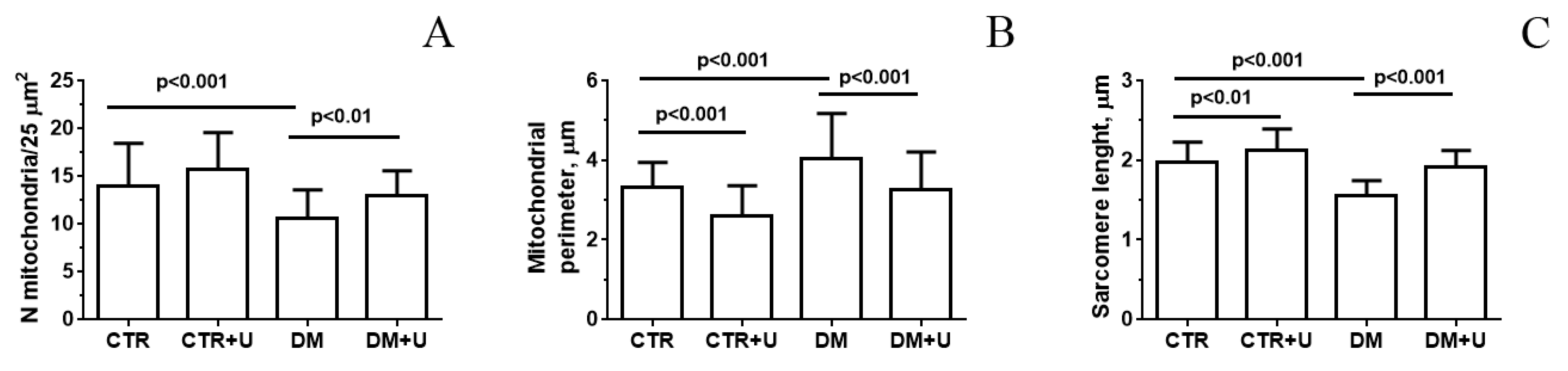
2.2. Effect of Uridine on the Ultrastructure of Heart Mitochondria in DM Mice
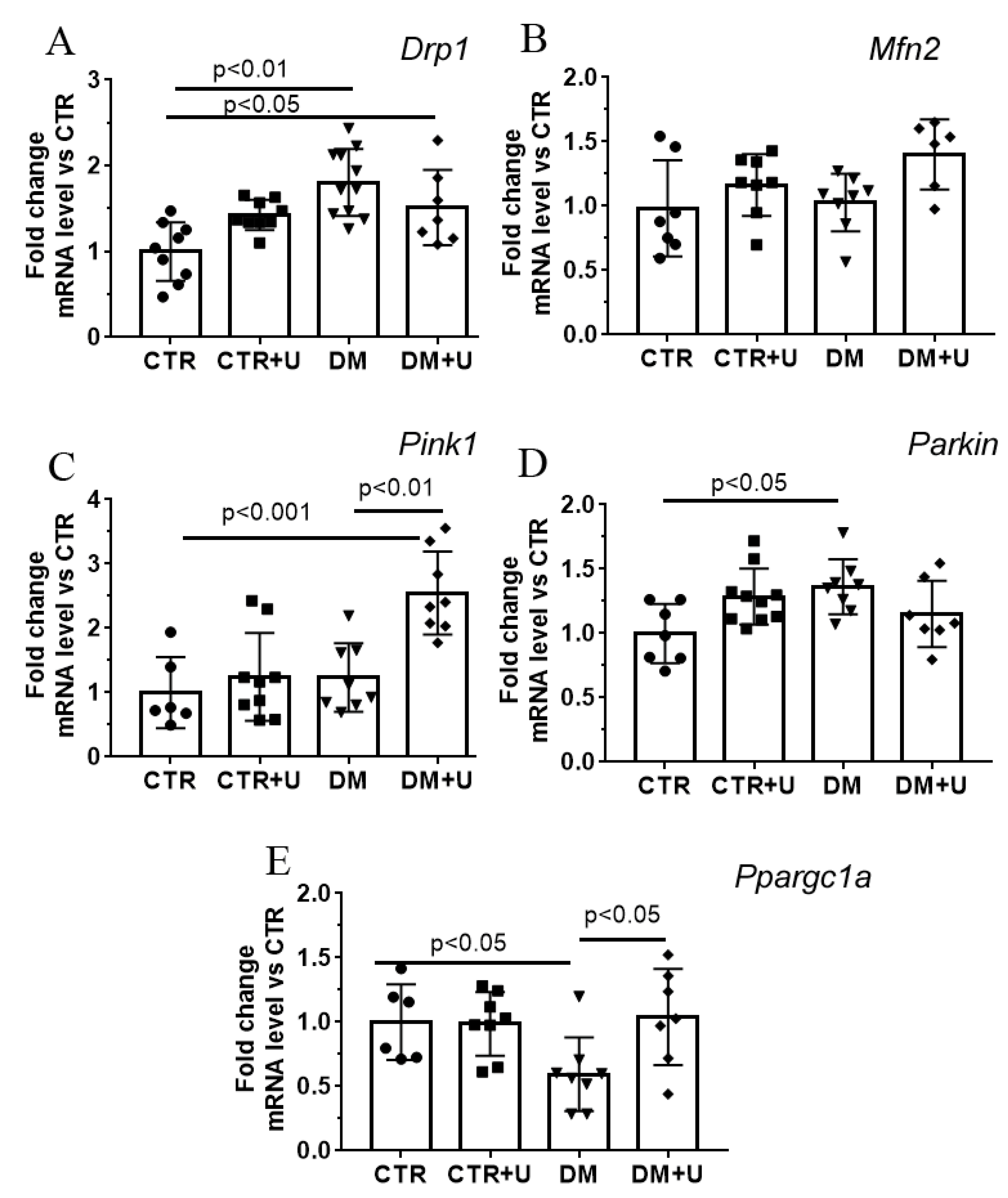
2.3. Effect of Uridine on DM-Induced Changes in the mRNA Expression of Proteins Responsible for Mitochondrial Quality Control
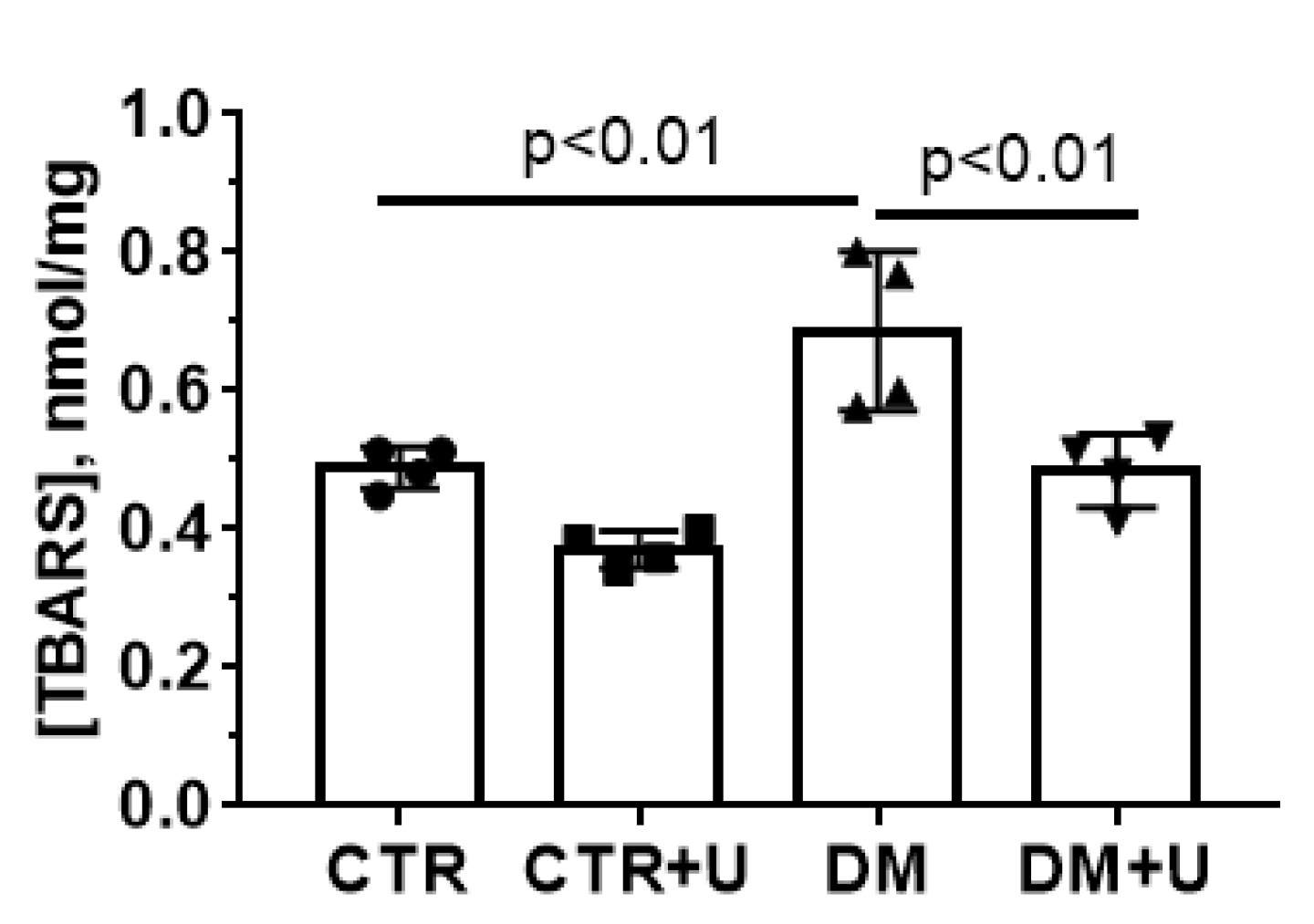
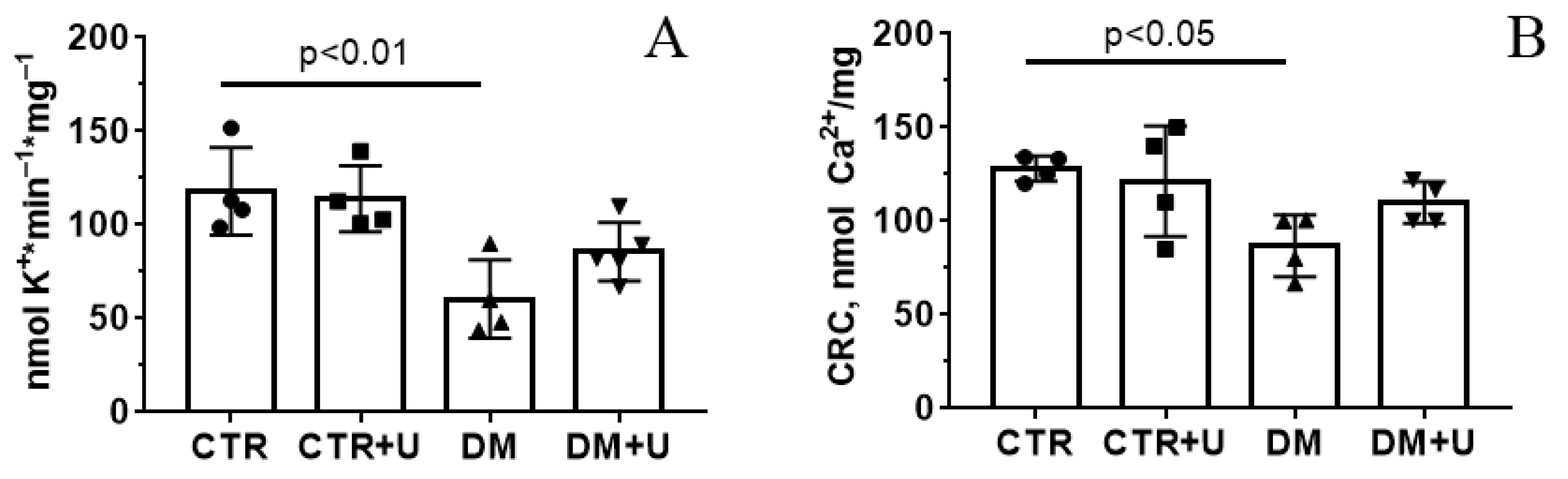
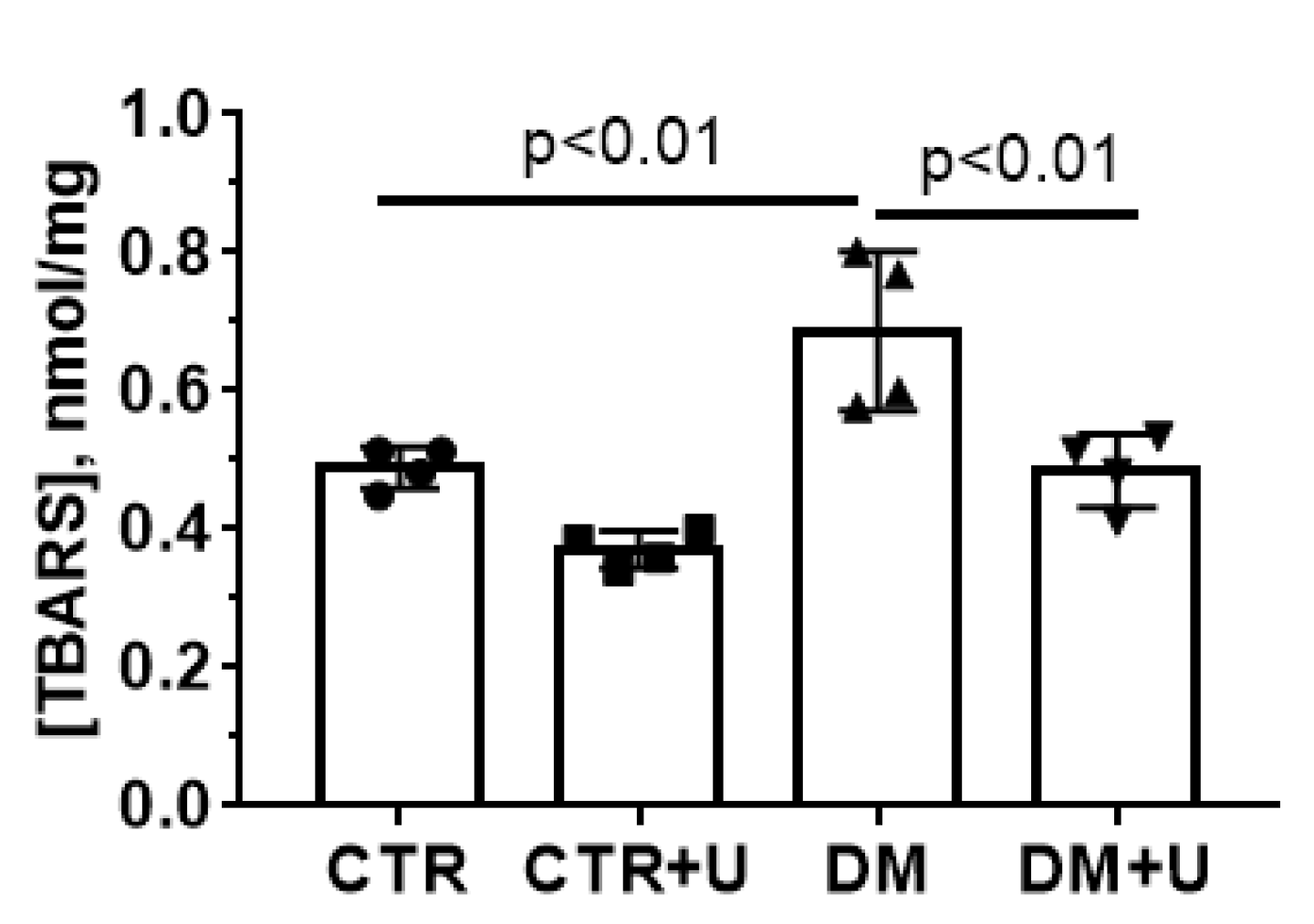
2.4. Effects of Uridine on DM-Induced Changes in the Functioning of Heart Mitochondria
3. Discussion
4. Materials and Methods
4.1. Experimental Animals, Induction and Validation of Diabetes
4.2. Transmission Electron Microscopy
4.3. RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR
4.4. Isolation of Heart Mitochondria and Assessment of Mitochondrial Functions
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Diabetes Association Professional Practice Committee 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45, S17–S38. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021. [Google Scholar]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, W.H. Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Gulsin, G.S.; Athithan, L.; McCann, G.P. Diabetic cardiomyopathy: Prevalence, determinants and potential treatments. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018819834869. [Google Scholar] [CrossRef] [PubMed]
- Gollmer, J.; Zirlik, A.; Bugger, H. Mitochondrial Mechanisms in Diabetic Cardiomyopathy. Diabetes Metab. J. 2020, 44, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Makrecka-Kuka, M.; Liepinsh, E.; Murray, A.; Lemieux, H.; Dambrova, M.; Tepp, K.; Puurand, M.; Käämbre, T.; Han, W.H.; De Goede, P.; et al. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. 2019, 228, e13430. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, S.; Xie, C.; Fang, J. Uridine Metabolism and Its Role in Glucose, Lipid, and Amino Acid Homeostasis. Biomed. Res. Int. 2020, 2020, 7091718. [Google Scholar] [CrossRef]
- Connolly, G.P.; Duley, J.A. Uridine and its nucleotides: Biological actions, therapeutic potentials. Trends Pharmacol. Sci. 1999, 20, 218–225. [Google Scholar] [CrossRef]
- Yamamoto, T.; Koyama, H.; Kurajoh, M.; Shoji, T.; Tsutsumi, Z.; Moriwaki, Y. Biochemistry of uridine in plasma. Clin. Chim. Acta 2011, 412, 1712–1724. [Google Scholar] [CrossRef]
- Le, T.T.; Ziemba, A.; Urasaki, Y.; Hayes, E.; Brotman, S.; Pizzorno, G. Disruption of uridine homeostasis links liver pyrimidine metabolism to lipid accumulation. J. Lipid. Res. 2013, 54, 1044–1057. [Google Scholar] [CrossRef] [PubMed]
- Lebrecht, D.; Vargas-Infante, Y.A.; Setzer, B.; Kirschner, J.; Walker, U.A. Uridine supplementation antagonizes zalcitabine-induced microvesicular steatohepatitis in mice. Hepatology 2007, 45, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Urasaki, Y.; Pizzorno, G. Uridine prevents fenofibrate-induced fatty liver. PLoS ONE 2014, 9, e87179. [Google Scholar] [CrossRef]
- Le, T.T.; Urasaki, Y.; Pizzorno, G. Uridine prevents tamoxifen-induced liver lipid droplet accumulation. BMC Pharmacol. Toxicol. 2014, 23, 15–27. [Google Scholar] [CrossRef]
- Bul’on, V.V.; Krylova, I.B.; Selina, E.N.; Rodionova, O.M.; Evdokimova, N.R.; Sapronov, N.S.; Mironova, G.D. Antiarrhythmic effect of uridine and uridine-5’-monophosphate in acute myocardial ischemia. Bull. Exp. Biol. Med. 2014, 157, 728–731. [Google Scholar] [CrossRef]
- Krylova, I.B.; Bulion, V.V.; Selina, E.N.; Mironova, G.D.; Sapronov, N.S. Effect of uridine on energy metabolism, LPO, and antioxidant system in the myocardium under conditions of acute coronary insufficiency. Bull. Exp. Biol. Med. 2012, 153, 644–646. [Google Scholar] [CrossRef]
- Krylova, I.B.; Kachaeva, E.V.; Rodionova, O.M.; Negoda, A.E.; Evdokimova, N.R.; Balina, M.I.; Sapronov, N.S.; Mironova, G.D. The cardioprotective effect of uridine and uridine-5’-monophosphate: The role of the mitochondrial ATP-dependent potassium channel. Exp. Gerontol. 2006, 41, 697–703. [Google Scholar] [CrossRef]
- Krylova, I.B.; Selina, E.N.; Bulion, V.V.; Rodionova, O.M.; Evdokimova, N.R.; Belosludtseva, N.V.; Shigaeva, M.I.; Mironova, G.D. Uridine treatment prevents myocardial injury in rat models of acute ischemia and ischemia/reperfusion by activating the mitochondrial ATP-dependent potassium channel. Sci. Rep. 2021, 11, 16999. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, Z.V.; Gordillo, R.; An, Y.; Zhang, C.; Liang, Q.; Yoshino, J.; Cautivo, K.M.; De Brabander, J.; Elmquist, J.K.; et al. An adipo-biliary-uridine axis that regulates energy homeostasis. Science 2017, 355, eaaf5375. [Google Scholar] [CrossRef]
- Urasaki, Y.; Pizzorno, G.; Le, T.T. Uridine affects liver protein glycosylation, insulin signaling, and heme biosynthesis. PLoS ONE 2014, 9, e99728. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, C.; Zhai, Z.; Deng, Z.Y.; De Jonge, H.R.; Wu, X.; Ruan, Z. Uridine attenuates obesity, ameliorates hepatic lipid accumulation and modifies the gut microbiota composition in mice fed with a high-fat diet. Food Funct. 2021, 12, 1829–1840. [Google Scholar] [CrossRef] [PubMed]
- Gallai, V.; Mazzotta, G.; Montesi, S.; Sarchielli, P.; Del Gatto, F. Effects of uridine in the treatment of diabetic neuropathy: An electrophysiological study. Acta Neurol. Scand. 1992, 86, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Urasaki, Y.; Pizzorno, G.; Le, T.T. Chronic Uridine Administration Induces Fatty Liver and Pre-Diabetic Conditions in Mice. PLoS ONE 2016, 11, e0146994. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Inokuchi, T.; Ka, T.; Yamamoto, A.; Takahashi, S.; Tsutsumi, Z.; Tamada, D.; Okuda, C.; Moriwaki, Y. Relationship between plasma uridine and insulin resistance in patients with non-insulin-dependent diabetes mellitus. Nucleosides Nucleotides Nucleic Acids 2010, 29, 504–508. [Google Scholar] [CrossRef]
- Dudzinska, W. Uridine correlates with the concentration of fructosamine and HbA1c in children with type 1 diabetes. Acta Paediatr. 2011, 100, 712–716. [Google Scholar] [CrossRef]
- Dai, W.; Lu, H.; Chen, Y.; Yang, D.; Sun, L.; He, L. The Loss of Mitochondrial Quality Control in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2021, 9, 706832. [Google Scholar] [CrossRef]
- Fujimaki, S.; Kuwabara, T. Diabetes-induced dysfunction of mitochondria and stem cells in skeletal muscle and the nervous system. Int. J. Mol. Sci. 2017, 18, E2147. [Google Scholar] [CrossRef]
- Taddeo, E.P.; Laker, R.C.; Breen, D.S.; Akhtar, Y.N.; Kenwood, B.M.; Liao, J.A.; Zhang, M.; Fazakerley, D.J.; Tomsig, J.L.; Harris, T.E.; et al. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol. Metab. 2013, 3, 124–134. [Google Scholar] [CrossRef]
- Benton, C.R.; Holloway, G.P.; Han, X.X.; Yoshida, Y.; Snook, L.A.; Lally, J.; Glatz, J.F.; Luiken, J.J.; Chabowski, A.; Bonen, A. Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1alpha) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia 2010, 53, 2008–2019. [Google Scholar] [CrossRef]
- Mironova, G.D.; Negoda, A.E.; Marinov, B.S.; Paucek, P.; Costa, A.D.; Grigoriev, S.M.; Skarga, Y.Y.; Garlid, K.D. Functional distinctions between the mitochondrial ATP-dependent K+ channel (mitoKATP) and its inward rectifier subunit (mitoKIR). J. Biol. Chem. 2004, 279, 32562–32568. [Google Scholar] [CrossRef] [Green Version]
- Baranova, O.V.; Skarga, Y.Y.; Negoda, A.E.; Mironova, G.D. Inhibition of 2,4-dinitrophenol-induced potassium efflux by adenine nucleotides in mitochondria. Biochemistry 2000, 65, 218–222. [Google Scholar] [PubMed]
- Oliveira, P.J.; Seiça, R.; Coxito, P.M.; Rolo, A.P.; Palmeira, C.; Santos, M.S.; Moreno, A.J. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003, 554, 511–514. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Starinets, V.S.; Mikheeva, I.B.; Serov, D.A.; Astashev, M.E.; Belosludtsev, M.N.; Dubinin, M.V.; Belosludtsev, K.N. Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice. Biology 2021, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Riojas-Hernández, A.; Bernal-Ramírez, J.; Rodríguez-Mier, D.; Marroquin, F.E.M.; Domínguez-Barragán, E.M.; Borja-Villa, C.; Rivera-Álvarez, I.; García-Rivas, G.; Altamirano, J.; García, N. Enhanced oxidative stress sensitizes the mitochondrial permeability transition pore to opening in heart from Zucker Fa/fa rats with type 2 diabetes. Life Sci. 2015, 141, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Guzmán Mentesana, G.; Báez, A.L.; Lo Presti, M.S.; Domínguez, R.; Córdoba, R.; Bazán, C.; Strauss, M.; Fretes, R.; Rivarola, H.W.; Paglini-Oliva, P. Functional and structural alterations of cardiac and skeletal muscle mitochondria in heart failure patients. Arch. Med. Res. 2014, 45, 237–246. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: Ischemia—reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 2001, 33, 1065–1089. [Google Scholar] [CrossRef]
- Chen, M.; Angeli, F.S.; Shen, Y.T.; Shannon, R.P. GLP-1 (7-36) amide restores myocardial insulin sensitivity and prevents the progression of heart failure in senescent beagles. Cardiovasc. Diabetol. 2014, 13, 115. [Google Scholar] [CrossRef]
- Williamson, C.L.; Dabkowski, E.R.; Baseler, W.A.; Croston, T.L.; Always, S.E.; Hollander, J.M. Enhanced apoptotic propensity in diabetic cardiac mitochondria: Influence of subcellular spatial location. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H633–H642. [Google Scholar] [CrossRef]
- Lu, X.; Thai, P.N.; Lu, S.; Pu, J.; Bers, D.M. Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes. J. Mol. Cell. Cardiol. 2019, 136, 72–84. [Google Scholar] [CrossRef]
- Fannin, S.W.; Lesnefsky, E.J.; Slabe, T.J.; Hassan, M.O.; Hoppel, C.L. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch. Biochem. Biophys. 1999, 372, 399–407. [Google Scholar] [CrossRef]
- Rajab, B.S.; Kassab, S.; Stonall, C.D.; Daghistani, H.; Gibbons, S.; Mamas, M.; Smith, D.; Mironov, A.; AlBalawi, Z.; Zhang, Y.H.; et al. Differential remodelling of mitochondrial subpopulations and mitochondrial dysfunction are a feature of early stage diabetes. Sci. Rep. 2022, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Croston, T.L.; Thapa, D.; Holden, A.A.; Tveter, K.J.; Lewis, S.E.; Shepherd, D.L.; Nichols, C.E.; Long, D.M.; Olfert, I.M.; Jagannathan, R.; et al. Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H54–H65. [Google Scholar] [CrossRef] [PubMed]
- Paucek, P.; Mironova, G.; Mahdi, F.; Beavis, A.D.; Woldegiorgis, G.; Garlid, K.D. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. J. Biol. Chem. 1992, 267, 26062–26069. [Google Scholar] [CrossRef] [PubMed]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabò, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtseva, N.V. Alisporivir Treatment Alleviates Mitochondrial Dysfunction in the Skeletal Muscles of C57BL/6NCrl Mice with High-Fat Diet/Streptozotocin-Induced Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 9524. [Google Scholar] [CrossRef] [PubMed]
- Starinets, V.S.; Serov, D.A.; Penkov, N.V.; Belosludtseva, N.V.; Dubinin, M.V.; Belosludtsev, K.N. Alisporivir Normalizes Mitochondrial Function of Primary Mouse Lung Endothelial Cells Under Conditions of Hyperglycemia. Biochemistry 2022, 87, 605–616. [Google Scholar] [CrossRef]
- Monaco, C.; Hughes, M.C.; Ramos, S.V.; Varah, N.E.; Lamberz, C.; Rahman, F.A.; McGlory, C.; Tarnopolsky, M.A.; Krause, M.P.; Laham, R.; et al. Altered mitochondrial bioenergetics and ultrastructure in the skeletal muscle of young adults with type 1 diabetes. Diabetologia 2018, 61, 1411–1423. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.; Belosludtseva, N.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Starinets, V.S.; Pavlik, L.L.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtsev, K.N. The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes. Biology 2020, 9, 309. [Google Scholar] [CrossRef]
- Gilbert, E.R.; Fu, Z.; Liu, D. Development of a nongenetic mouse model of type 2 diabetes. Exp. Diabetes Res. 2011, 2011, 416254. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Ghosh, S.K.; Choudhury, Y. A murine model of type 2 diabetes mellitus developed using a combination of high fat diet and multiple low doses of streptozotocin treatment mimics the metabolic characteristics of type 2 diabetes mellitus in humans. J. Pharmacol. Toxicol. Methods 2017, 84, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Kosareva, E.A.; Talanov, E.Y.; Gudkov, S.V.; Dubinin, M.V. Itaconic acid impairs the mitochondrial function by the inhibition of complexes II and IV and induction of the permeability transition pore opening in rat liver mitochondria. Biochimie 2020, 176, 150–157. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTR | CTR + U | DM | DM + U | |
|---|---|---|---|---|
| BG (fed state), mM | 10.1 ± 0.2 | 9.2 ± 0.7 | 14.8 ± 0.6 * | 11.3 ± 1.5 # |
| Triglycerides, mM | 1.56 ± 0.05 | 1.63 ± 0.15 | 2.18 ± 0.24 * | 1.34 ± 0.25 # |
| Insulin, μIU/mL | 14.0 ± 0.2 | 14.7 ± 0.3 | 15.9 ± 0.3 * | 15.0 ± 0.4 |
| Group | V Respiration, nmol O2 * min−1 * mg−1 Protein | RCR | |||
|---|---|---|---|---|---|
| State 2 | State 3 | State 4 | State 3UDNP | ||
| CTR | 18.3 ± 1.7 | 45.9 ± 1.9 | 18.6 ± 0.6 | 43.9 ± 3.8 | 2.5 ± 0.1 |
| CTR + U | 15.5 ± 0.7 | 41.8 ± 1.3 | 15.1 ± 1.3 | 41.7 ± 1.9 | 2.8 ± 0.2 |
| DM | 13.5 ± 0.9 | 29.6 ± 0.4 * | 15.4 ± 0.3 | 28.9 ± 1.5 * | 1.9 ± 0.1 * |
| DM + U | 16.8 ± 1.1 | 36.3 ± 1.1 *# | 14.0 ± 0.4 * | 35.0 ± 1.1 * | 2.6 ± 0.1 # |
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Pink1 | TTGCCCCACACCCTAACATC | GCAGGGTACAGGGGTAGTTCT |
| Parkin | AGCCAGAGGTCCAGCAGTTA | GAGGGTTGCTTGTTTGCAGG |
| Drp1 | TTACAGCACACAGGAATTGT | TTGTCACGGGCAACCTTTTA |
| Mfn2 | CACGCTGATGCAGACGGAGAA | ATCCCAGCGGTTGTTCAGG |
| Ppargc1a | CTGCCATTGTTAAGACCGAG | GTGTGAGGAGGGTCATCGTT |
| Rplp2 | CGGCTCAACAAGGTCATCAGTGA | AGCAGAAACAGCCACAGCCCCAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosludtseva, N.V.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, M.N.; Dubinin, M.V.; Mironova, G.D.; Belosludtsev, K.N. Effect of Chronic Treatment with Uridine on Cardiac Mitochondrial Dysfunction in the C57BL/6 Mouse Model of High-Fat Diet–Streptozotocin-Induced Diabetes. Int. J. Mol. Sci. 2022, 23, 10633. https://doi.org/10.3390/ijms231810633
Belosludtseva NV, Starinets VS, Mikheeva IB, Belosludtsev MN, Dubinin MV, Mironova GD, Belosludtsev KN. Effect of Chronic Treatment with Uridine on Cardiac Mitochondrial Dysfunction in the C57BL/6 Mouse Model of High-Fat Diet–Streptozotocin-Induced Diabetes. International Journal of Molecular Sciences. 2022; 23(18):10633. https://doi.org/10.3390/ijms231810633
Chicago/Turabian StyleBelosludtseva, Natalia V., Vlada S. Starinets, Irina B. Mikheeva, Maxim N. Belosludtsev, Mikhail V. Dubinin, Galina D. Mironova, and Konstantin N. Belosludtsev. 2022. "Effect of Chronic Treatment with Uridine on Cardiac Mitochondrial Dysfunction in the C57BL/6 Mouse Model of High-Fat Diet–Streptozotocin-Induced Diabetes" International Journal of Molecular Sciences 23, no. 18: 10633. https://doi.org/10.3390/ijms231810633