
TMAO Suppresses Megalin Expression and Albumin Uptake in Human Proximal Tubular Cells Via PI3K and ERK Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
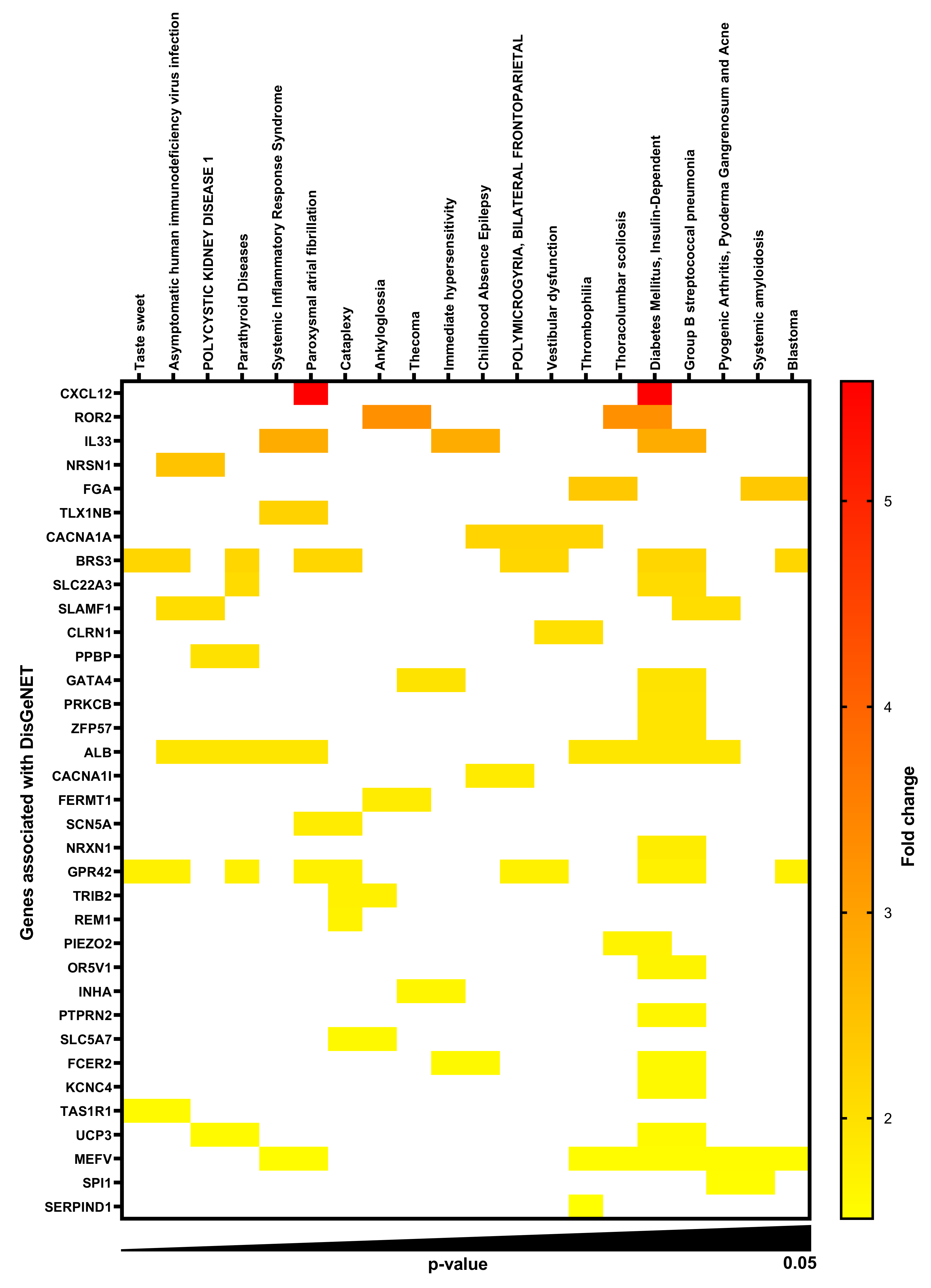
2.1. Gene-Disease Associations in TMAO-Treated Human Renal Proximal Tubular Cells
2.2. TMAO Decreases Protein Expression of Megalin, but Not Cubilin, in Proximal Tubular Cells
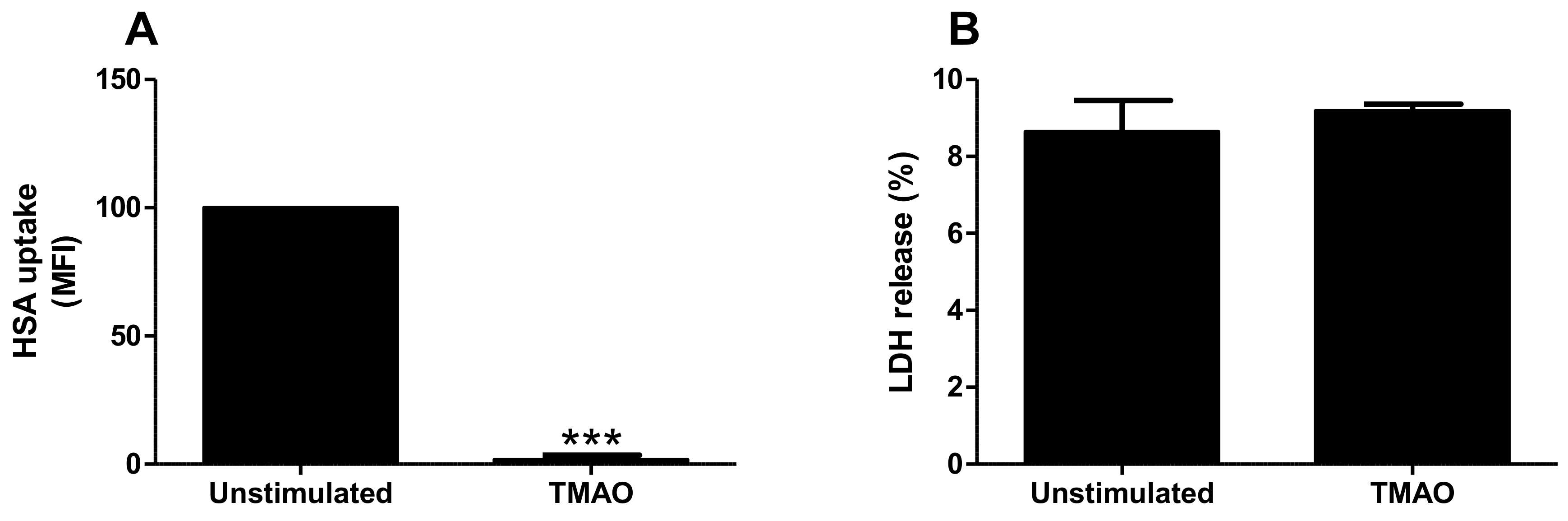
2.3. TMAO Reduces Albumin Uptake by Proximal Tubular Cells
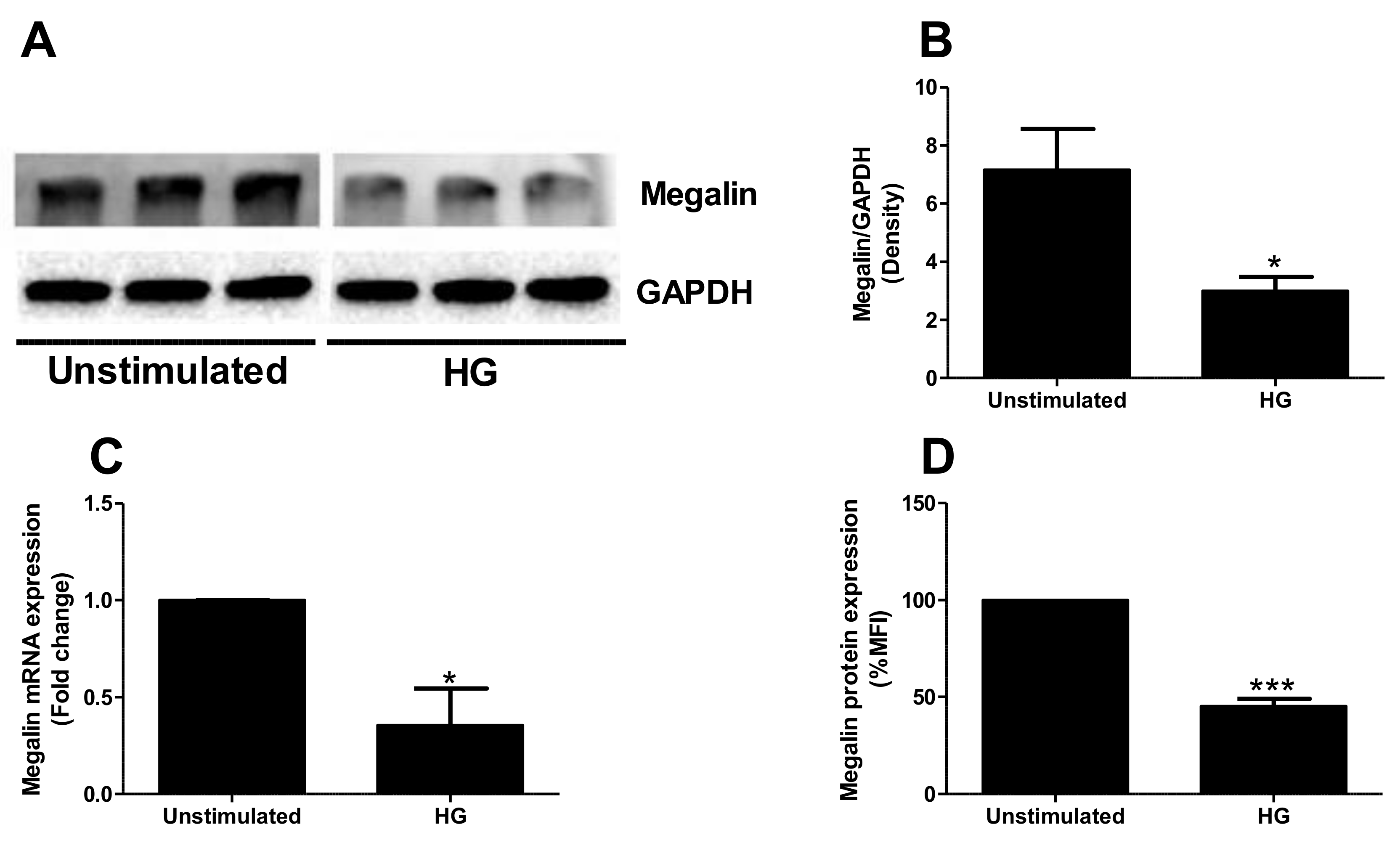
2.4. High Glucose Exposure Reduces Megalin Expression
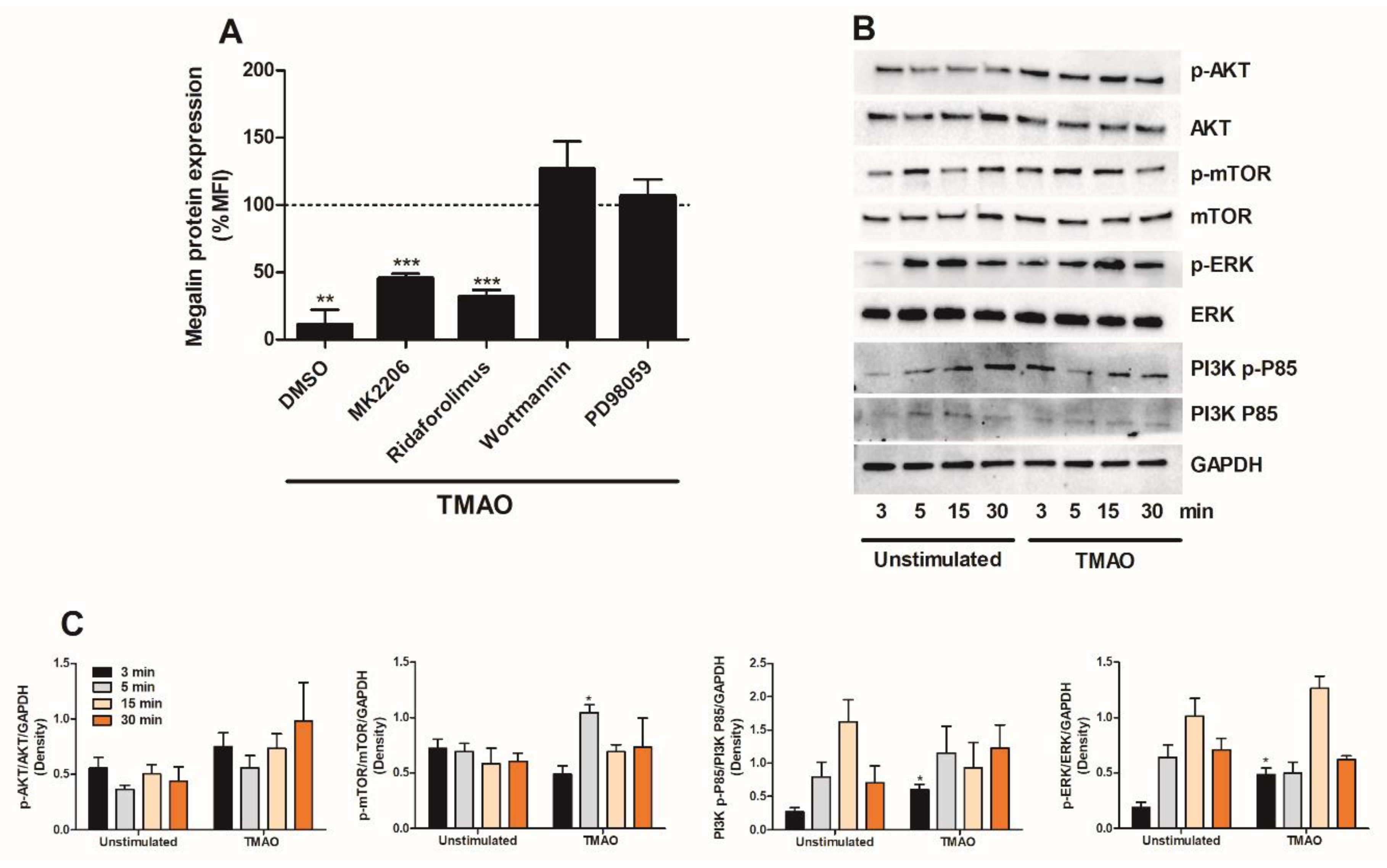
2.5. TMAO Suppresses Megalin Expression in Proximal Tubular Cells via PI3K and ERK
2.6. Candesartan, Dapagliflozin and Enalaprilat Counteract TMAOs Effect on Megalin
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Stimulation of Renal Proximal Tubule Epithelial Cells
4.3. Western Blot Analysis
4.4. RNA Isolation and Real Time RT-PCR
4.5. Microarray Analysis
4.6. Albumin Uptake and Cell Viability Assay
4.7. Flow Cytometry
4.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Golestaneh, L.; Alvarez, P.J.; Reaven, N.L.; Funk, S.E.; McGaughey, K.J.; Romero, A.; Brenner, M.S.; Onuigbo, M. All-cause costs increase exponentially with increased chronic kidney disease stage. Am. J. Manag. Care 2017, 23 (Suppl. 10), S163–S172. [Google Scholar] [PubMed]
- Wang, V.; Vilme, H.; Maciejewski, M.L.; Boulware, L.E. The Economic Burden of Chronic Kidney Disease and End-Stage Renal Disease. Semin. Nephrol. 2016, 36, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Elshahat, S.; Cockwell, P.; Maxwell, A.P.; Griffin, M.; O’Brien, T.; O’Neill, C. The impact of chronic kidney disease on developed countries from a health economics perspective: A systematic scoping review. PLoS ONE 2020, 15, e0230512. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Du, J.; Chen, Z.; Shi, D.; Qu, H. Effects of Microbiota-Driven Therapy on Circulating Trimethylamine-N-Oxide Metabolism: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2021, 8, 710567. [Google Scholar] [CrossRef]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered microbiome in chronic kidney disease: Systemic effects of gut-derived uremic toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Gruppen, E.G.; Garcia, E.; Connelly, M.A.; Jeyarajah, E.J.; Otvos, J.D.; Bakker, S.J.L.; Dullaart, R.P.F. TMAO is Associated with Mortality: Impact of Modestly Impaired Renal Function. Sci. Rep. 2017, 7, 13781. [Google Scholar] [CrossRef]
- Mueller, D.M.; Allenspach, M.; Othman, A.; Saely, C.H.; Muendlein, A.; Vonbank, A.; Drexel, H.; von Eckardstein, A. Plasma levels of trimethylamine-N-oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis 2015, 243, 638–644. [Google Scholar] [CrossRef]
- Missailidis, C.; Hällqvist, J.; Qureshi, A.R.; Barany, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.B.; Morse, B.L.; Djurdjev, O.; Tang, M.; Muirhead, N.; Barrett, B.; Holmes, D.T.; Madore, F.; Clase, C.M.; Rigatto, C.; et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 2016, 89, 1144–1152. [Google Scholar] [CrossRef]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Tang, Z.; You, L.; Wu, Y.; Liu, J.; Xue, J. Trimethylamine-N-oxide is an independent risk factor for hospitalization events in patients receiving maintenance hemodialysis. Ren. Fail. 2020, 42, 580–586. [Google Scholar] [CrossRef]
- Rodríguez-Iturbe, B.; García García, G. The role of tubulointerstitial inflammation in the progression of chronic renal failure. Nephron. Clin. Pract. 2010, 116, c81–c88. [Google Scholar] [CrossRef]
- Nast, C.C. The Renal Tubulointerstitium. Adv. Chronic. Kidney Dis. 2017, 24, 55–56. [Google Scholar] [CrossRef]
- Kapetanaki, S.; Kumawat, A.K.; Persson, K.; Demirel, I. The Fibrotic Effects of TMAO on Human Renal Fibroblasts Is Mediated by NLRP3, Caspase-1 and the PERK/Akt/mTOR Pathway. Int. J. Mol. Sci. 2021, 22, 11864. [Google Scholar] [CrossRef]
- Hodgkins, K.S.; Schnaper, H.W. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr. Nephrol. 2012, 27, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Dickson, L.E.; Wagner, M.C.; Sandoval, R.M.; Molitoris, B.A. The proximal tubule and albuminuria: Really! J. Am. Soc. Nephrol. 2014, 25, 443–453. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, G.; Bazzi, C. Pathophysiology of proteinuria. Kidney Int. 2003, 63, 809–825. [Google Scholar] [CrossRef] [Green Version]
- Baines, R.J.; Brunskill, N.J. Tubular toxicity of proteinuria. Nat. Rev. Nephrol. 2011, 7, 177–180. [Google Scholar] [CrossRef]
- Peruchetti, D.B.; Silva-Aguiar, R.P.; Siqueira, G.M.; Dias, W.B.; Caruso-Neves, C. High glucose reduces megalin-mediated albumin endocytosis in renal proximal tubule cells through protein kinase B O-GlcNAcylation. J. Biol. Chem. 2018, 293, 11388–11400. [Google Scholar] [CrossRef] [Green Version]
- Dong, F.; Jiang, S.; Tang, C.; Wang, X.; Ren, X.; Wei, Q.; Tian, J.; Hu, W.; Guo, J.; Fu, X.; et al. Trimethylamine N-oxide promotes hyperoxaluria-induced calcium oxalate deposition and kidney injury by activating autophagy. Free Radic. Biol. Med. 2022, 179, 288–300. [Google Scholar] [CrossRef]
- Zhang, W.; Miikeda, A.; Zuckerman, J.; Jia, X.; Charugundla, S.; Zhou, Z.; Kaczor-Urbanowicz, K.E.; Magyar, C.; Guo, F.; Wang, Z.; et al. Inhibition of microbiota-dependent TMAO production attenuates chronic kidney disease in mice. Sci. Rep. 2021, 11, 518. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, N.; Jia, J.; Gu, L.; Du, Y.; Tang, G.; Wang, X.; Yang, M.; Yuan, W. Trimethylamine N-oxide mediated Y-box binding protein-1 nuclear translocation promotes cell cycle progression by directly downregulating Gadd45a expression in a cellular model of chronic kidney disease. Life Sci. 2021, 271, 119173. [Google Scholar] [CrossRef]
- Christensen, E.I.; Birn, H. Megalin and cubilin: Synergistic endocytic receptors in renal proximal tubule. Am. J. Physiol. Renal. Physiol. 2001, 280, F562–F573. [Google Scholar] [CrossRef]
- Christensen, E.I.; Birn, H.; Storm, T.; Weyer, K.; Nielsen, R. Endocytic receptors in the renal proximal tubule. Physiology 2012, 27, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, R.; Yammani, R.; Bauer, J.A.; Kalra, S.; Seetharam, S.; Seetharam, B. Interactions of cubilin with megalin and the product of the amnionless gene (AMN): Effect on its stability. Biochem. J. 2008, 410, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Yammani, R.R.; Seetharam, S.; Seetharam, B. Cubilin and megalin expression and their interaction in the rat intestine: Effect of thyroidectomy. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E900–E907. [Google Scholar] [CrossRef]
- Moestrup, S.K.; Kozyraki, R.; Kristiansen, M.; Kaysen, J.H.; Rasmussen, H.H.; Brault, D.; Pontillon, F.; Goda, F.O.; Christensen, E.I.; Hammond, T.G.; et al. The intrinsic factor-vitamin B12 receptor and target of teratogenic antibodies is a megalin-binding peripheral membrane protein with homology to developmental proteins. J. Biol. Chem. 1998, 273, 5235–5242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gansevoort, R.T.; de Jong, P.E. The case for using albuminuria in staging chronic kidney disease. J. Am. Soc. Nephrol. 2009, 20, 465–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Q.; Zheng, B.; Liu, N.; Liu, J.; Liu, W.; Huang, X.; Zeng, X.; Chen, L.; Li, Z.; Ouyang, D. Trimethylamine N-Oxide Exacerbates Renal Inflammation and Fibrosis in Rats With Diabetic Kidney Disease. Front. Physiol. 2021, 12, 682482. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.S.; Ramasamy, T.S.; Murphy, N.; Holt, M.K.; Czapiewski, R.; Wei, S.K.; Cui, W. PI3K/mTORC2 regulates TGF-β/Activin signalling by modulating Smad2/3 activity via linker phosphorylation. Nat. Commun. 2015, 6, 7212. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Su, P.; Wang, L.; Chen, J.; Zimmermann, M.; Genbacev, O.; Afonja, O.; Horne, M.C.; Tanaka, T.; Duan, E.; et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7840–7845. [Google Scholar] [CrossRef] [Green Version]
- Paling, N.R.; Wheadon, H.; Bone, H.K.; Welham, M.J. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J. Biol. Chem. 2004, 279, 48063–48070. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.M.; Reynolds, D.; Cliff, T.; Ohtsuka, S.; Mattheyses, A.L.; Sun, Y.; Menendez, L.; Kulik, M.; Dalton, S. Signaling network crosstalk in human pluripotent cells: A Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem. Cell 2012, 10, 312–326. [Google Scholar] [CrossRef] [Green Version]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell Physiol. 2019, 234, 2373–2385. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Margaria, J.P.; Campa, C.C.; De Santis, M.C.; Hirsch, E.; Franco, I. The PI3K/Akt/mTOR pathway in polycystic kidney disease: A complex interaction with polycystins and primary cilium. Cell Signal 2020, 66, 109468. [Google Scholar] [CrossRef]
- Miricescu, D.; Balan, D.G.; Tulin, A.; Stiru, O.; Vacaroiu, I.A.; Mihai, D.A.; Popa, C.C.; Papacocea, R.I.; Enyedi, M.; Sorin, N.A.; et al. PI3K/AKT/mTOR signalling pathway involvement in renal cell carcinoma pathogenesis (Review). Exp. Ther. Med. 2021, 21, 540. [Google Scholar] [CrossRef]
- Lan, A.; Du, J. Potential role of Akt signaling in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Athavale, A.; Roberts, D.M. Management of proteinuria: Blockade of the renin-angiotensin-aldosterone system. Aust. Prescr. 2020, 43, 121–125. [Google Scholar] [CrossRef]
- Hosojima, M.; Sato, H.; Yamamoto, K.; Kaseda, R.; Soma, T.; Kobayashi, A.; Suzuki, A.; Kabasawa, H.; Takeyama, A.; Ikuyama, K.; et al. Regulation of megalin expression in cultured proximal tubule cells by angiotensin II type 1A receptor- and insulin-mediated signaling cross talk. Endocrinology 2009, 150, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Corrêa, J.W.N.; Boaro, K.R.; Sene, L.B.; Polidoro, J.Z.; Salles, T.A.; Martins, F.L.; Bendhack, L.M.; Girardi, A.C.C. Antiproteinuric and Hyperkalemic Mechanisms Activated by Dual Versus Single Blockade of the RAS in Renovascular Hypertensive Rats. Front. Physiol. 2021, 12, 656460. [Google Scholar] [CrossRef]
- Kjeldsen, S.E.; Stålhammar, J.; Hasvold, P.; Bodegard, J.; Olsson, U.; Russell, D. Effects of losartan vs candesartan in reducing cardiovascular events in the primary treatment of hypertension. J. Hum. Hypertens. 2010, 24, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Van Liefde, I.; Vauquelin, G. Sartan-AT1 receptor interactions: In vitro evidence for insurmountable antagonism and inverse agonism. Mol. Cell Endocrinol. 2009, 302, 237–243. [Google Scholar] [CrossRef]
- Fabiani, M.E.; Dinh, D.T.; Nassis, L.; Casley, D.J.; Johnston, C.I. In vivo inhibition of angiotensin receptors in the rat kidney by candesartan cilexetil: A comparison with losartan. Am. J. Hypertens. 2000, 13, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Ojima, M.; Inada, Y.; Shibouta, Y.; Wada, T.; Sanada, T.; Kubo, K.; Nishikawa, K. Candesartan (CV-11974) dissociates slowly from the angiotensin AT1 receptor. Eur. J. Pharmacol. 1997, 319, 137–146. [Google Scholar] [CrossRef]
- Fuchs, B.; Breithaupt-Grögler, K.; Belz, G.G.; Roll, S.; Malerczyk, C.; Herrmann, V.; Spahn-Langguth, H.; Mutschler, E. Comparative pharmacodynamics and pharmacokinetics of candesartan and losartan in man. J. Pharm. Pharmacol. 2000, 52, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, H.; Hayashi, K.; Homma, K.; Yoshioka, K.; Kanda, T.; Takamatsu, I.; Tatematsu, S.; Wakino, S.; Saruta, T. Differing anti-proteinuric action of candesartan and losartan in chronic renal disease. Hypertens. Res. 2003, 26, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Klarström Engström, K.; Zhang, B.; Demirel, I. Human renal fibroblasts are strong immunomobilizers during a urinary tract infection mediated by uropathogenic Escherichia coli. Sci. Rep. 2019, 9, 2296. [Google Scholar] [CrossRef]
- Demirel, I.; Persson, A.; Brauner, A.; Särndahl, E.; Kruse, R.; Persson, K. Activation of the NLRP3 Inflammasome Pathway by Uropathogenic Escherichia coli Is Virulence Factor-Dependent and Influences Colonization of Bladder Epithelial Cells. Front. Cell Infect. Microbiol. 2018, 8, 81. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapetanaki, S.; Kumawat, A.K.; Persson, K.; Demirel, I. TMAO Suppresses Megalin Expression and Albumin Uptake in Human Proximal Tubular Cells Via PI3K and ERK Signaling. Int. J. Mol. Sci. 2022, 23, 8856. https://doi.org/10.3390/ijms23168856
Kapetanaki S, Kumawat AK, Persson K, Demirel I. TMAO Suppresses Megalin Expression and Albumin Uptake in Human Proximal Tubular Cells Via PI3K and ERK Signaling. International Journal of Molecular Sciences. 2022; 23(16):8856. https://doi.org/10.3390/ijms23168856
Chicago/Turabian StyleKapetanaki, Stefania, Ashok Kumar Kumawat, Katarina Persson, and Isak Demirel. 2022. "TMAO Suppresses Megalin Expression and Albumin Uptake in Human Proximal Tubular Cells Via PI3K and ERK Signaling" International Journal of Molecular Sciences 23, no. 16: 8856. https://doi.org/10.3390/ijms23168856