
Ligand-Based Virtual Screening and Molecular Docking of Benzimidazoles as Potential Inhibitors of Triosephosphate Isomerase Identified New Trypanocidal Agents
 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
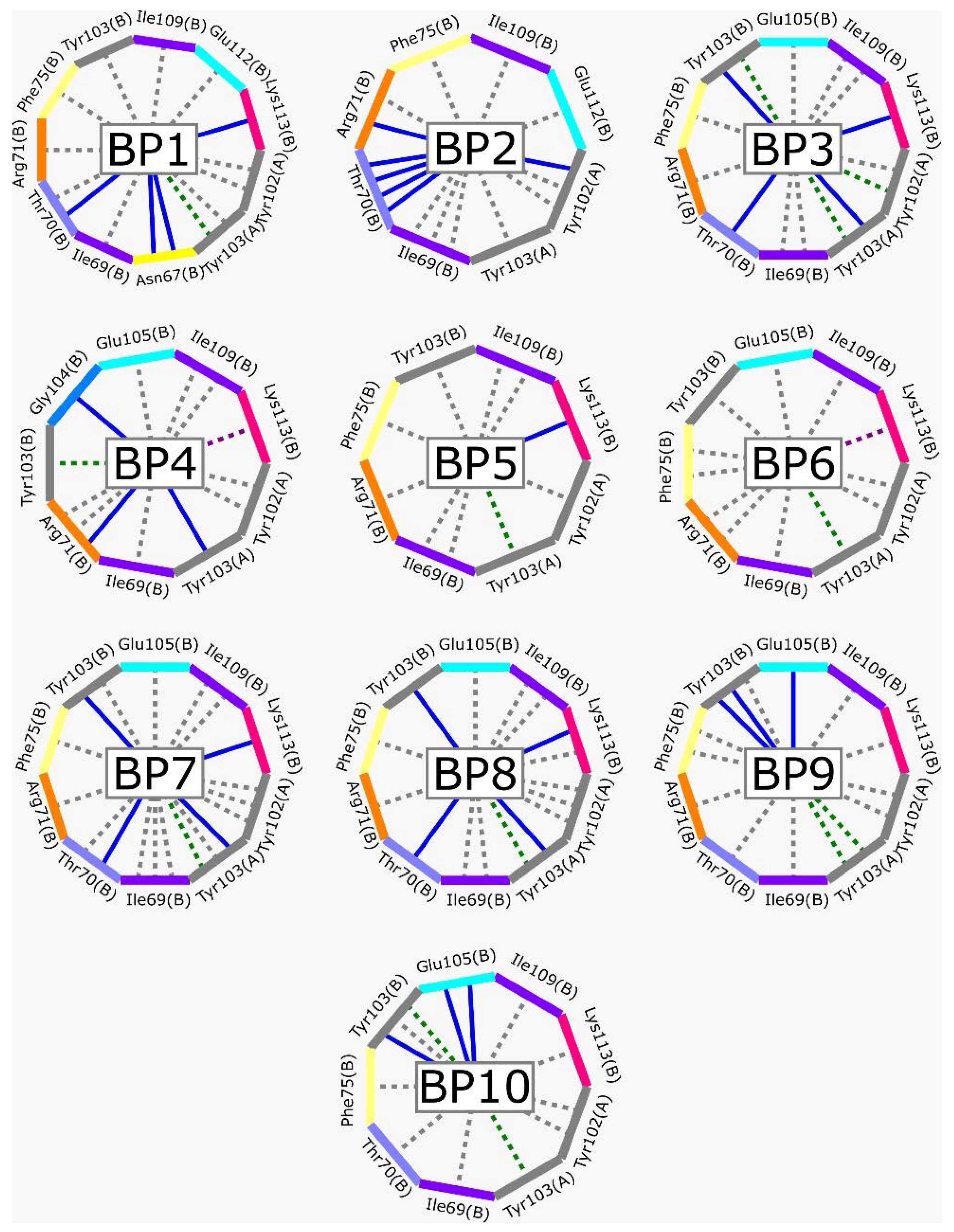
2.1. TcTIM Inhibitors Analysis
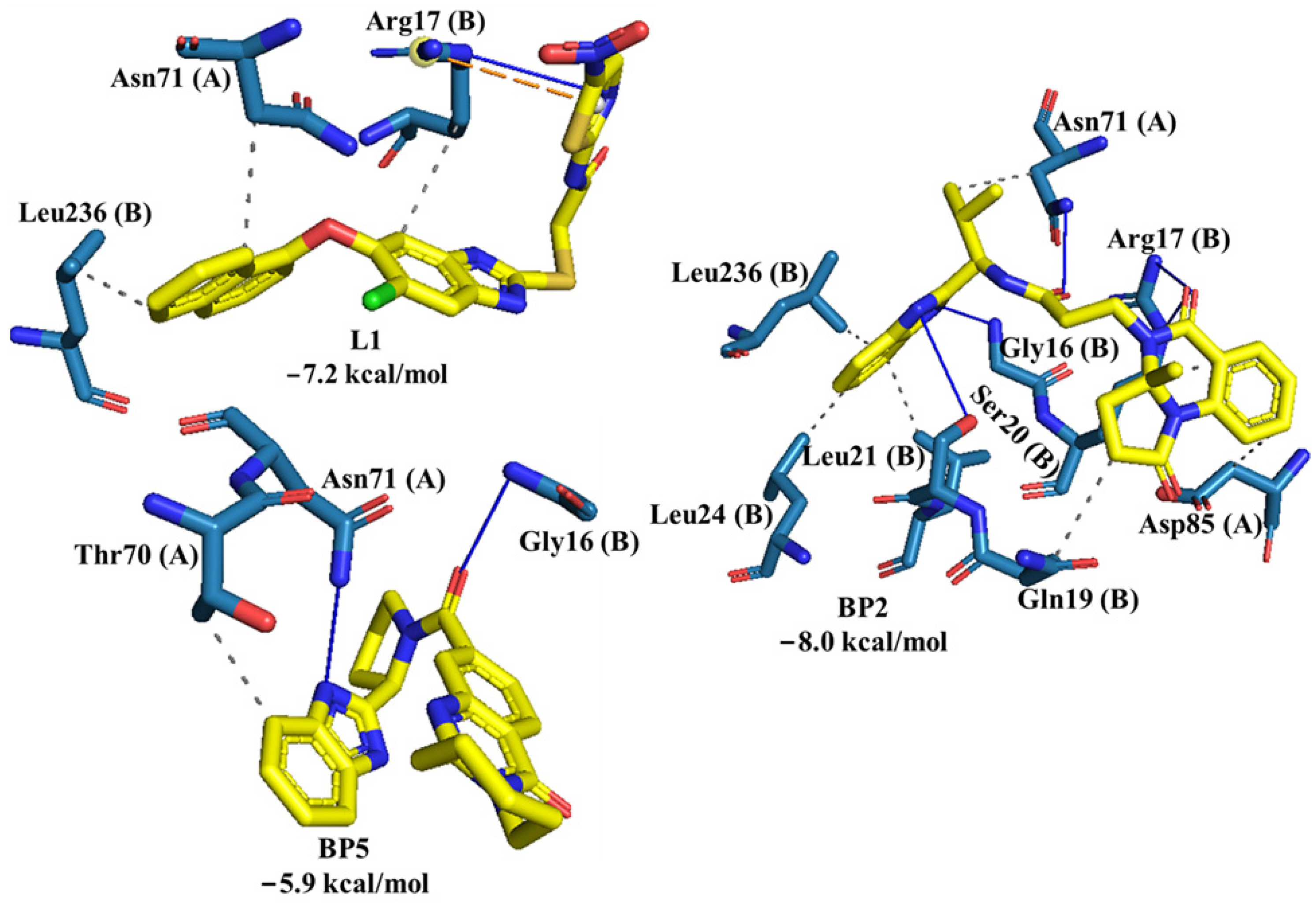
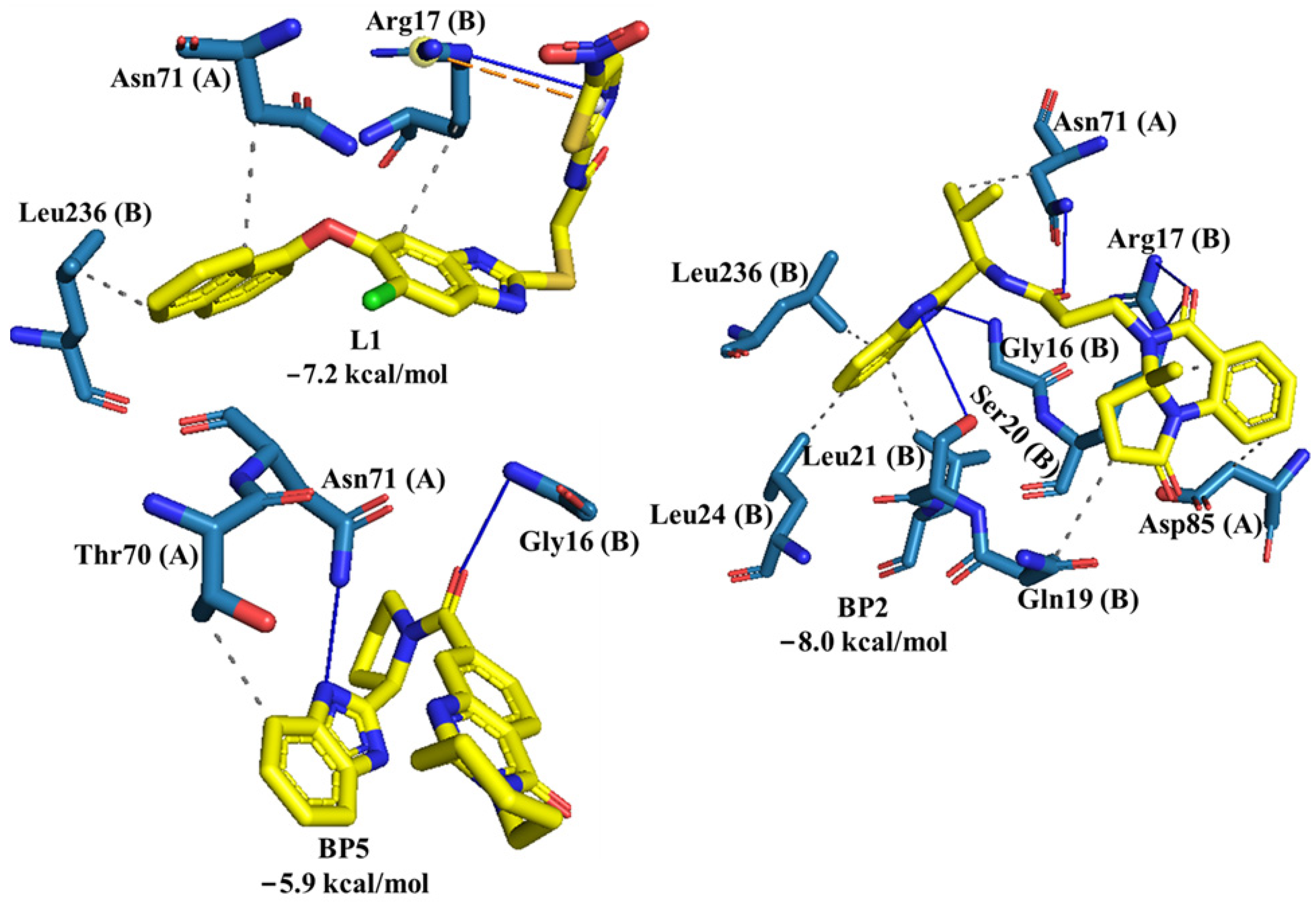
2.2. Molecular Docking of Control Ligands at the TcTIM Interface
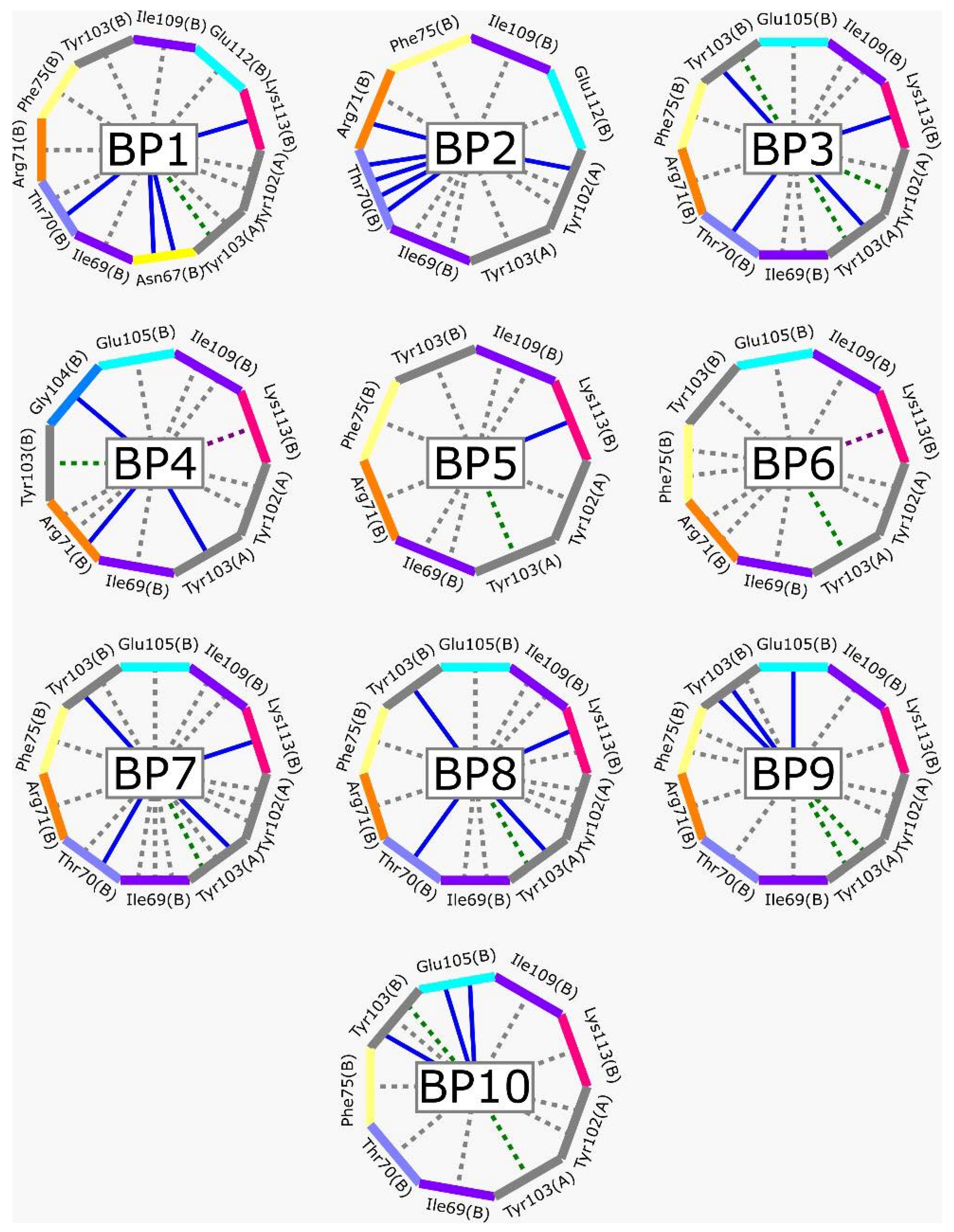
2.3. LBVS from ZINC15 Database and Molecular Docking Analysis
2.4. Trypanocidal Activity
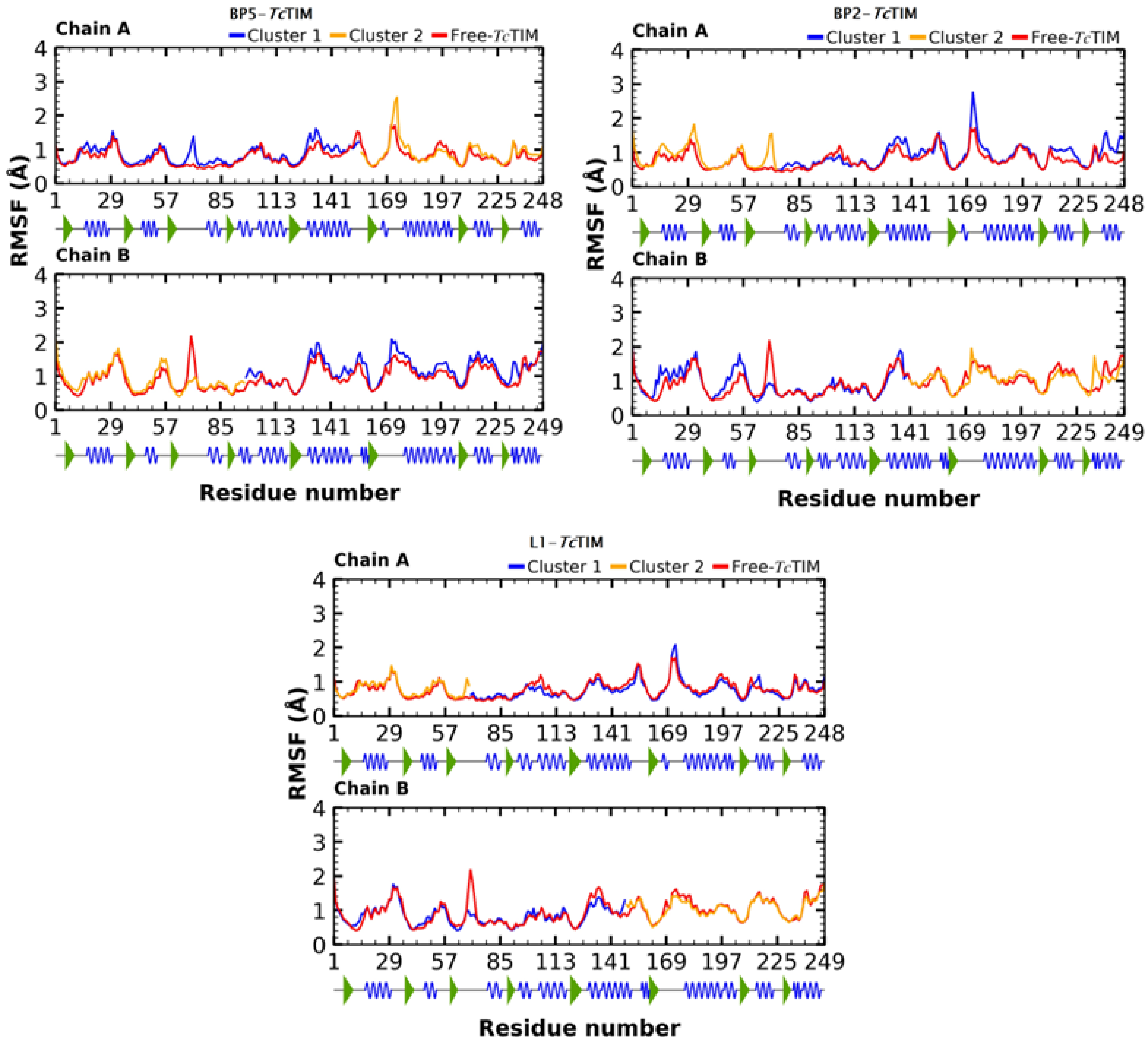
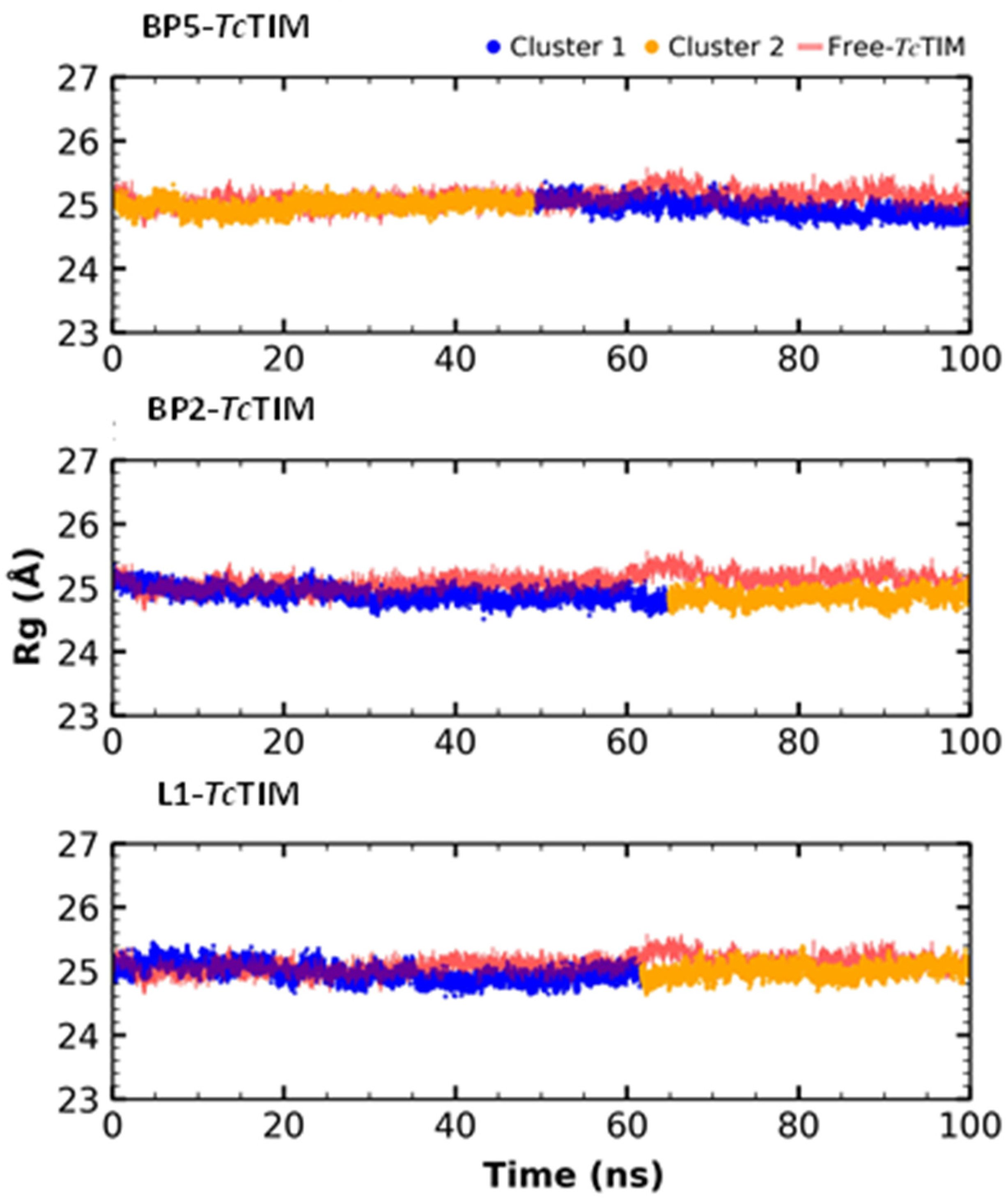
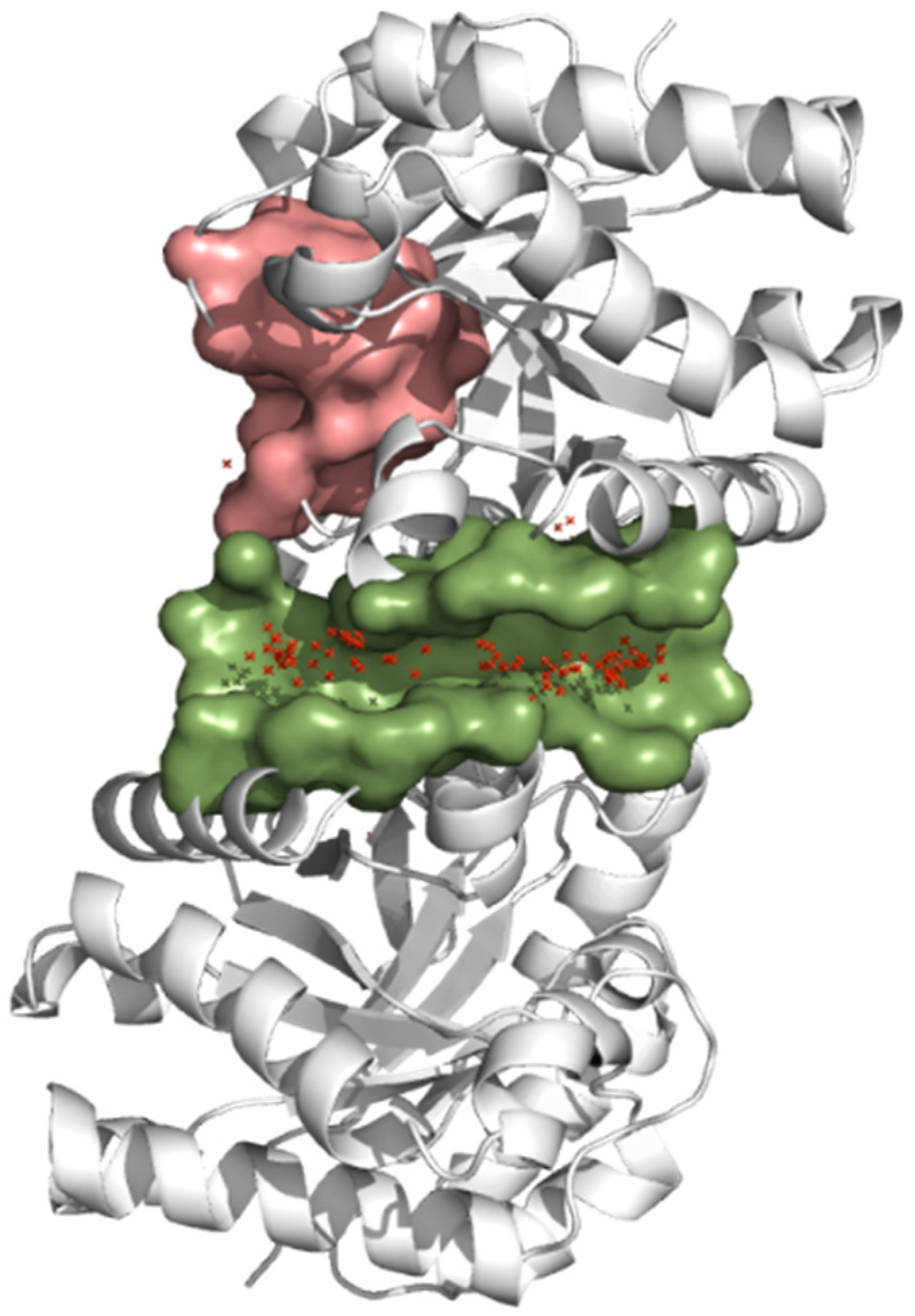
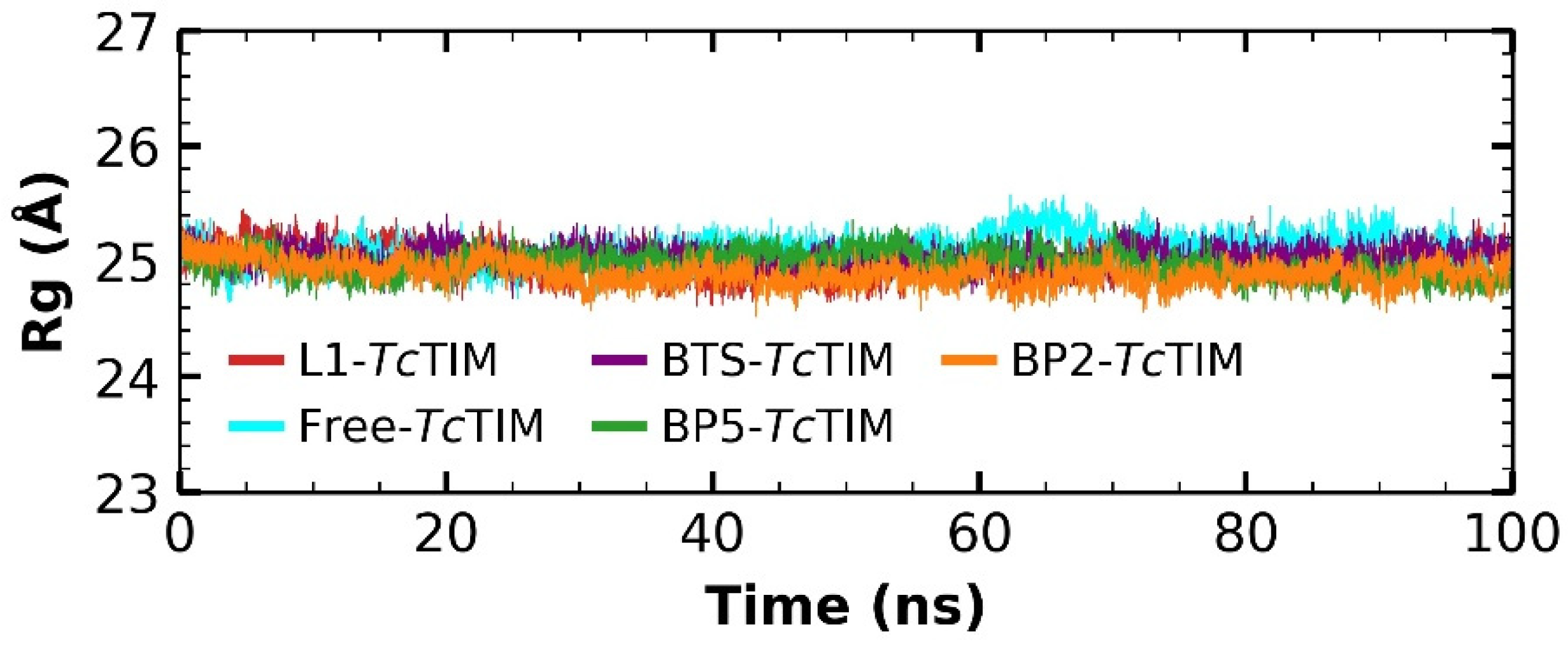
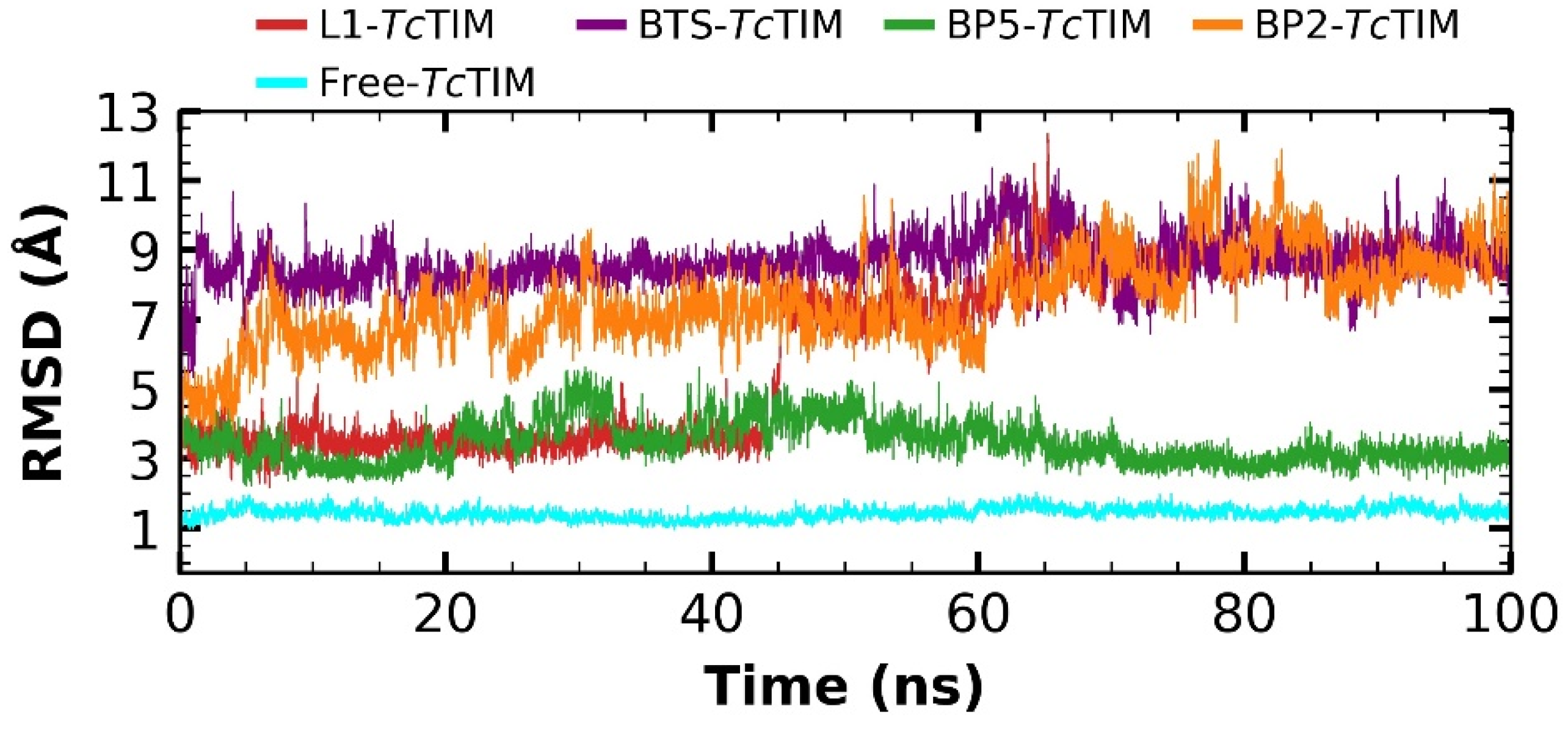
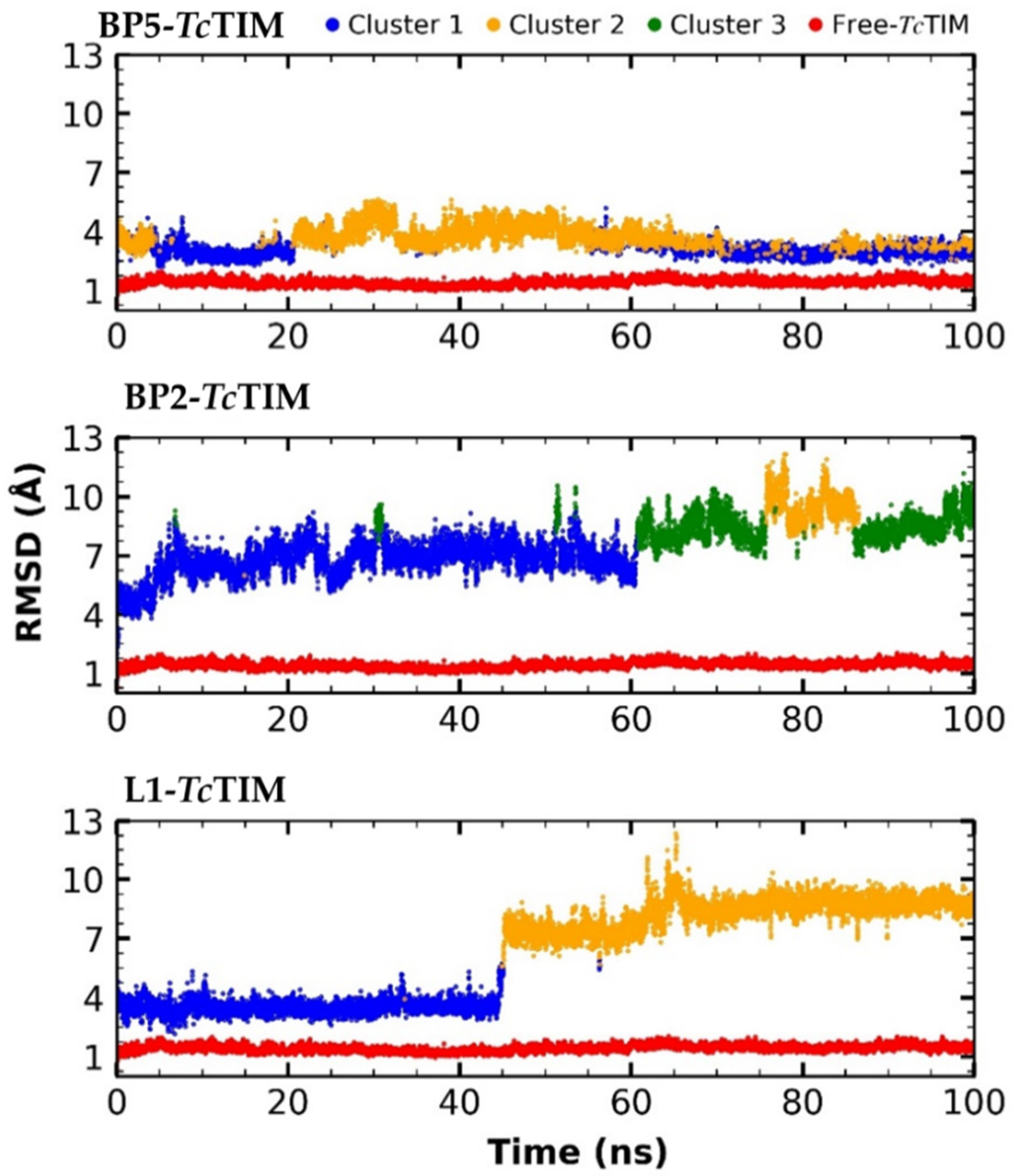
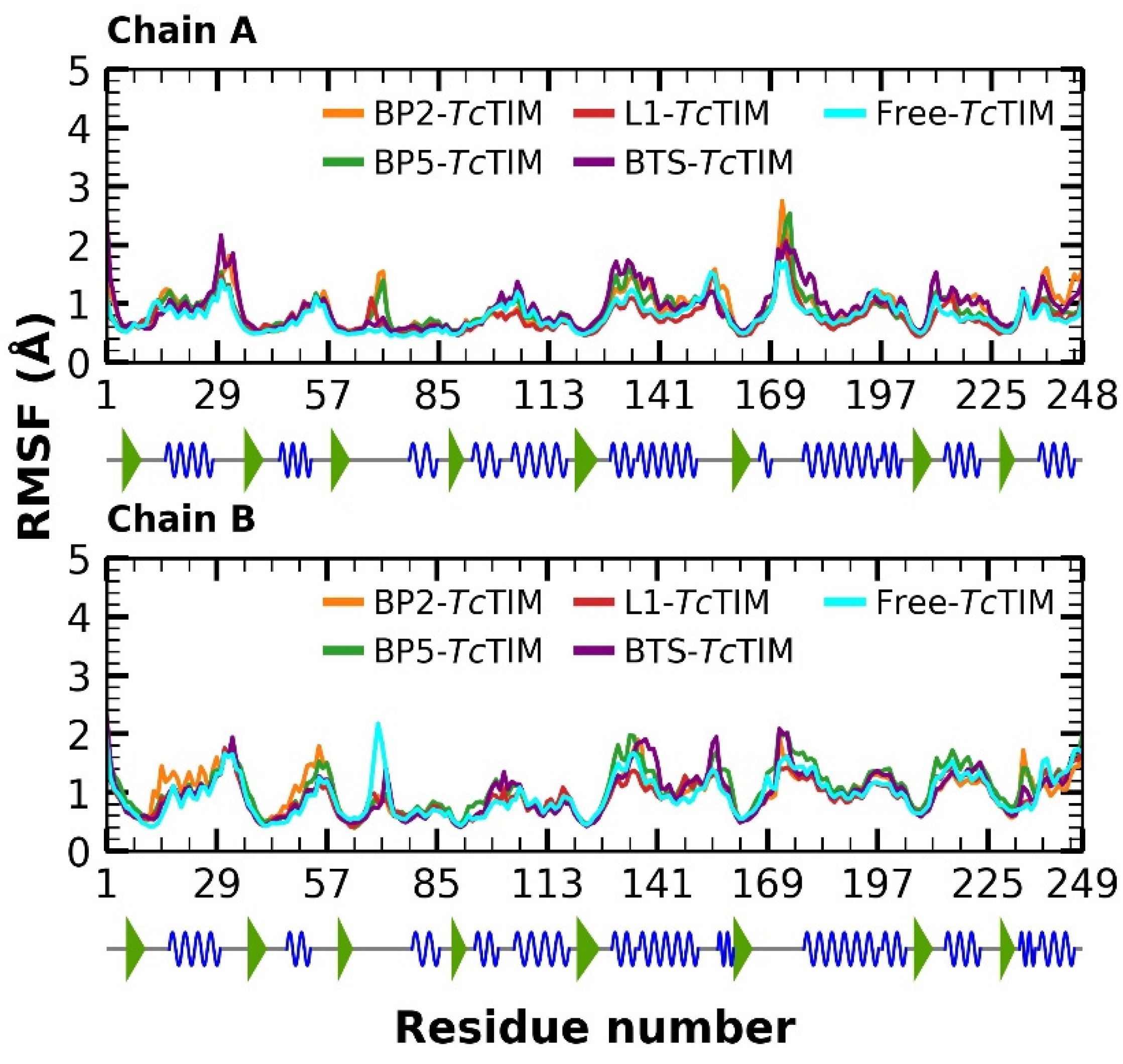
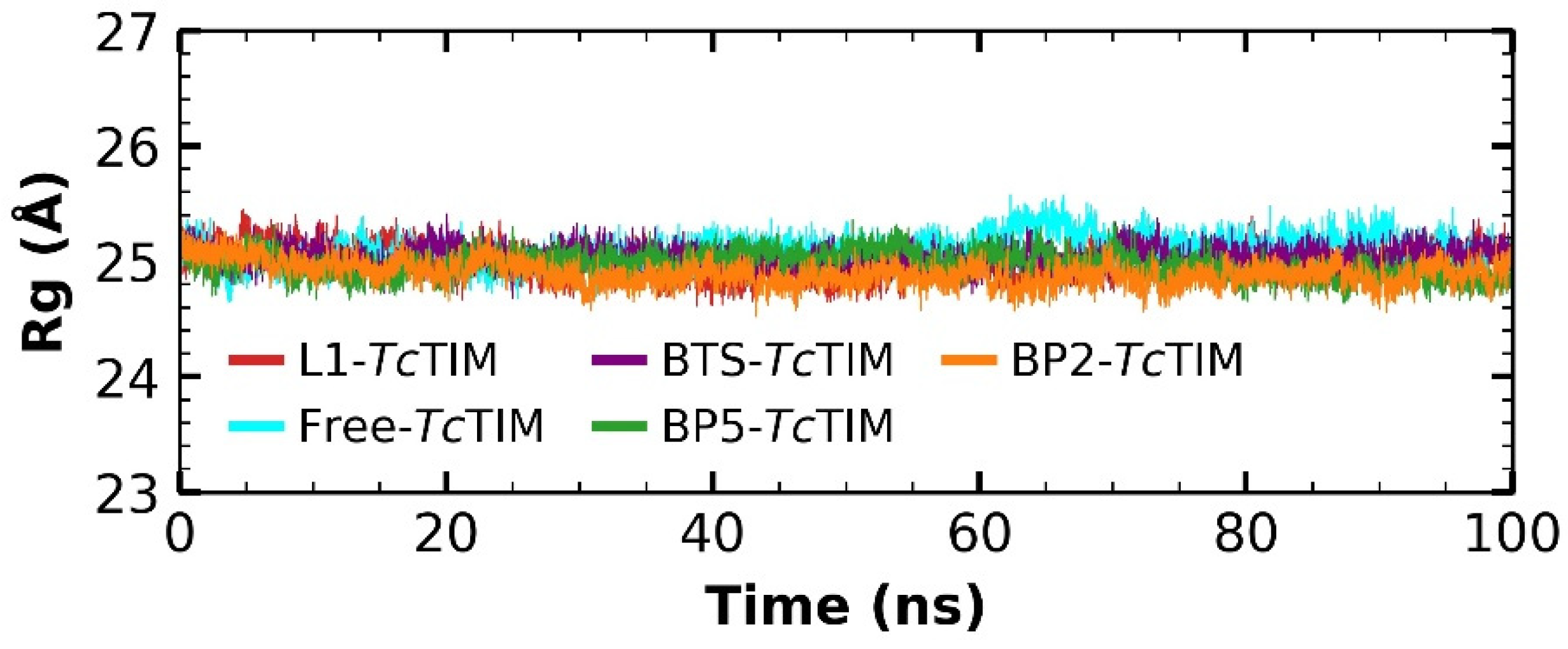
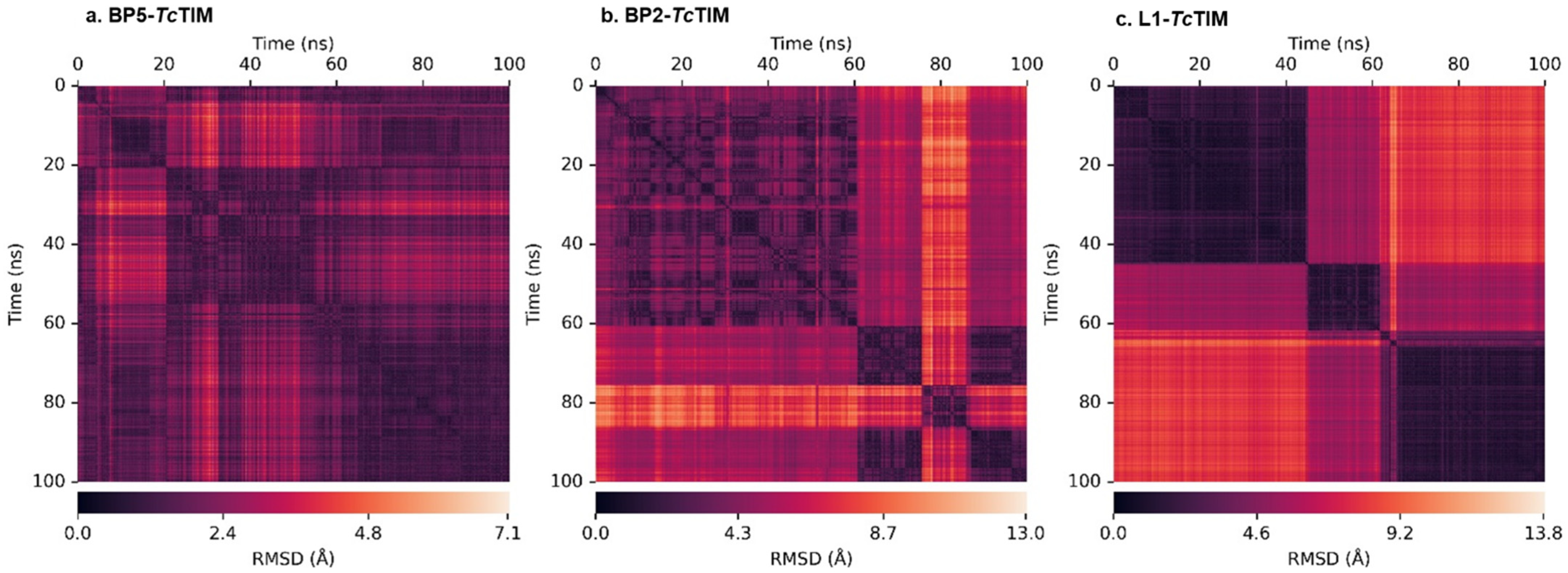
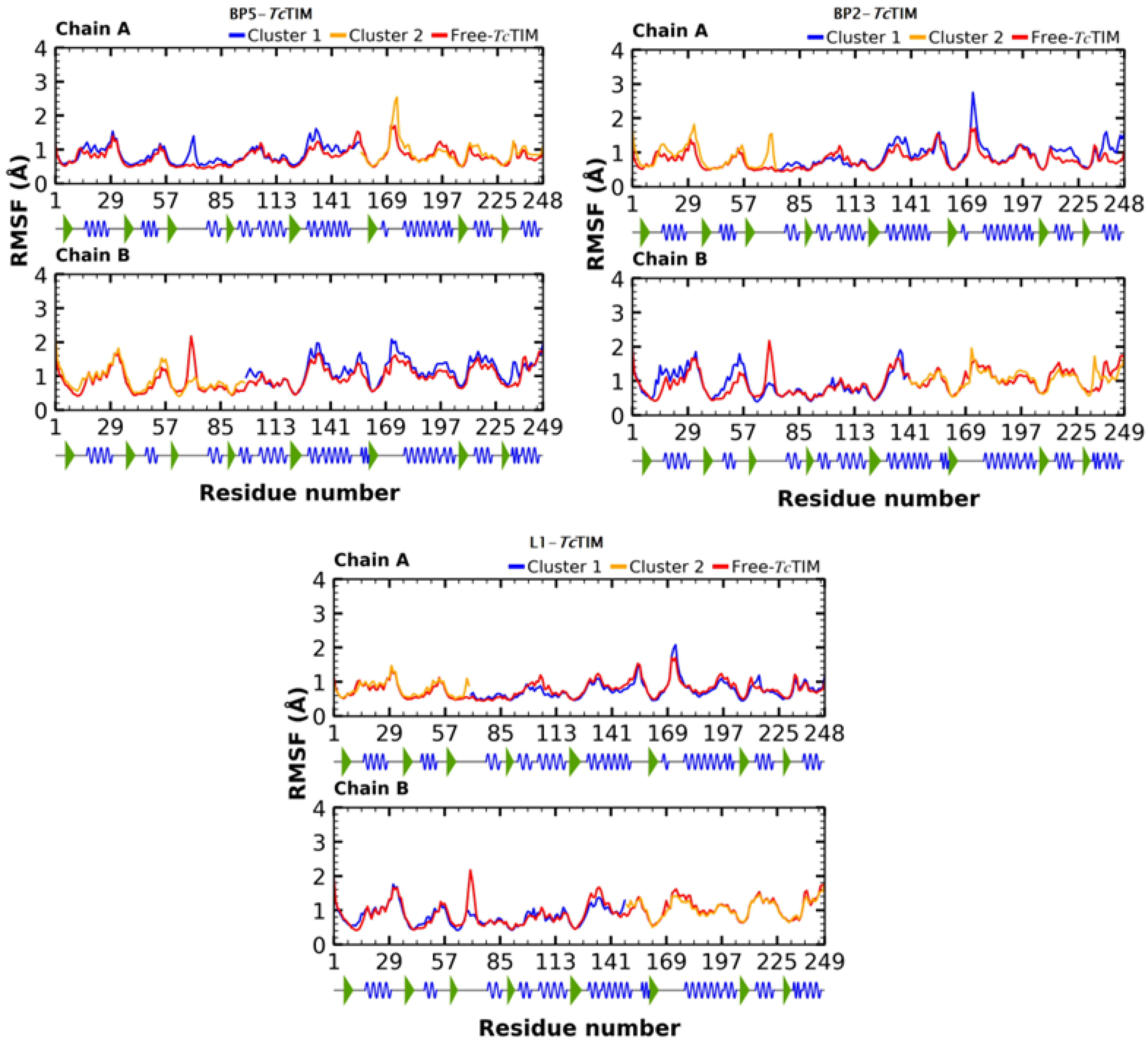
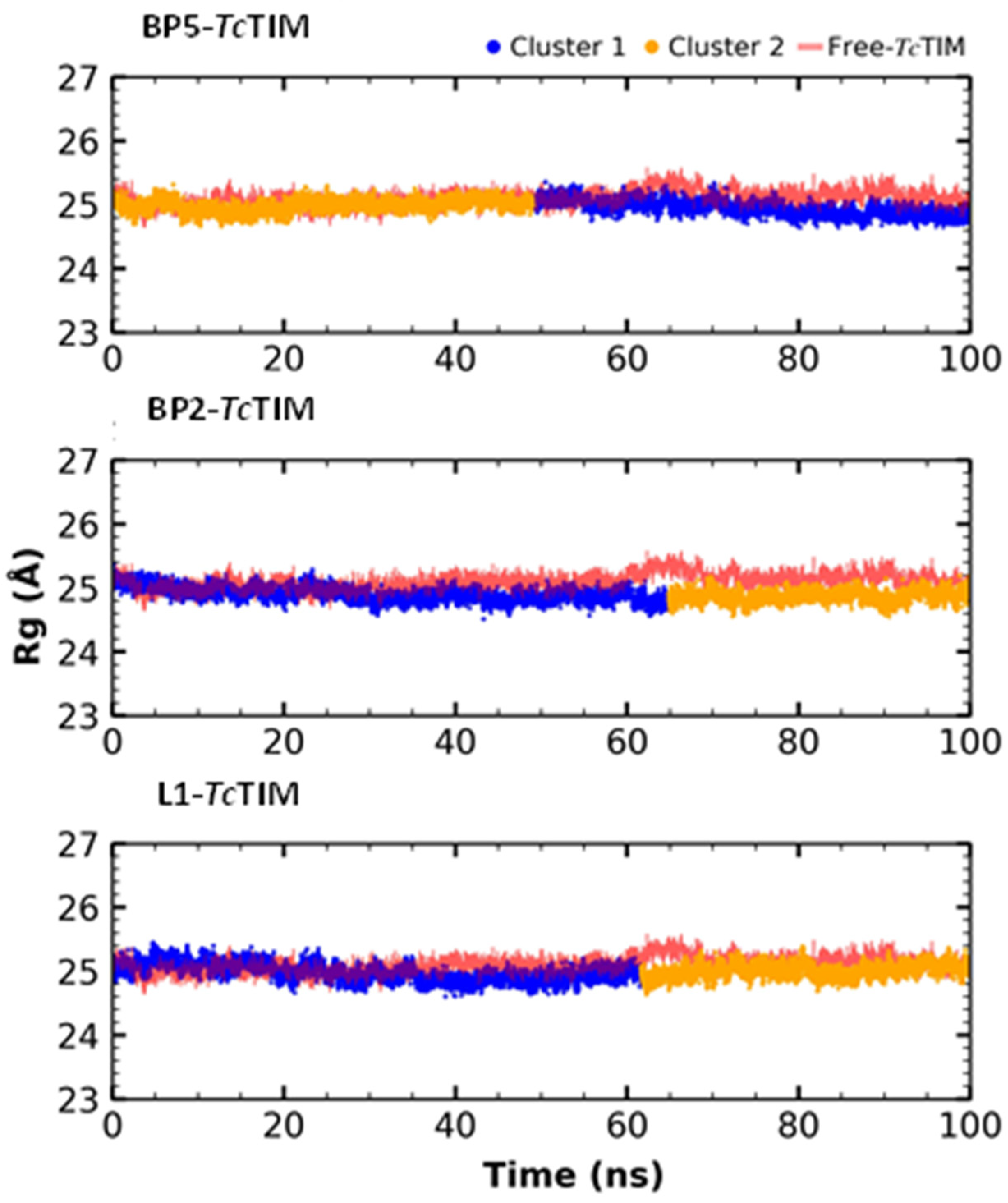
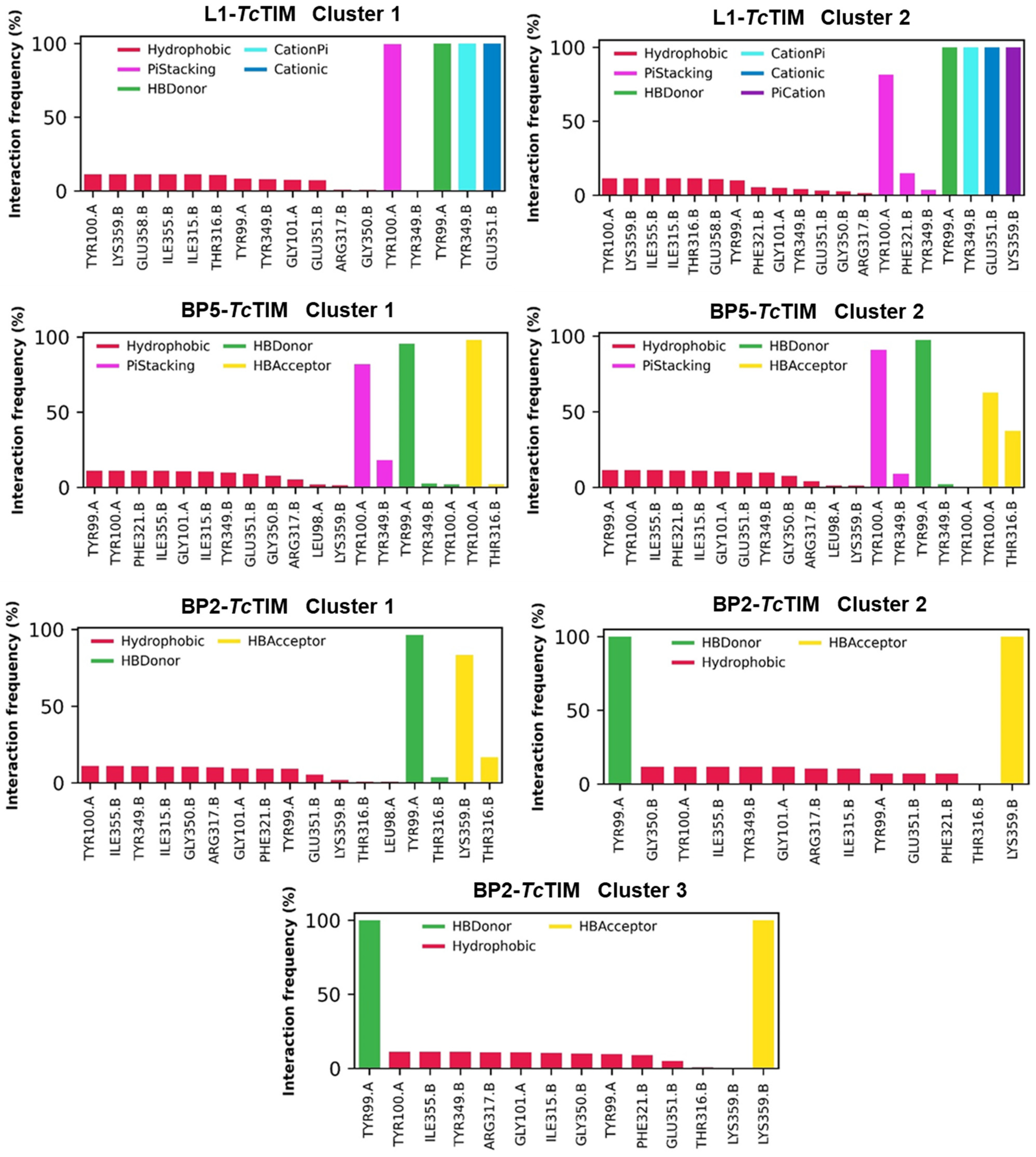
2.5. Molecular Dynamics Simulation
2.6. Analysis of Molecular Physicochemical Properties
2.7. Molecular Docking at the HsTIM Interface
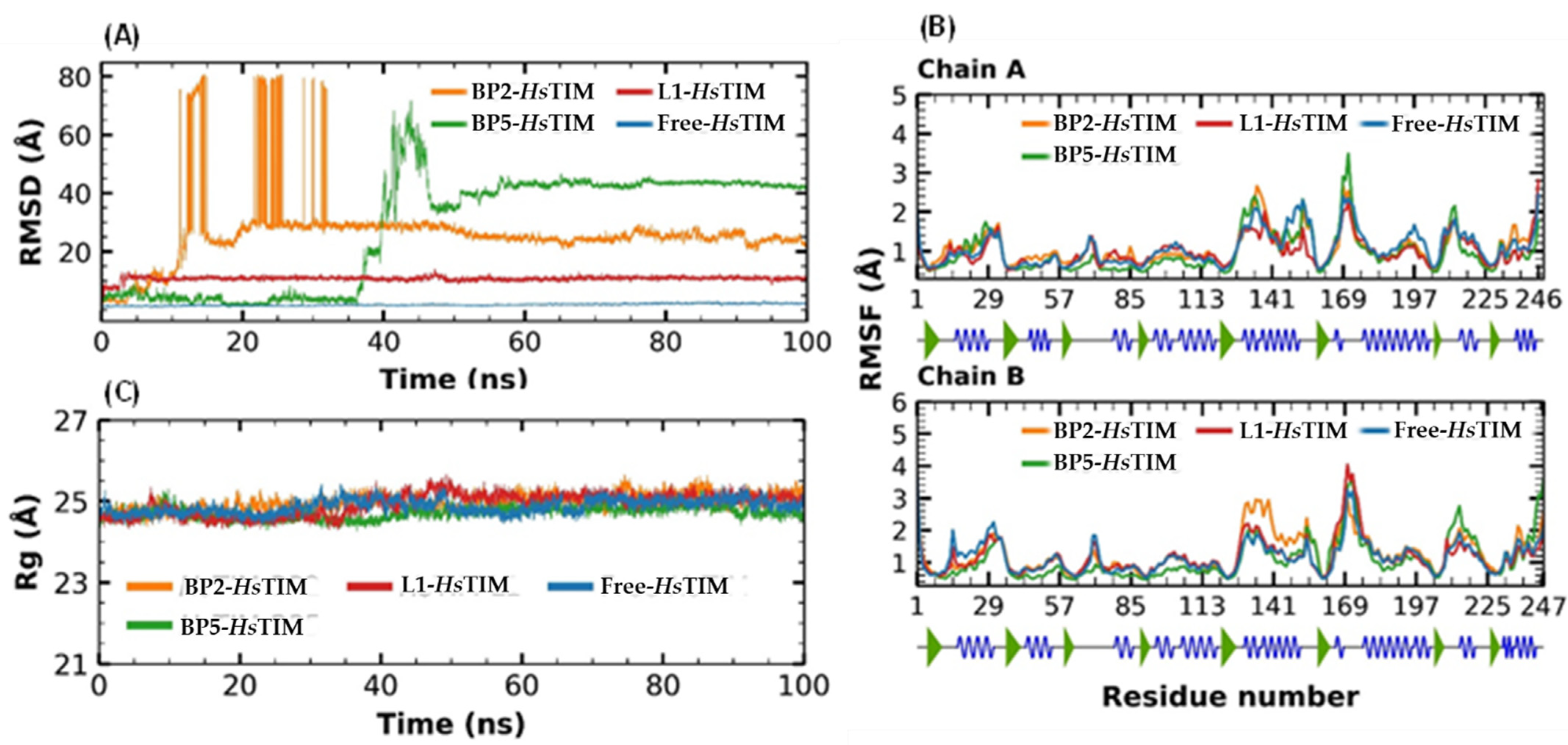
2.8. Molecular Dynamics Simulation at the HsTIM Interface
3. Discussion
3.1. TcTIM Inhibitors Analysis
3.2. LBVS from ZINC15 Database and Molecular Docking Analysis
3.3. Trypanocidal Activity
3.4. Molecular Dynamics Simulation at the TcTIM Interface
3.5. Analysis of Molecular Physicochemical Properties
3.6. Molecular Docking at the HsTIM Interface
4. Materials and Methods
4.1. Data Preparation
4.2. Molecular Docking
4.3. Virtual Screening
4.4. Trypanocidal Assay
4.5. Molecular Dynamics Analysis
4.6. Analysis of Molecular Physicochemical Properties
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


References
- Zamora-Ledesma, S.; Hernández-Camacho, N.; Sánchez-Moreno, M.; Ruiz-Piña, H.; Villagrán-Herrera, M.E.; Marín-Sánchez, C.; Carrillo-Angeles, I.G.; Jones, R.W.; Camacho-Macías, B. Seropositivity for Trypanosoma cruzi and Leishmania mexicana in dogs from a metropolitan region of Central México. Vet. Parasitol. Reg. Stud. Rep. 2020, 22, 100459. [Google Scholar] [CrossRef] [PubMed]
- PAHO Pan-American Health Organization. Available online: http://www.paho.org/hq/index.php?option=com_topics&view=article&id=10&Itemid=40743 (accessed on 15 April 2021).
- Nunes, M.C.; Dones, W.; Morillo, C.A.; Encina, J.J.; Ribeiro, A.L. Chagas disease: An overview of clinical and epidemiological aspects. J. Am. Coll. Cardiol. 2013, 62, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Illana, V.; Riveros-Rosas, H.; Cabrera, N.; de Gómez-Puyou, M.; Pérez-Montfort, R.; Costas, M.; Gómez-Puyou, A. A guide to the effects of a large portion of the residues of triosephosphate isomerase on catalysis, stability, druggability, and human disease. Proteins 2017, 85, 1190–1211. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Raygoza, A.; Cano-González, L.; Velázquez-Martínez, I.; Trejo-Soto, P.J.; Castillo, R.; Hernández-Campos, A.; Hernández-Luis, F.; Oria-Hernández, J.; Castillo-Villanueva, A.; Avitia-Domínguez, C.; et al. Species-Specific Inactivation of Triosephosphate Isomerase from Trypanosoma brucei: Kinetic and Molecular Dynamics Studies. Molecules 2017, 22, 2055. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Illana, V.; Pérez-Montfort, R.; López-Calahorra, F.; Costas, M.; Rodríguez-Romero, A.; Tuena de Gómez-Puyou, M.; Gómez Puyou, A. Structural differences in triosephosphate isomerase from different species and discovery of a multitrypanosomatid inhibitor. Biochemistry 2006, 45, 2556–2560. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, E.L.; Deinema, M.S.; Kearns, S.P.; Frey, T.A.; Tanghe, S.; Perry, K.; Roy, T.A.; Gracz, H.S.; Rodriguez, A.; D’Antonio, J. Structure-based Approach to the Identification of a Novel Group of Selective Glucosamine Analogue Inhibitors of Trypanosoma Cruzi Glucokinase. Mol. Biochem. Parasitol. 2015, 204, 64–76. [Google Scholar] [CrossRef]
- Maldonado, E.; Soriano-García, M.; Moreno, A.; Cabrera, N.; Garza-Ramos, G.; Tuena de Gómez-Puyou, M.; Gómez-Puyou, A.; Pérez-Montfort, R. Differences in the intersubunit contacts in triosephosphate isomerase from two closely related pathogenic trypanosomes. J. Mol. Biol. 1998, 283, 193–203. [Google Scholar] [CrossRef]
- Zomosa-Signoret, V.; Hernández-Alcántara, G.; Reyes-Vivas, H.; Martínez-Martínez, E.; Garza-Ramos, G.; Pérez-Montfort, R.; de Gómez-Puyou, M.T.; Gómez-Puyou, A. Control of the reactivation kinetics of homodimeric triosephosphate isomerase from unfolded monomers. Biochemistry 2003, 42, 3311–3318. [Google Scholar] [CrossRef]
- Pérez-Montfort, R.; Garza-Ramos, G.; Alcántara, G.; Reyes-Vivas, H.; Gao, X.G.; Maldonado, E.; de Gómez-Puyou, M.T.; Gómez-Puyou, A. Derivatization of the Interface Cysteine of Triosephosphate Isomerase from Trypanosoma Brucei and Trypanosoma Cruzi as Probe of the Interrelationship Between the Catalytic Sites and the Dimer Interface. Biochemistry 1999, 38, 4114–4120. [Google Scholar] [CrossRef]
- Álvarez, G.; Aguirre-López, B.; Varela., J.; Cabrera., M.; Merlino., A.; López., G.V.; Lavaggi., M.L.; Porcal, W.; Di-Maio, R.; González, M.; et al. Massive screening yields novel and selective Trypanosoma cruzi triosephosphate isomerase dimer-interface irreversible inhibitors with anti-trypanosomal activity. Eur. J. Med. Chem. 2010, 45, 5767–5772. [Google Scholar] [CrossRef]
- Flores-Sandoval, C.A.; Cuevas-Hernández, R.I.; CorreaBasurto, J.; Beltrán-Conde, H.I.; Padilla-Martínez, I.I.; Farfán-García, J.N.; Nogueda-Torres, B.; Trujillo-Ferrara, J.G. Synthesis and theoretic calculations of benzoxazoles and docking studies of their interactions with triosephosphate isomerase. Med. Chem. Res. 2013, 22, 2768–2777. [Google Scholar] [CrossRef]
- Cuevas-Hernández, R.I.; Correa-Basurto, J.; FloresSandoval, C.A.; Padilla-Martínez, I.I.; Nogueda-Torres, B.; Villa-Tanaca, M.L.; Tamay-Cach, F.; Nolasco-Fidencio, J.J.; Trujillo-Ferrara, J.G. Fluorine-containing benzothiazole as a novel trypanocidal agent: Design, in silico study, synthesis and activity evaluation. Med. Chem. Res. 2016, 25, 211–224. [Google Scholar] [CrossRef]
- Kurkcuoglu, Z.; Findik, D.; Akten, E.D.; Doruker, P. How an Inhibitor Bound to Subunit Interface Alters Triosephosphate Isomerase Dynamics. Biophys. J. 2015, 109, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, R.K.; Noble, M.E.; Vriend, G.; Nauche, S.; Hol, W.G. Refined 1.83 A structure of trypanosomal triosephosphate isomerase crystallized in the presence of 2.4 M-ammonium sulphate. A comparison with the structure of the trypanosomal triosephosphate isomerase-glycerol-3-phosphate complex. J. Mol. Biol. 1991, 220, 995–1015. [Google Scholar] [CrossRef]
- Wierenga, R.K.; Noble, M.E. Comparison of the refined crystal structures of liganded and unliganded chicken, yeast and trypanosomal triosephosphate isomerase. J. Mol. Biol. 1992, 224, 1115–1126. [Google Scholar] [CrossRef]
- Álvarez, G.; Martínez, J.; Varela, J.; Birriel, E.; Cruces, E.; Gabay, M.; Leal, S.M.; Escobar, P.; Aguirre-López, B.; Cabrera, N.; et al. Development of bis-thiazoles as inhibitors of triosephosphate isomerase from Trypanosoma cruzi. Identification of new non-mutagenic agents that are active in vivo. Eur. J. Med. Chem. 2015, 100, 246–256. [Google Scholar] [CrossRef]
- Shaaban, M.R.; Saleh, T.S.; Mayhoub, A.S.; Mansour, A.; Farag, A.M. Synthesis and analgesic/anti-inflammatory evaluation of fused heterocyclic ring systems incorporating phenylsulfonyl moiety. Bioorg. Med. Chem. 2008, 16, 6344–6352. [Google Scholar] [CrossRef]
- El Rashedy, A.A.; Aboul-Enein, H.Y. Benzimidazole derivatives as potential anticancer agents. Mini Rev. Med. Chem. 2013, 13, 399–407. [Google Scholar] [CrossRef]
- Song, D.; Shutao, M. Recent Development of Benzimidazole-Containing Antibacterial Agents. Chem. Med. Chem. 2016, 11, 646–659. [Google Scholar] [CrossRef]
- Francesconi, V.; Cichero, E.; Schenone, E.; Naesens, L.; Tonelli, M. Synthesis and Biological Evaluation of Novel (thio) semicarbazone-Based Benzimidazoles as Antiviral Agents against Human Respiratory Viruses. Molecules 2020, 25, 1487. [Google Scholar] [CrossRef] [Green Version]
- Romo-Mancillas, A.; Téllez-Valencia, A.; Yépez-Mulia, L.; Hernández-Luis, F.; Hernández-Campos, A.; Castillo, R. The design and inhibitory profile of new benzimidazole derivatives against triosephosphate isomerase from Trypanosoma cruzi: A problem of residue motility. J. Mol. Graph. Model. 2011, 30, 90–99. [Google Scholar] [CrossRef]
- Oh, S.; Kim, S.; Kong, S.; Yang, G.; Lee, N.; Han, D.; Goo, J.; Siqueira-Neto, J.L.; Freitas-Junior, L.H.; Song, R. Synthesis and biological evaluation of 2,3-dihydroimidazo[1,2-a]benzimidazole derivatives against Leishmania donovani and Trypanosoma cruzi. Eur. J. Med. Chem. 2014, 84, 395–403. [Google Scholar] [CrossRef]
- Melchor-Doncel de la Torre, S.; Vázquez, C.; González-Chávez, Z.; Yépez-Mulia, L.; Nieto-Meneses, R.; Jasso-Chávez, R.; Saavedra, M.; Hernández-Luis, F. Synthesis and biological evaluation of 2-methyl-1H-benzimidazole-5-carbohydrazides derivatives as modifiers of redox homeostasis of Trypanosoma cruzi. Bioorg. Med. Chem. Lett. 2017, 27, 3403–3407. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-López, J.M.; Hernández-Campos, A.; Yépez-Mulia, L.; Téllez-Valencia, A.; Flores-Carrillo, P.; Nieto-Meneses, R.; Castillo, R. Synthesis and trypanocidal activity of novel benzimidazole derivatives. Eur. J. Med. Chem. Lett. 2016, 26, 4377–4381. [Google Scholar] [CrossRef] [PubMed]
- Bezabeh, S.; Mackey, A.C.; Kluetz, P.; Jappar, D.; Korvick, J. Accumulating Evidence for a Drug-Drug Interaction between Methotrexate and Proton Pump Inhibitors. Oncologist 2012, 17, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Han, X.; Zhou, Z. New Substituted Benzimidazole Derivatives: A Patent Review (2013–2014). Expert Opin. Ther. Pat. 2015, 25, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.J.; Sultan, M.Z.; Rashid, M.A.; Kuddus, M.R. Does Rabeprazole Sodium Alleviate the Anti-diabetic Activity of Linagliptin? Drug-Drug Interaction Analysis by In Vitro and In Vivo Methods. Drug Res. 2020, 70, 519–527. [Google Scholar] [CrossRef]
- Hossain, M.J.; Sultan, M.Z.; Rashid, M.A.; Kuddus, M.R. Interactions of Linagliptin, Rabeprazole Sodium, and Their Formed Complex with Bovine Serum Albumin: Computational Docking and Fluorescence Spectroscopic Methods. Anal. Sci. Adv. 2021, 7, 202000153. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]; Anthelmintic Agents; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Kurkcuoglu, Z.; Ural, G.; Demet Akten, E.; Doruker, P. Blind Dockings of Benzothiazoles to Multiple Receptor Conformations of Triosephosphate Isomerase from Trypanosoma cruzi and Human. Mol. Inform. 2011, 30, 986–995. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Abraham, A.; Pedregosa, F.; Eickenberg, M.; Gervais, P.; Mueller, A.; Kossaifi, J.; Gramfort, A.; Thirion, B.; Varoquaux, G. Machine learning for neuroimaging with scikit-learn. Front. Neuroinform. 2014, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- De Paris, R.; Quevedo, C.V.; Ruiz, D.D.; Norberto de Souza, O. An effective approach to cluster inhA molecular dynamics pathway using substrate-binding cavity features. PLoS ONE 2015, 10, e0133172. [Google Scholar] [CrossRef] [PubMed]
- De Paris, R.; Quevedo, C.V.; Ruiz, D.D.; Norberto de Souza, O.; Barros, R.C. Clustering molecular dynamics trajectories for optimizing docking experiments. Comput. Intell. Neurosci. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Kirschner, K.N. Principal component and clustering analysis on molecular dynamics data of the ribosomal L11·23S subdomain. J. Mol. Model. 2013, 19, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Singh, S. Dynamics of heroin molecule inside the lipid membrane: A molecular dynamics study. J. Mol. Model. 2019, 25, 121. [Google Scholar] [CrossRef]
- Minini, L.; Álvarez, G.; González, M.; Cerecetto, H.; Merlino, A. Molecular docking and molecular dynamics simulation studies of Trypanosoma cruzi triosephosphate isomerase inhibitors. Insights into the inhibition mechanism and selectivity. J. Mol. Graph. Model. 2015, 58, 40–49. [Google Scholar] [CrossRef]
- Téllez-Valencia, A.; Olivares-Illana, V.; Hernandez-Santoyo, A.; Perez-Montfort, R.; Costas, M.; Rodriguez-Romero, A.; López-Calahorra, F.; de Gómez-Puyou, M.T.; Gómez-Puyou, A. Inactivation of triosephosphate isomerase from Trypanosoma cruzi by an agent that perturbs its dimer interface. J. Mol. Biol. 2004, 341, 1355–1365. [Google Scholar] [CrossRef]
- Juárez-Saldivar, A.; Barbosa-Cabrera, E.; Lara-Ramírez, E.E.; Paz-González, A.D.; Martínez-Vázquez, A.V.; Bocanegra-García, V.; Palos, I.; Campillo, N.E.; Rivera, G. Virtual Screening of FDA-Approved Drugs against Triose Phosphate Isomerase from Entamoeba histolytica and Giardia lamblia Identifies Inhibitors of Their Trophozoite Growth Phase. Int. J. Mol. Sci. 2021, 22, 5943. [Google Scholar] [CrossRef]
- Téllez-Valencia, A.; Avila-Ríos, S.; Pérez-Montfort, R.; Rodríguez-Romero, A.; de Gómez-Puyou, M.T.; López-Calahorra, F.; Gómez-Puyou, A. Highly Specific Inactivation of Triosephosphate Isomerase from Trypanosoma Cruzi. Biochem. Biophys Res. Commun. 2002, 295, 958–963. [Google Scholar] [CrossRef]
- Unni, S.; Aouti, S.; Thiyagarajan, S.; Padmanabhan, B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. biosci. 2020, 45, 130. [Google Scholar] [CrossRef]
- Espinosa-Bustos, C.; Ortiz-Pérez, M.; González-González, A.; Zarate, A.M.; Rivera, G.; Belmont-Díaz, J.A.; Saavedra, E.; Cuellar, M.A.; Vázquez, K.; Salas, C.O. New Amino Naphthoquinone Derivatives as Anti-Trypanosoma cruzi Agents Targeting Trypanothione Reductase. Pharmaceutics 2022, 14, 1121. [Google Scholar] [CrossRef] [PubMed]
- Perilla, J.R.; Goh, B.C.; Cassidy, C.K.; Liu, B.; Bernardi, R.C.; Rudack, T.; Yu, H.; Wu, Z.; Schulten, K. Molecular dynamics simulations of large macromolecular complexes. Curr. Opin. Struct. Biol. 2015, 31, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Álvarez, D.; Herrera-Mayorga, D.; Juárez-Saldivar, A.; Paz-González, A.D.; Ortiz-Pérez, E.; Bandyopadhyay, D.; Pérez-Sánchez, H.; Rivera, G. Ligand-based virtual screening, molecular docking, and molecular dynamics of eugenol analogs as potential acetylcholinesterase inhibitors with biological activity against Spodoptera frugiperda. Mol. Divers. 2021, 26, 2025–2037. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, J.; Wang, H.; Wang, Y.; Wang, J.; Zhao, D. Molecular docking, 3D-QSAR, and molecular dynamics simulations of thieno [3, 2-b] pyrrole derivatives against anticancer targets of KDM1A/LSD1. J. Biomole. Struct. Dyn. 2020, 59, 4035–4061. [Google Scholar] [CrossRef]
- Luise, C.; Robaa, D.; Regenass, P.; Maurer, D.; Ostrovskyi, D.; Seifert, L.; Bacher, J.; Burgahn, T.; Wagner, T.; Seitz, J.; et al. Structure-Based Design, Docking and Binding Free Energy Calculations of A366 Derivatives as Spindlin1 Inhibitors. Int. J. Mol. Sci. 2021, 22, 5910. [Google Scholar] [CrossRef]
- Williams, J.C.; McDermott, A.E. Dynamics of the flexible loop of triosephosphate isomerase: The loop motion is not ligand gated. Biochemistry 1995, 34, 8309–8319. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of green tea polyphenols as novel corona virus (SARS-CoV-2) main protease (Mpro) inhibitors-an in silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2021, 39, 4362–4374. [Google Scholar] [CrossRef]
- Niu, Y.; Pan, D.; Shi, D.; Bai, Q.; Liu, H.; Yao, X. Influence of Chirality of Crizotinib on Its MTH1 Protein Inhibitory Activity: Insight from Molecular Dynamics Simulations and Binding Free Energy Calculations. PLoS ONE 2015, 10, e0145219. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, Z.Y.; Uzairu, A.; Shallangwa, G.A.; Abechi, S.E. Application of QSAR Method in the Design of Enhanced Antimalarial Derivatives of Azetidine-2-carbonitriles, their Molecular Docking, Drug-likeness, and SwissADME Properties. Iran J. Pharm. Res. 2021, 20, 254–270. [Google Scholar] [CrossRef]
- Mollazadeh, S.; Sahebkar, A.; Hadizadeh, F.; Behravan, J.; Arabzadeh, S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. 2018, 214, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Glaeser, H. Importance of P-glycoprotein for drug-drug interactions. Handb. Exp. Pharmacol. 2011, 201, 285–297. [Google Scholar] [CrossRef]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Berman, H.M.; Kleywegt, G.J.; Nakamura, H.; Markley, J.L. How community has shaped the Protein Data Bank. Structure 2013, 21, 1485–1491. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Joachim-Haupt, V.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–Ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- López-López, E.; Naveja, J.J.; Medina-Franco, J.L. DataWarrior: An evaluation of the open-source drug discovery tool. Expert Opin. Drug Discov. 2019, 14, 335–341. [Google Scholar] [CrossRef]
- Chacón-Vargas, K.F.; Nogueda-Torres, B.; Sánchez-Torres, L.E.; Suarez-Contreras, E.; Villalobos-Rocha, J.C.; Torres-Martinez, Y.; Lara-Ramírez, E.E.; Fiorani, G.; Krauth-Siegel, R.L.; Bolognesi, M.L.; et al. Trypanocidal Activity of Quinoxaline 1,4 Di-N-oxide Derivatives as Trypanothione Reductase Inhibitors. Molecules 2017, 22, 220. [Google Scholar] [CrossRef]
- Becerra, N.A.; Espinosa-Bustos, C.; Vázquez, K.; Rivera, G.; Paulino, M.; Cantero, J.; Nogueda, B.; Chacón-Vargas, F.; Castillo-Velazquez, U.; Elizondo Rodríguez, A.F.; et al. Expanding the chemical space of aryloxy-naphthoquinones as potential anti-Chagasic agents: Synthesis and trypanosomicidal activity. Med. Chem. Res. 2021, 30, 2256–2265. [Google Scholar] [CrossRef]
- Van-Der-Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Sousa-da-Silva, A.W.; Wim Vranken, F. ACPYPE AnteChamber PYthon Parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Bouysset, C.; Fiorucci, S. ProLIF: A library to encode molecular interactions as fingerprints. J. Cheminform. 2021, 13, 1–9. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, P.; Hamacher, K. Biotite: A unifying open-source computational biology framework in Python. BMC Bioinform. 2018, 19, 1–8. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Compound | Antiparasitic Activity | Enzymatic Activity | Docking Score Kcal/mol | Hydrophobic Interactions | Hydrogen Bonds | π-Stacking Interactions |
|---|---|---|---|---|---|---|---|
| L1 |  | - | % inhibition = 48 at 200 µM [20] | −8.9 | Tyr102 (A) Ile69 (B) Arg71 (B) Phe75 (B) | Glu75 (A) Tyr102 (A) Arg71 (B) | Tyr103 (A) |
| L2 | 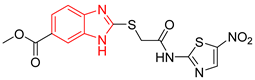 | - | % inhibition = 52 at 200 µM [21] | −6.8 | Tyr102 (A) Ile69 (B) | Leu101 (A) Tyr102 (A) Tyr103 (A) Arg71 (B) | Tyr103 (A) |
| L3 | 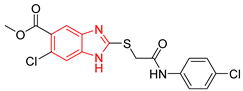 | Epimastigotes IC50 = 186.23 μM [25] | % inhibition = 65 at 100 µM [21] | −6.9 | Tyr102 (A) Ile69 (B) | Tyr103 (A) Arg71 (B) | Tyr103 (A) Tyr103 (B) |
| L4 |  | Epimastigotes IC50 = 48.91 μM [25] | % inhibition = 59 at 200 µM [25] | −7.3 | Tyr102 (A) Ile69 (B) | Tyr103 (A) Arg71 (B) | Tyr103 (A) Tyr103 (B) |
| L5 | 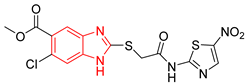 | Epimastigotes IC50 = 42.51 μM [25] | % inhibition = 69 at 200 µM [25] | −7.3 | Tyr102 (A) Ile69 (B) | Tyr103 (A) Arg71 (B) | Tyr103 (A) |
| L6 |  | Epimastigotes IC50 = 28.67 μM [25] | % de inhibición = 68 at 200 µM [25] | −7.2 | Tyr102 (A) Ile69 (B) Phe75 (B) | Tyr103 (A) Tyr103 (B) Gly104 (B) | Tyr103 (A) |
| L7 |  | - | IC50 = 8.0 µM; % inhibition = 95 at 250 µM [26,27] | −6.9 | Tyr102 (A) Ile69 (B) Glu108 (B) Ile109 (B) | Tyr103 (A) Glu112 (B) | Tyr103 (A) |
| L8 |  | Epimastigotes IC50 = 5.8 µM [28] | IC50 = 3.5 µM [28] | −6.9 | Tyr102 (A) Ile69 (B) Phe75 (B) | Tyr102 (A) Tyr103 (B) | Tyr103 (A) |
| L9 |  | Epimastigotes IC50 = 0.6 µM [29] | IC50 = 0.086 µM [29] | −7.2 | Tyr102 (A) Tyr103 (A) Ile69 (B) Arg71 (B) Phe75 (B) Tyr103 (B) Glu105 (B) | Arg71 (B) | Tyr102 (A) |
| BTS | 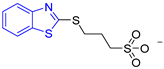 | - | semi-maximal inhibition at 35 μM [31] | −6.2 | Tyr102 (A) Ile69 (B) Phe75 (B) | Tyr103 (B) | Tyr103 (A) |
| Group | Compounds in the Group | ID Best Composite of Each Group | Docking Score (Kcal/mol) | Structure |
|---|---|---|---|---|
| 1 | 167 | BP1 (ZINC000150134991) | −10.2 | 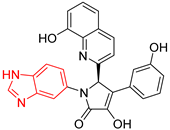 |
| 2 | 135 | BP2 (ZINC000040071949) | −10.4 |  |
| 3 | 206 | BP3 (ZINC000030009841) | −10.3 | 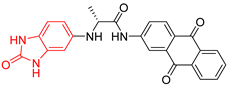 |
| 4 | 171 | BP4 (ZINC000150010278) | −10.0 | 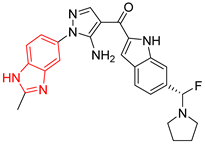 |
| 5 | 142 | BP5 (ZINC000040013445) | −10.2 | 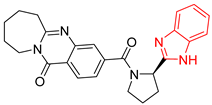 |
| 6 | 103 | BP6 (ZINC000140212311) | −10.2 | 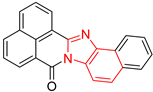 |
| 7 | 181 | BP7 (ZINC000040170214) | −10.5 |  |
| 8 | 162 | BP8 (ZINC000040170215) | −10.6 | 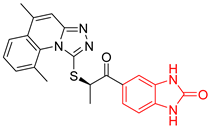 |
| 9 | 191 | BP9 (ZINC000170014382) | −10.4 |  |
| 10 | 146 | BP10 (ZINC000170072839) | −10.4 | 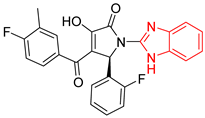 |
| Compound | LC50 (µM) * | ||
|---|---|---|---|
| NINOA | INC-5 | ||
| BP2 | 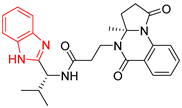 | 155.86 ± 3.4 | 226.30 ± 15.4 |
| BP5 | 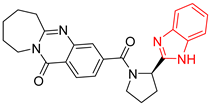 | 179.55 ± 19.7 | 179.71 ± 19.0 |
| Nfx | 70.41 ± 8.0 | 139.37 ± 3.0 | |
| Bzn | 130.72 ± 8.8 | 191.28 ± 1.8 | |
| Complexes | ΔEvdw (kcal/mol) | ΔEele (kcal/mol) | ΔGpolar (kcal/mol) | ΔGSA (kcal/mol) | ΔGb (kcal/mol) |
|---|---|---|---|---|---|
| L1-TcTIM | −33.97 ± 0.15 | −3.85 ± 0.06 | 16.99 ± 0.12 | −3.89 ± 0.01 | −24.73 ± 0.15 |
| BP2-TcTIM | −29.73 ± 0.24 | −3.78 ± 0.11 | 17.77 ± 0.20 | −3.28 ± 0.03 | −19.03 ± 0.16 |
| BP5-TcTIM | −46.58 ± 0.2 | −5.21 ± 0.1 | 30.61 ± 0.13 | −4.49 ± 0.01 | −25.68 ± 0.20 |
| BTS-TcTIM | −24.53 ± 0.23 | −14.93 ± 0.37 | 28.28 ± 0.21 | −2.93 ± 0.01 | −14.12 ± 0.23 |
| L1-HsTIM | −32.19 ± 0.44 | −11.71 ± 0.37 | 34.74 ± 0.67 | −3.56 ± 0.03 | −12.72 ± 0.25 |
| BP2-HsTIM | −30.68 ± 0.24 | −0.09 ± 0.12 | 14.02 ± 0.2 | −2.98 ± 0.02 | −19.72 ± 0.21 |
| BP5-HsTIM | −23.04 ± 0.18 | −5.22 ± 0.18 | 15.63 ± 0.83 | −2.26 ± 0.02 | −14.89 ± 0.84 |
| Physicochemical Properties | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | MW (g/mol) < 500 | Nb < 10 | Nhba < 10 | Nhbd < 5 | TPSA (Å2) < 140 | Log P < 5 | Log S 1 | |||||||
| BP2 | 459.54 | 7 | 4 | 2 | 98.40 | 2.70 | Moderately soluble | |||||||
| BP5 | 427.50 | 3 | 4 | 1 | 88.88 | 3.20 | Moderately soluble | |||||||
| Probability of pharmacokinetic properties | ||||||||||||||
| Compound | Blood-brain permeability | Human gastrointestinal absorption 2 | P-glycoprotein substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | Hepatotoxicity | |||||
| BP2 | No | Soluble | Yes | No | Yes | Yes | Yes | Yes | Inactive | |||||
| BP5 | No | Soluble | Yes | No | Yes | Yes | Yes | Yes | Inactive | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Jiménez, L.K.; Juárez-Saldivar, A.; Gómez-Escobedo, R.; Delgado-Maldonado, T.; Méndez-Álvarez, D.; Palos, I.; Bandyopadhyay, D.; Gaona-Lopez, C.; Ortiz-Pérez, E.; Nogueda-Torres, B.; et al. Ligand-Based Virtual Screening and Molecular Docking of Benzimidazoles as Potential Inhibitors of Triosephosphate Isomerase Identified New Trypanocidal Agents. Int. J. Mol. Sci. 2022, 23, 10047. https://doi.org/10.3390/ijms231710047
Vázquez-Jiménez LK, Juárez-Saldivar A, Gómez-Escobedo R, Delgado-Maldonado T, Méndez-Álvarez D, Palos I, Bandyopadhyay D, Gaona-Lopez C, Ortiz-Pérez E, Nogueda-Torres B, et al. Ligand-Based Virtual Screening and Molecular Docking of Benzimidazoles as Potential Inhibitors of Triosephosphate Isomerase Identified New Trypanocidal Agents. International Journal of Molecular Sciences. 2022; 23(17):10047. https://doi.org/10.3390/ijms231710047
Chicago/Turabian StyleVázquez-Jiménez, Lenci K., Alfredo Juárez-Saldivar, Rogelio Gómez-Escobedo, Timoteo Delgado-Maldonado, Domingo Méndez-Álvarez, Isidro Palos, Debasish Bandyopadhyay, Carlos Gaona-Lopez, Eyra Ortiz-Pérez, Benjamín Nogueda-Torres, and et al. 2022. "Ligand-Based Virtual Screening and Molecular Docking of Benzimidazoles as Potential Inhibitors of Triosephosphate Isomerase Identified New Trypanocidal Agents" International Journal of Molecular Sciences 23, no. 17: 10047. https://doi.org/10.3390/ijms231710047