
JIB-04, a Pan-Inhibitor of Histone Demethylases, Targets Histone-Lysine-Demethylase-Dependent AKT Pathway, Leading to Cell Cycle Arrest and Inhibition of Cancer Stem-Like Cell Properties in Hepatocellular Carcinoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. JIB-04 Caused Reduced Cell Proliferation and Cell Cycle Arrest in HCC Cells
2.2. JIB-04 Treatment Interrupted HCC Cell Migration and Invasion
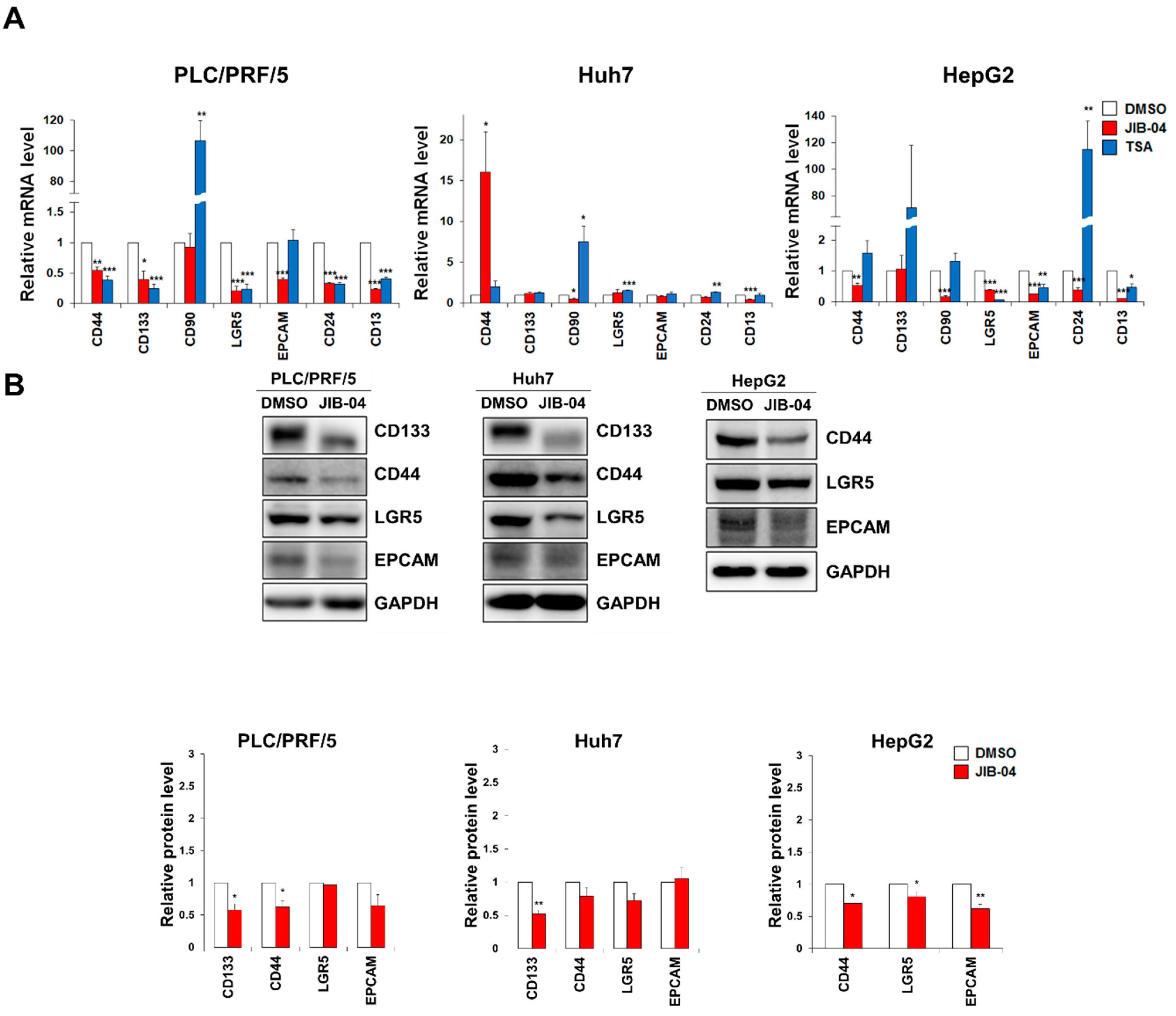
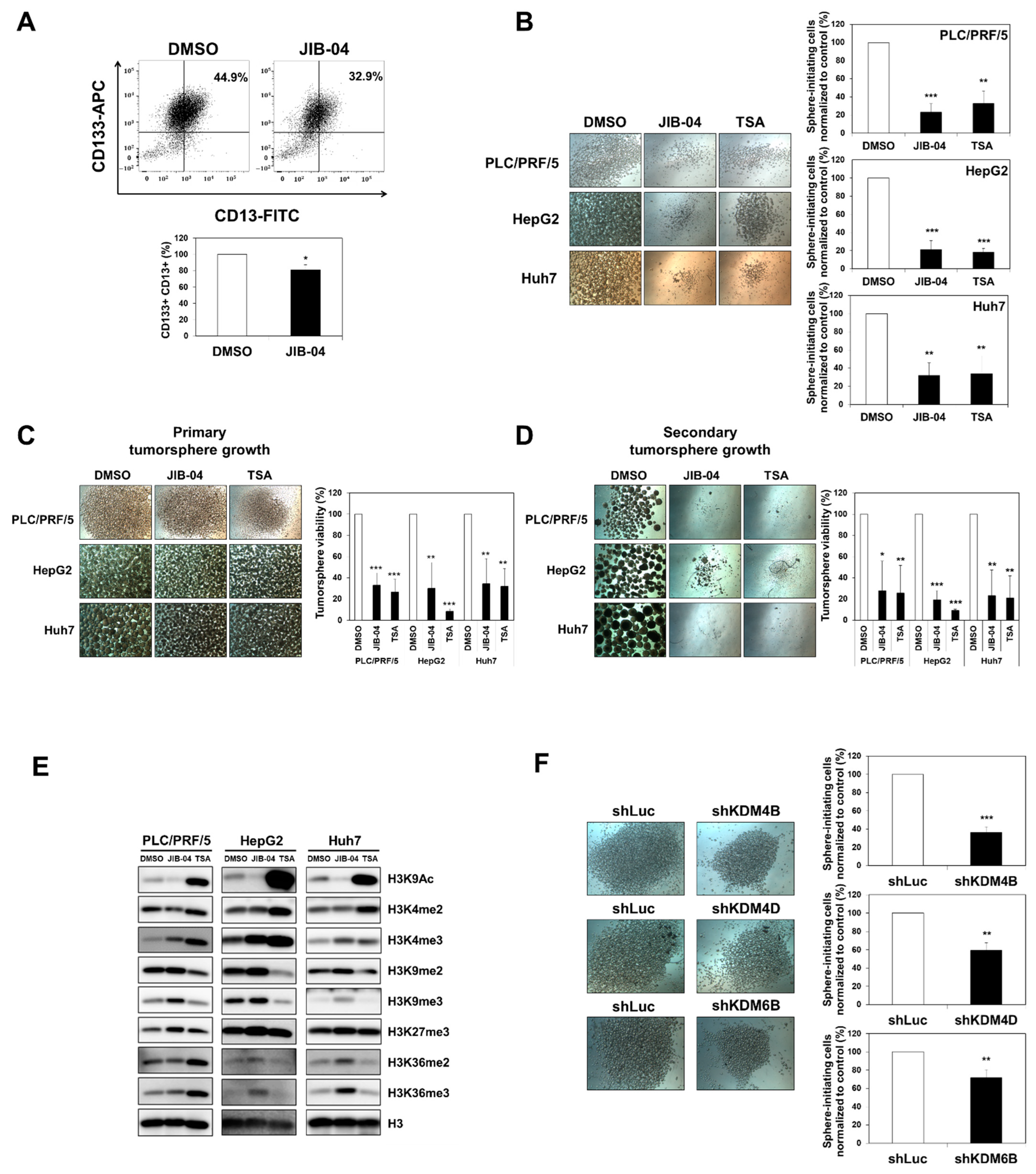
2.3. JIB-04 Treatment Reduced the Expression Levels of CSC Markers
2.4. JIB-04 Diminished the Tumor Initiation, Growth, and Relapse Abilities of Tumorspheres Derived from Liver CSCs
2.5. Transcriptome Analysis Revealed JIB-04-Targeted Pathways in HCC Cells
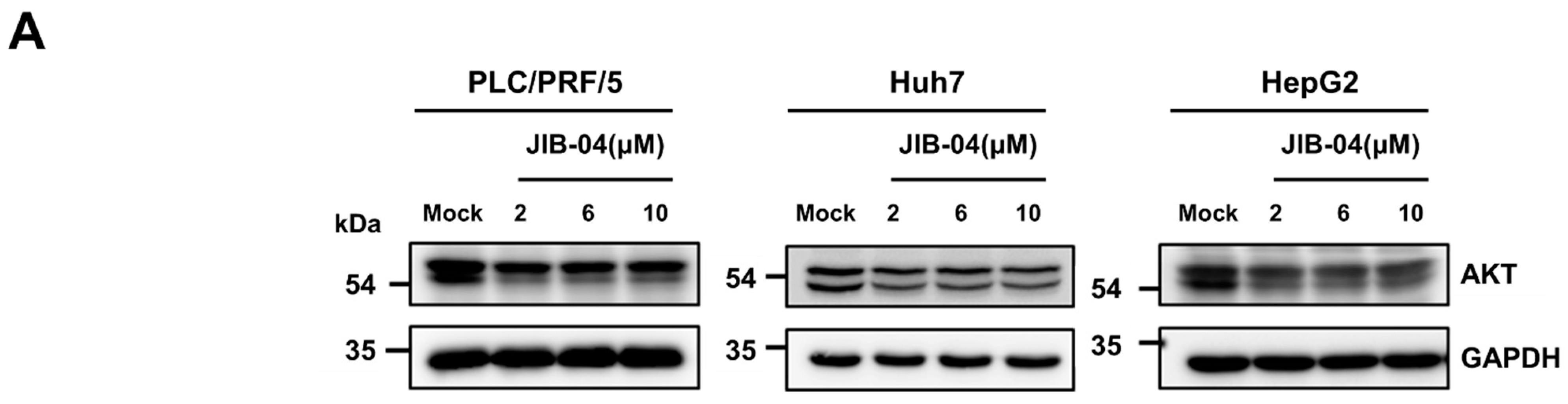
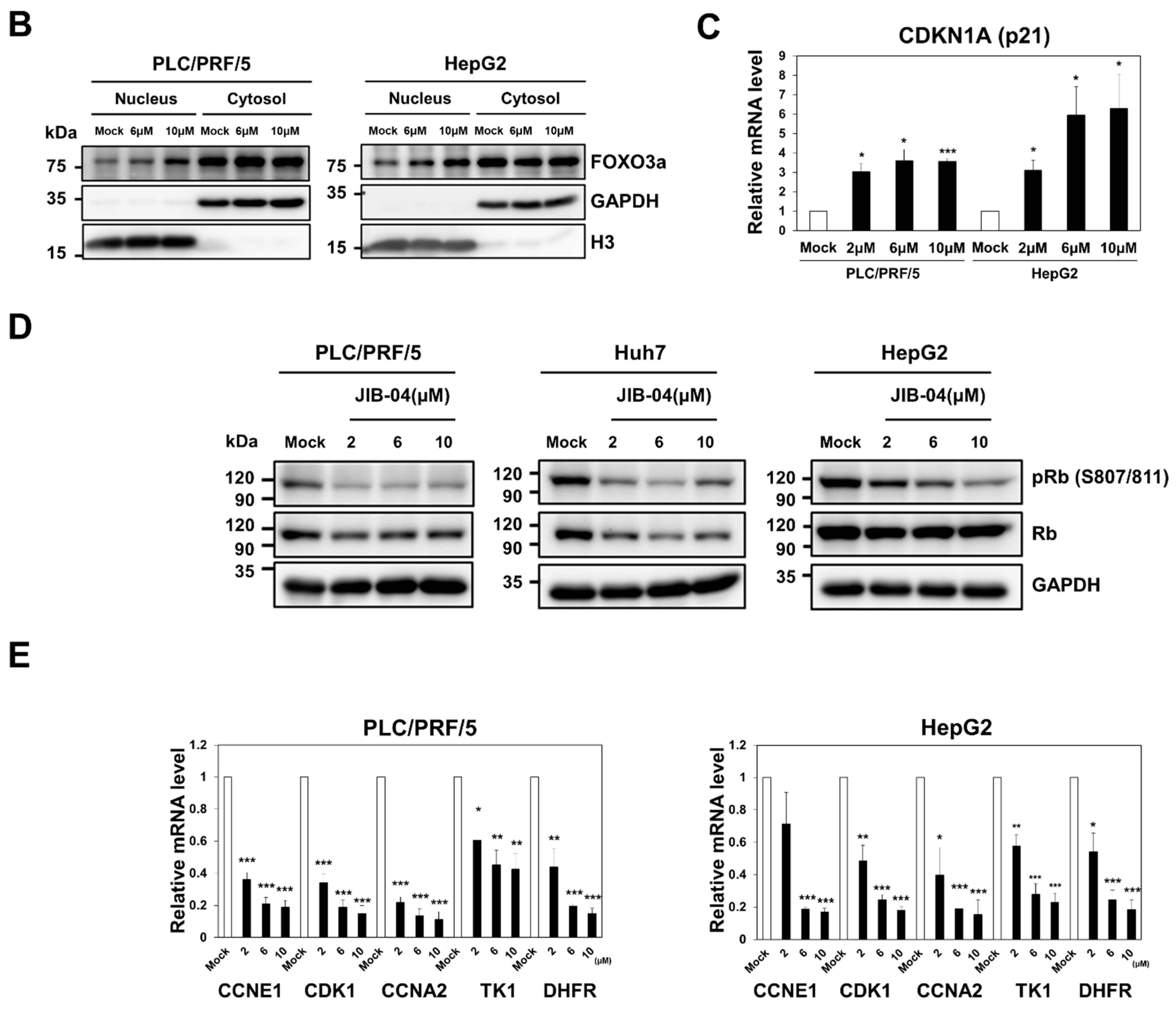
2.6. JIB-04 Induced Anti-Cancer Effects by Targeting AKT-FOXO3a-p21-RB-E2F Axis
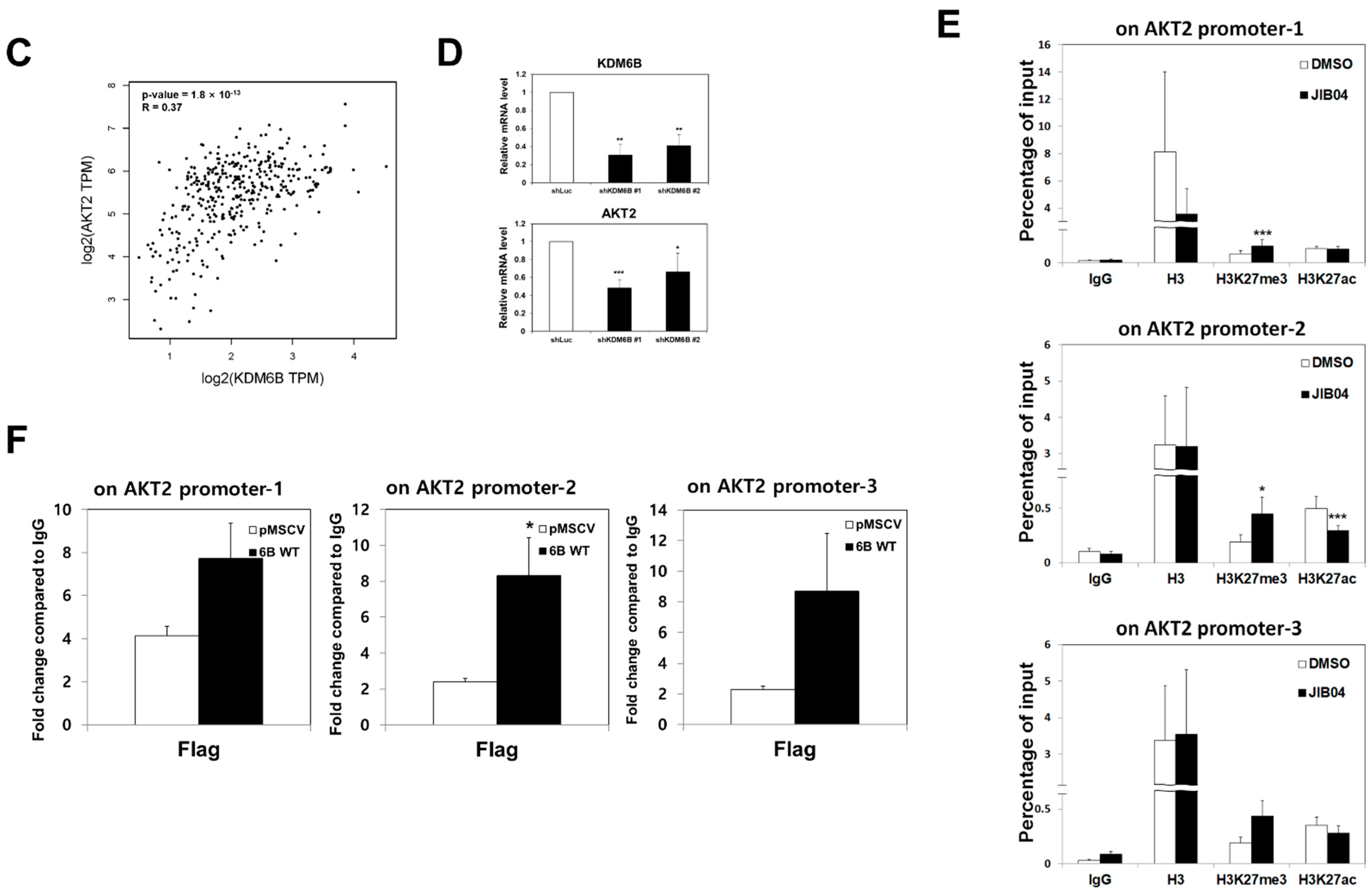
2.7. KDM6B Regulated AKT2 Expression through H3K27me3 Modifications
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Drug Treatment
4.2. Lentiviral Production and Infection
4.3. Retroviral Production and Infection
4.4. Cell Growth Assay
4.5. Cell Cycle Analysis
4.6. Western Blotting
4.7. Fluorescent Immunocytochemistry
4.8. Chromatin Isolation Assay
4.9. ChIP Assay
4.10. Isolation of RNA and Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR) Analysis
4.11. Cell Migration and Invasion Assay
4.12. Wound-Healing Assay
4.13. Tumorsphere Formation
4.14. Histone Preparation
4.15. FACS Double-Staining
4.16. RNA Sequencing
4.17. Gene Correlation Analysis
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Con sent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Laursen, L. A preventable cancer. Nature 2014, 516, S2–S3. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma. Hepatology 2005, 42, 1208–1236. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Takayama, T.; Geschwind, J.; Marrero, J.A.; Bronowicki, J. Current Approaches to the Treatment of Early Hepatocellular Carcinoma. Oncologist 2010, 15, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.K.; Lam, C.T.; Poon, R.T.P.; Fan, S.T. Significance of CD90+ Cancer Stem Cells in Human Liver Cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.M.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukowati, C.H.C.; Rosso, N.; Crocè, L.S.; Tiribelli, C. Hepatic cancer stem cells and drug resistance: Relevance in targeted therapies for hepatocellular carcinoma. World J. Hepatol. 2010, 2, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Peña-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035. [Google Scholar] [CrossRef]
- Parrish, J.K.; McCann, T.S.; Sechler, M.; Sobral, L.M.; Ren, W.; Jones, K.L.; Tan, A.C.; Jedlicka, P. The Jumonji-domain histone demethylase inhibitor JIB-04 deregulates oncogenic programs and increases DNA damage in Ewing Sarcoma, resulting in impaired cell proliferation and survival, and reduced tumor growth. Oncotarget 2018, 9, 33110–33123. [Google Scholar] [CrossRef] [Green Version]
- Banelli, B.; Daga, A.; Forlani, A.; Allemanni, G.; Marubbi, D.; Pia Pistillo, M.; Profumo, A.; Romani, M. Small molecules targeting histone demethylase genes (KDMs) inhibit growth of Temozolomide-resistant glioblastoma cells. Oncotarget 2017, 8, 34896–34910. [Google Scholar] [CrossRef] [Green Version]
- Dalvi, M.P.; Wang, L.; Zhong, R.; Kollipara, R.K.; Park, H.; Bayo, J.; Yenerall, P.; Zhou, Y.; Timmons, B.C.; Rodriguez-Canales, J.; et al. Taxane-Platin-Resistant Lung Cancers Co-develop Hypersensitivity to JumonjiC Demethylase Inhibitors. Cell Rep. 2017, 19, 1669–1684. [Google Scholar] [CrossRef]
- Kim, M.S.; Cho, H.I.; Yoon, H.J.; Ahn, Y.-H.; Park, E.J.; Jin, Y.H.; Jang, Y.K. JIB-04, A Small Molecule Histone Demethylase Inhibitor, Selectively Targets Colorectal Cancer Stem Cells by Inhibiting the Wnt/βCatenin Signaling Pathway. Sci. Rep. 2018, 8, 2035. [Google Scholar] [CrossRef]
- Hers, I.; Vincent, E.E.; Tavaré, J.M. Akt signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhu, J.; Du, W.; Ning, W.; Zhang, Y.; Zeng, Y.; Liu, Z.; Huang, J.A. AKT2 drives cancer progression and is negatively modulated by miR-124 in human lung adenocarcinoma. Respir. Res. 2020, 21, 227. [Google Scholar] [CrossRef] [PubMed]
- Rychahou, P.G.; Kang, J.; Gulhati, P.; Doan, H.Q.; Chen, L.A.; Xiao, S.Y.; Chung, D.H.; Evers, B.M. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 20315–20320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringel, M.D.; Hayre, N.; Saito, J.; Saunier, B.; Schuppert, F.; Burch, H.; Bernet, V.; Burman, K.D.; Kohn, L.D.; Saji, M. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001, 61, 6105–6111. [Google Scholar]
- Sahlberg, S.H.; Gustafsson, A.S.; Pendekanti, P.N.; Glimelius, B.; Stenerlöw, B. The influence of AKT isoforms on radiation sensitivity and DNA repair in colon cancer cell lines. Tumor Biol. 2014, 35, 3525–3534. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, M.; Wu, S.; Ye, M.; Lin, Z.; Shun, T.; Duan, C. Microrna-137-regulated akt serine/threonine kinase 2 inhibits tumor growth and sensitizes cisplatin in patients with non-small cell lung cancer. Oncol. Lett. 2018, 16, 1876–1884. [Google Scholar] [CrossRef] [Green Version]
- Honjo, S.; Ajani, J.A.; Scott, A.W.; Chen, Q.; Skinner, H.D.; Stroehlein, J.; Johnson, R.L.; Song, S. Metformin sensitizes chemotherapy by targeting cancer stem cells and the mTOR pathway in esophageal cancer. Int. J. Oncol. 2014, 45, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Dubrovska, A.; Kim, S.; Salamone, R.J.; Walker, J.R.; Maira, S.-M.; García-Echeverría, C.; Schultz, P.G.; Reddy, V.A. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl. Acad. Sci. USA 2009, 106, 268–273. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Jin, Y.; Jin, S.; Tan, Z.; Peng, Z.; Kuang, Y. Arsenite inhibits the function of CD133+ CD13+ liver cancer stem cells by reducing PML and Oct4 protein expression. Tumor Biol. 2016, 37, 14103–14115. [Google Scholar] [CrossRef]
- Liang, J.; Slingerland, J.M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003, 2, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, K.; Nakamura, A.; Sugimoto, Y.; Tsuruo, T.; Fujita, N. FOXO transcription factor-dependent p15INK4b and p19 INK4d expression. Oncogene 2008, 27, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Gomis, R.R.; Alarcon, C.; He, W.; Wang, Q.; Seoane, J.; Lash, A.; Massague, J. A FoxO-Smad synexpression group in human keratinocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 12747–12752. [Google Scholar] [CrossRef] [Green Version]
- Seoane, J.; Le, H.; Shen, L.; Anderson, S.A.; Massague, J. Integration of Smad and Forkhead Pathways in the Control of Neuroepithelial and Glioblastoma Cell Proliferation Joan. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Medema, R.H.; Kops, G.J.P.L.; Bos, J.L.; Burgering, B.M.T. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27(kip1). Nature 2000, 404, 782–787. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Knudsen, K.E. Tailoring to RB: Tumour suppressor status and therapeutic response. Nat. Rev. Cancer 2008, 8, 714–724. [Google Scholar] [CrossRef]
- Jiao, W.; Datta, J.; Lin, H.M.; Dundr, M.; Rane, S.G. Nucleocytoplasmic shuttling of the retinoblastoma tumor suppressor protein via Cdk phosphorylation-dependent nuclear export. J. Biol. Chem. 2006, 281, 38098–38108. [Google Scholar] [CrossRef] [Green Version]
- Rotili, D.; Mai, A. Targeting histone demethylases: A new avenue for the fight against cancer. Genes Cancer 2011, 2, 663–679. [Google Scholar] [CrossRef]
- Ma, S. Deciphering ZIC2/OCT4 signaling as a vulnerability in liver cancer stem cells. Transl. Cancer Res. 2016, 5, 722–724. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Wang, X. Cancer stem cells in the development of liver cancer. J. Clin. Investig. 2013, 123, 1911–1918. [Google Scholar] [CrossRef]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Ohkuma, M.; Kim, H.M.; Akita, H.; Takiuchi, D.; Hatano, H.; Nagano, H.; et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 2010, 120, 3326–3339. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.K.W.; Castilho, A.; Cheung, V.C.H.; Tang, K.H.; Ma, S.; Ng, I.O.L. CD24 + Liver Tumor-Initiating Cells Drive Self-Renewal and Tumor Initiation through STAT3-Mediated NANOG Regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.J.; Wang, J.; Xiao, H.L.; Guo, Y.; Wang, T.; Li, Q.; Liu, L.; Luo, X.; Fan, L.L.; Lin, L.; et al. Lysine-specific demethylase 1 promotes the stemness and chemoresistance of Lgr5+liver cancer initiating cells by suppressing negative regulators of β-catenin signaling. Oncogene 2015, 34, 3188–3198. [Google Scholar] [CrossRef]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.W.; Wo, J.Y.H.; Ng, I.O.L.; Zheng, B.J.; Guan, X.Y. Identification and Characterization of Tumorigenic Liver Cancer Stem/Progenitor Cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef]
- Mima, K.; Okabe, H.; Ishimoto, T.; Hayashi, H.; Nakagawa, S.; Kuroki, H.; Watanabe, M.; Beppu, T.; Tamada, M.; Nagano, O.; et al. CD44s regulates the TGF-β-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. 2012, 72, 3414–3423. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. EpCAM-Positive Hepatocellular Carcinoma Cells Are Tumor-Initiating Cells with Stem/Progenitor Cell Features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Evangelisti, C.; Follo, M.Y.; Ramazzotti, G.; Fini, M.; Giardino, R.; Manzoli, L.; McCubrey, J.A.; Cocco, L. Targeting the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin Signaling Network in Cancer Stem Cells. Curr. Med. Chem. 2011, 18, 2715–2726. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, J.; Zhang, X.; Zhou, H.; Liu, G.; Li, Q. Cancer Stem Cells: A Potential Breakthrough in HCC-Targeted Therapy. Front. Pharmacol. 2020, 11, 198. [Google Scholar] [CrossRef]
- Carmona, F.J.; Montemurro, F.; Kannan, S.; Rossi, V.; Verma, C.; Baselga, J.; Scaltriti, M. AKT signaling in ERBB2-amplified breast cancer. Pharmacol. Ther. 2016, 158, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grǎdinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT kinases in cancer: Implications for therapeutic targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar] [CrossRef]
- Fresno Vara, J.Á.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. P13K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Gener, P.; Rafael, D.; Seras-franzoso, J.; Perez, A.; Pindado, L.A.; Casas, G.; Arango, D.; Fernández, Y.; Díaz-riascos, Z.V.; Abasolo, I.; et al. Pivotal role of AKT2 during dynamic phenotypic change of breast cancer stem cells. Cancers 2019, 11, 1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargini, R.; Cerliani, J.P.; Escoll, M.; Antõn, I.M.; Wandosell, F. Cancer stem cell-like phenotype and survival are coordinately regulated by Akt/FoxO/bim pathway. Stem Cells 2015, 33, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, M.S.; Park, S.H.; Jang, Y.K. Tousled-like kinase 1 is a negative regulator of core transcription factors in murine embryonic stem cells. Sci. Rep. 2018, 8, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Yu, S.E.; Chai, Y.G.; Jang, Y.K. CDK2-dependent phosphorylation of Suv39H1 is involved in control of heterochromatin replication during cell cycle progression. Nucleic Acids Res. 2014, 42, 6196–6207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, M.S.; Kim, M.A.; Jang, Y.K. Calmidazolium chloride inhibits growth of murine embryonal carcinoma cells, a model of cancer stem-like cells. Toxicol. Vitr. 2016, 35, 86–92. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [Green Version]
- Bahmad, H.F.; Cheaito, K.; Chalhoub, R.M.; Hadadeh, O.; Monzer, A.; Ballout, F.; El-Hajj, A.; Mukherji, D.; Liu, Y.N.; Daoud, G.; et al. Sphere-Formation Assay: Three-dimensional in vitro culturing of prostate cancer stem/Progenitor sphere-forming cells. Front. Oncol. 2018, 8, 347. [Google Scholar] [CrossRef] [Green Version]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Sphere Culture of Murine Lung Cancer Cell Lines Are Enriched with Cancer Initiating Cells. PLoS ONE 2012, 7, e49752. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Kim, J.-S.; Cho, H.-I.; Jo, S.-R.; Jang, Y.-K. JIB-04, a Pan-Inhibitor of Histone Demethylases, Targets Histone-Lysine-Demethylase-Dependent AKT Pathway, Leading to Cell Cycle Arrest and Inhibition of Cancer Stem-Like Cell Properties in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2022, 23, 7657. https://doi.org/10.3390/ijms23147657
Lee J, Kim J-S, Cho H-I, Jo S-R, Jang Y-K. JIB-04, a Pan-Inhibitor of Histone Demethylases, Targets Histone-Lysine-Demethylase-Dependent AKT Pathway, Leading to Cell Cycle Arrest and Inhibition of Cancer Stem-Like Cell Properties in Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences. 2022; 23(14):7657. https://doi.org/10.3390/ijms23147657
Chicago/Turabian StyleLee, Jina, Ji-Soo Kim, Hye-In Cho, So-Ra Jo, and Yeun-Kyu Jang. 2022. "JIB-04, a Pan-Inhibitor of Histone Demethylases, Targets Histone-Lysine-Demethylase-Dependent AKT Pathway, Leading to Cell Cycle Arrest and Inhibition of Cancer Stem-Like Cell Properties in Hepatocellular Carcinoma Cells" International Journal of Molecular Sciences 23, no. 14: 7657. https://doi.org/10.3390/ijms23147657