
Measuring Molecular Diffusion in Dynamic Subcellular Nanostructures by Fast Raster Image Correlation Spectroscopy and 3D Orbital Tracking

 , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Fast RICS Captures Molecular Diffusion in Dynamic Nanostructures: Validation Tests in Solution
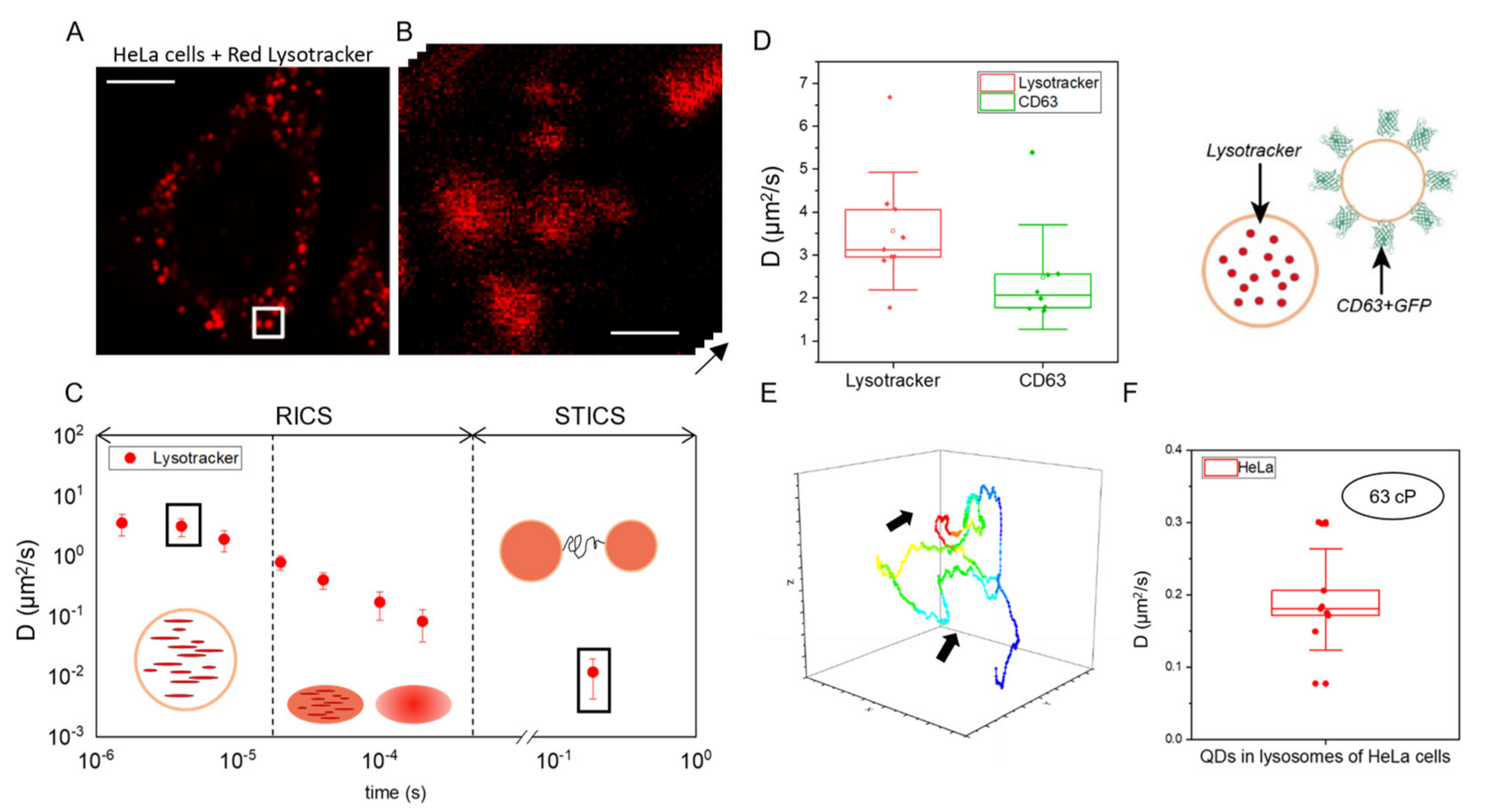
2.2. Fast RICS Captures Molecular Diffusion in Dynamic Nanostructures: Validation Tests in Living Cells
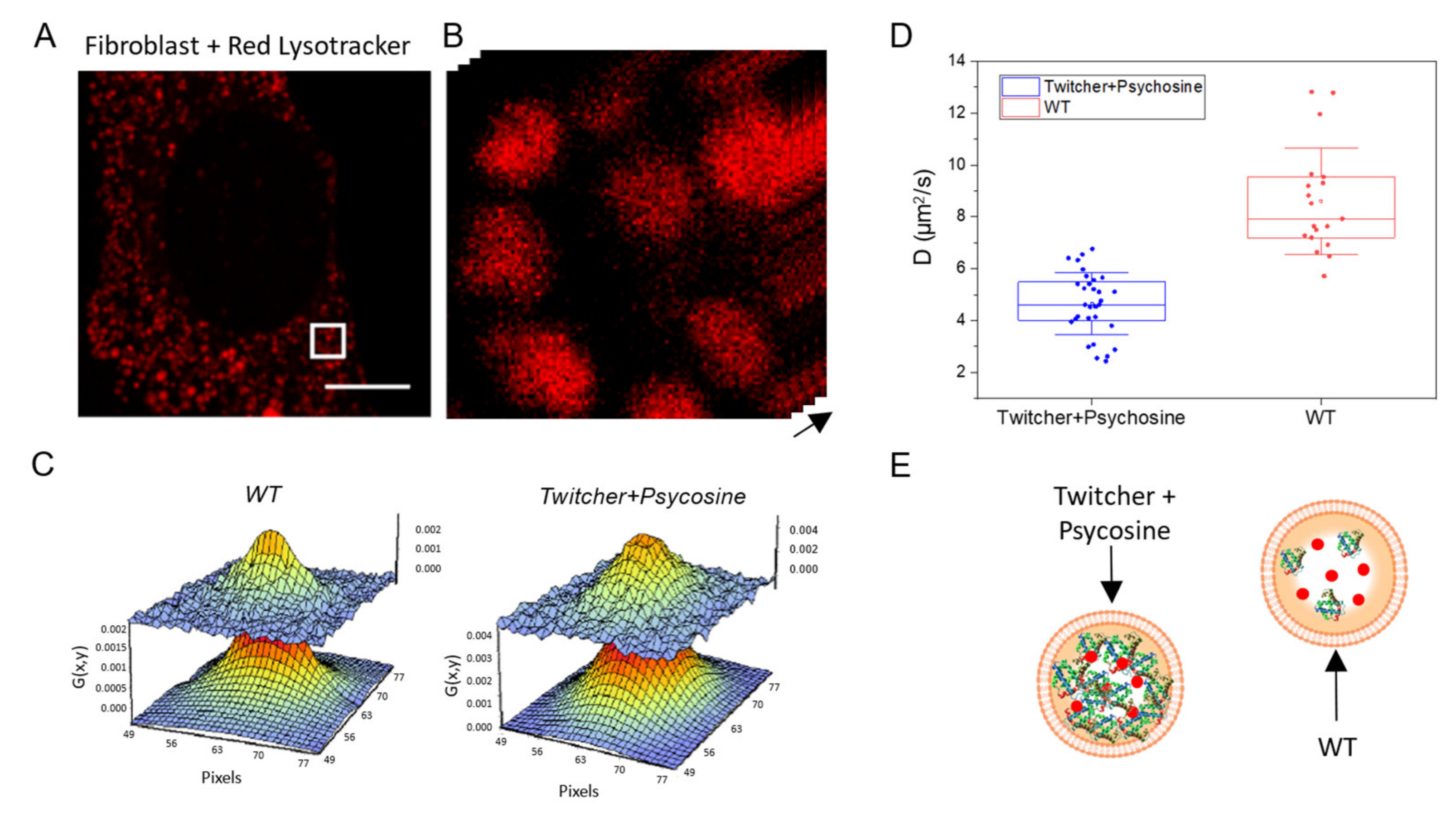
2.3. Measuring Diffusion to Probe Viscosity of Intracellular Dynamic Nanostructures: Application to Lysosomes in Krabbe Disease
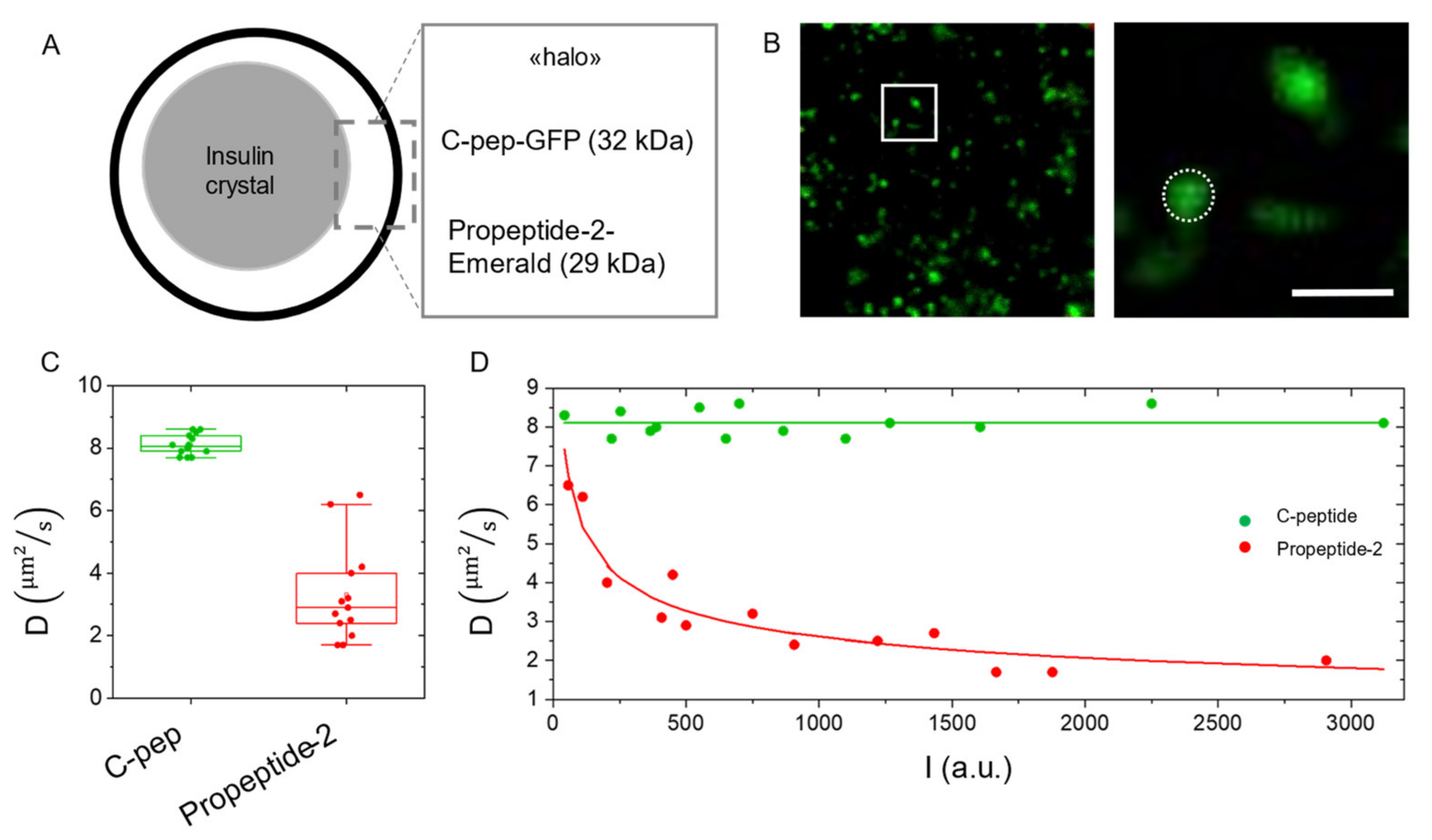
2.4. Measuring Diffusion to Probe Molecular Aggregation within Subcellular Nanostructures: Application to the Insulin Secretory Granule
3. Materials and Methods
3.1. Preparation of Fluorophore Solutions
3.2. Preparation of Liposomes and Liposome-Based Samples
3.3. Cell Culture and Treatments
3.4. Raster Image Correlation Spectroscopy in Tunable Timescales: Measurement and Data Analysis
3.5. Spatio-Temporal Image Correlation Spectroscopy: Measurement and Data Analysis
3.6. D Orbital Tracking Experiments
3.7. Calibration of Hydrodynamic Radius of QDs in Buffer Solution
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Kroug, M. Ground-state-depletion fluorscence microscopy: A concept for breaking the diffraction resolution limit. Appl. Phys. B 1995, 60, 495–497. [Google Scholar] [CrossRef]
- Hell, S.W.; Jakobs, S.; Kastrup, L. Imaging and writing at the nanoscale with focused visible light through saturable optical transitions. Appl. Phys. A 2003, 77, 859–860. [Google Scholar] [CrossRef]
- Chung, E.; Kim, D.; Cui, Y.; Kim, Y.-H.; So, P.T.C. Two-Dimensional Standing Wave Total Internal Reflection Fluorescence Microscopy: Superresolution Imaging of Single Molecular and Biological Specimens. Biophys. J. 2007, 93, 1747–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, M.G.L.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-Dimensional Resolution Doubling in Wide-Field Fluorescence Microscopy by Structured Illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelrod, D. Evanescent Excitation and Emission in Fluorescence Microscopy. Biophys. J. 2013, 104, 1401–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [Green Version]
- Mukamel, E.A.; Babcock, H.; Zhuang, X. Statistical Deconvolution for Superresolution Fluorescence Microscopy. Biophys. J. 2012, 102, 2391–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [Green Version]
- Sharonov, A.; Hochstrasser, R.M. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. USA 2006, 103, 18911–18916. [Google Scholar] [CrossRef] [Green Version]
- Giannone, G.; Hosy, E.; Levet, F.; Constals, A.; Schulze, K.; Sobolevsky, A.I.; Rosconi, M.P.; Gouaux, E.; Tampé, R.; Choquet, D.; et al. Dynamic Superresolution Imaging of Endogenous Proteins on Living Cells at Ultra-High Density. Biophys. J. 2010, 99, 1303–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.; Zahn, J.; Maglione, M.; Sigrist, S.J.; Marquard, J.; Chojnacki, J.; Kräusslich, H.-G.; Sahl, S.J.; Engelhardt, J.; Hell, S.W. Ultrafast, temporally stochastic STED nanoscopy of millisecond dynamics. Nat. Methods 2015, 12, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eilers, Y.; Ta, H.; Gwosch, K.C.; Balzarotti, F.; Hell, S.W. MINFLUX monitors rapid molecular jumps with superior spatiotemporal resolution. Proc. Natl. Acad. Sci. USA 2018, 115, 6117–6122. [Google Scholar] [CrossRef] [Green Version]
- Gwosch, K.C.; Pape, J.K.; Balzarotti, F.; Hoess, P.; Ellenberg, J.; Ries, J.; Hell, S.W. MINFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nat. Methods 2020, 17, 217–224. [Google Scholar] [CrossRef]
- Schmidt, R.; Weihs, T.; Wurm, C.A.; Jansen, I.; Rehman, J.; Sahl, S.J.; Hell, S.W. MINFLUX nanometer-scale 3D imaging and microsecond-range tracking on a common fluorescence microscope. Nat. Commun. 2021, 12, 1478. [Google Scholar] [CrossRef]
- Di Rienzo, C.; Gratton, E.; Beltram, F.; Cardarelli, F. Spatiotemporal Fluctuation Analysis: A Powerful Tool for the Future Nanoscopy of Molecular Processes. Biophys. J. 2016, 111, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Digman, M.A.; Sengupta, P.; Wiseman, P.W.; Brown, C.M.; Horwitz, A.R.; Gratton, E. Fluctuation Correlation Spectroscopy with a Laser-Scanning Microscope: Exploiting the Hidden Time Structure. Biophys. J. 2005, 88, L33–L36. [Google Scholar] [CrossRef] [Green Version]
- Digman, M.A.; Gratton, E. Analysis of diffusion and binding in cells using the RICS approach. Microsc. Res. Tech. 2009, 72, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Di Rienzo, C.; Piazza, V.; Gratton, E.; Beltram, F.; Cardarelli, F. Probing short-range protein Brownian motion in the cytoplasm of living cells. Nat. Commun. 2014, 5, 5891. [Google Scholar] [CrossRef]
- Di Rienzo, C.; Gratton, E.; Beltram, F.; Cardarelli, F. Fast spatiotemporal correlation spectroscopy to determine protein lateral diffusion laws in live cell membranes. Proc. Natl. Acad. Sci. USA 2013, 110, 12307–12312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, V.; Ruan, Q.; Gratton, E. 3-D Particle Tracking in a Two-Photon Microscope: Application to the Study of Molecular Dynamics in Cells. Biophys. J. 2005, 88, 2919–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Digiacomo, L.; D’Autilia, F.; Durso, W.; Tentori, P.M.; Caracciolo, G.; Cardarelli, F. Dynamic fingerprinting of sub-cellular nanostructures by image mean square displacement analysis. Sci. Rep. 2017, 7, 14836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, G.; Digiacomo, L.; Lavagnino, Z.; Occhipinti, M.; Bugliani, M.; Cappello, V.; Caracciolo, G.; Marchetti, P.; Piston, D.W.; Cardarelli, F. Insulin secretory granules labelled with phogrin-fluorescent proteins show alterations in size, mobility and responsiveness to glucose stimulation in living β-cells. Sci. Rep. 2019, 9, 2890. [Google Scholar] [CrossRef] [PubMed]
- Hebert, B.; Costantino, S.; Wiseman, P.W. Spatiotemporal Image Correlation Spectroscopy (STICS) Theory, Verification, and Application to Protein Velocity Mapping in Living CHO Cells. Biophys. J. 2005, 88, 3601–3614. [Google Scholar] [CrossRef] [Green Version]
- Begarani, F.; D’Autilia, F.; Signore, G.; Del Grosso, A.; Cecchini, M.; Gratton, E.; Beltram, F.; Cardarelli, F. Capturing Metabolism-Dependent Solvent Dynamics in the Lumen of a Trafficking Lysosome. ACS Nano 2019, 13, 1670–1682. [Google Scholar] [CrossRef]
- Wang, L.; Xiao, Y.; Tian, W.; Deng, L. Activatable Rotor for Quantifying Lysosomal Viscosity in Living Cells. J. Am. Chem. Soc. 2013, 135, 2903–2906. [Google Scholar] [CrossRef]
- Voccoli, V.; Tonazzini, I.; Signore, G.; Caleo, M.; Cecchini, M. Role of extracellular calcium and mitochondrial oxygen species in psychosine-induced oligodendrocyte cell death. Cell Death Dis. 2014, 5, e1529. [Google Scholar] [CrossRef] [Green Version]
- Duchen, L.W.; Eicher, E.M.; Jacobs, J.M.; Scaravilli, F.; Teixeira, F. Hereditary leucodystrophy in the mouse: The new mutant twitcher. Brain 1980, 103, 695–710. [Google Scholar] [CrossRef]
- Cappello, V.; Marchetti, L.; Parlanti, P.; Landi, S.; Tonazzini, I.; Cecchini, M.; Piazza, V.; Gemmi, M. Ultrastructural Characterization of the Lower Motor System in a Mouse Model of Krabbe Disease. Sci. Rep. 2016, 6, 1. [Google Scholar] [CrossRef]
- Devany, J.; Chakraborty, K.; Krishnan, Y. Subcellular Nanorheology Reveals Lysosomal Viscosity as a Reporter for Lysosomal Storage Diseases. Nano Lett. 2018, 18, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Germanos, M.; Gao, A.; Taper, M.; Yau, B.; Kebede, M.A. Inside the Insulin Secretory Granule. Metabolites 2021, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.D.; Hope, M.J.; Cullis, P.R.; Janoff, A.S. Solute distributions and trapping efficiencies observed in freeze-thawed multilamellar vesicles. Biochim. Biophys. Acta (BBA)-Biomembr. 1985, 817, 193–196. [Google Scholar] [CrossRef]
- Lira, R.B.; Steinkühler, J.; Knorr, R.L.; Dimova, R.; Riske, K.A. Posing for a picture: Vesicle immobilization in agarose gel. Sci. Rep. 2016, 6, 25254. [Google Scholar] [CrossRef] [Green Version]
- Asfari, M.; Janjic, D.; Meda, P.; Li, G.; Halban, P.A.; Wollheim, C.B. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 1992, 130, 167–178. [Google Scholar] [CrossRef]
- Rizzo, M.A.; Magnuson, M.A.; Drain, P.F.; Piston, D.W. A Functional Link between Glucokinase Binding to Insulin Granules and Conformational Alterations in Response to Glucose and Insulin. J. Biol. Chem. 2002, 277, 34168–34175. [Google Scholar] [CrossRef] [Green Version]
- Barg, S.; Olofsson, C.S.; Schriever-Abeln, J.; Wendt, A.; Gebre-Medhin, S.; Renström, E.; Rorsman, P. Delay between Fusion Pore Opening and Peptide Release from Large Dense-Core Vesicles in Neuroendocrine Cells. Neuron 2002, 33, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Parlanti, P.; Cappello, V.; Brun, F.; Tromba, G.; Rigolio, R.; Tonazzini, I.; Cecchini, M.; Piazza, V.; Gemmi, M. Size and specimen-dependent strategy for x-ray micro-ct and tem correlative analysis of nervous system samples. Sci. Rep. 2017, 7, 2858. [Google Scholar] [CrossRef] [Green Version]
- Digman, M.A.; Brown, C.M.; Sengupta, P.; Wiseman, P.W.; Horwitz, A.R.; Gratton, E. Measuring Fast Dynamics in Solutions and Cells with a Laser Scanning Microscope. Biophys. J. 2005, 89, 1317–1327. [Google Scholar] [CrossRef] [Green Version]
- Scipioni, L.; Gratton, E.; Diaspro, A.; Lanzanò, L. Phasor Analysis of Local ICS Detects Heterogeneity in Size and Number of Intracellular Vesicles. Biophys. J. 2016, 111, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Digman, M.A.; Dalal, R.; Horwitz, A.F.; Gratton, E. Mapping the Number of Molecules and Brightness in the Laser Scanning Microscope. Biophys. J. 2008, 94, 2320–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Begarani, F.; D’Autilia, F.; Ferri, G.; Pesce, L.; Azzarello, F.; De Lorenzi, V.; Durso, W.; Del Grosso, A.; Cecchini, M.; Cardarelli, F. Measuring Molecular Diffusion in Dynamic Subcellular Nanostructures by Fast Raster Image Correlation Spectroscopy and 3D Orbital Tracking. Int. J. Mol. Sci. 2022, 23, 7623. https://doi.org/10.3390/ijms23147623
Begarani F, D’Autilia F, Ferri G, Pesce L, Azzarello F, De Lorenzi V, Durso W, Del Grosso A, Cecchini M, Cardarelli F. Measuring Molecular Diffusion in Dynamic Subcellular Nanostructures by Fast Raster Image Correlation Spectroscopy and 3D Orbital Tracking. International Journal of Molecular Sciences. 2022; 23(14):7623. https://doi.org/10.3390/ijms23147623
Chicago/Turabian StyleBegarani, Filippo, Francesca D’Autilia, Gianmarco Ferri, Luca Pesce, Fabio Azzarello, Valentina De Lorenzi, William Durso, Ambra Del Grosso, Marco Cecchini, and Francesco Cardarelli. 2022. "Measuring Molecular Diffusion in Dynamic Subcellular Nanostructures by Fast Raster Image Correlation Spectroscopy and 3D Orbital Tracking" International Journal of Molecular Sciences 23, no. 14: 7623. https://doi.org/10.3390/ijms23147623
APA StyleBegarani, F., D’Autilia, F., Ferri, G., Pesce, L., Azzarello, F., De Lorenzi, V., Durso, W., Del Grosso, A., Cecchini, M., & Cardarelli, F. (2022). Measuring Molecular Diffusion in Dynamic Subcellular Nanostructures by Fast Raster Image Correlation Spectroscopy and 3D Orbital Tracking. International Journal of Molecular Sciences, 23(14), 7623. https://doi.org/10.3390/ijms23147623