
Caveolin-1 Alleviates Acetaminophen—Induced Hepatotoxicity in Alcoholic Fatty Liver Disease by Regulating the Ang II/EGFR/ERK Axis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. APAP Aggravated Lipid Deposition and Liver Injury in Ethanol-Induced AFLD Mice
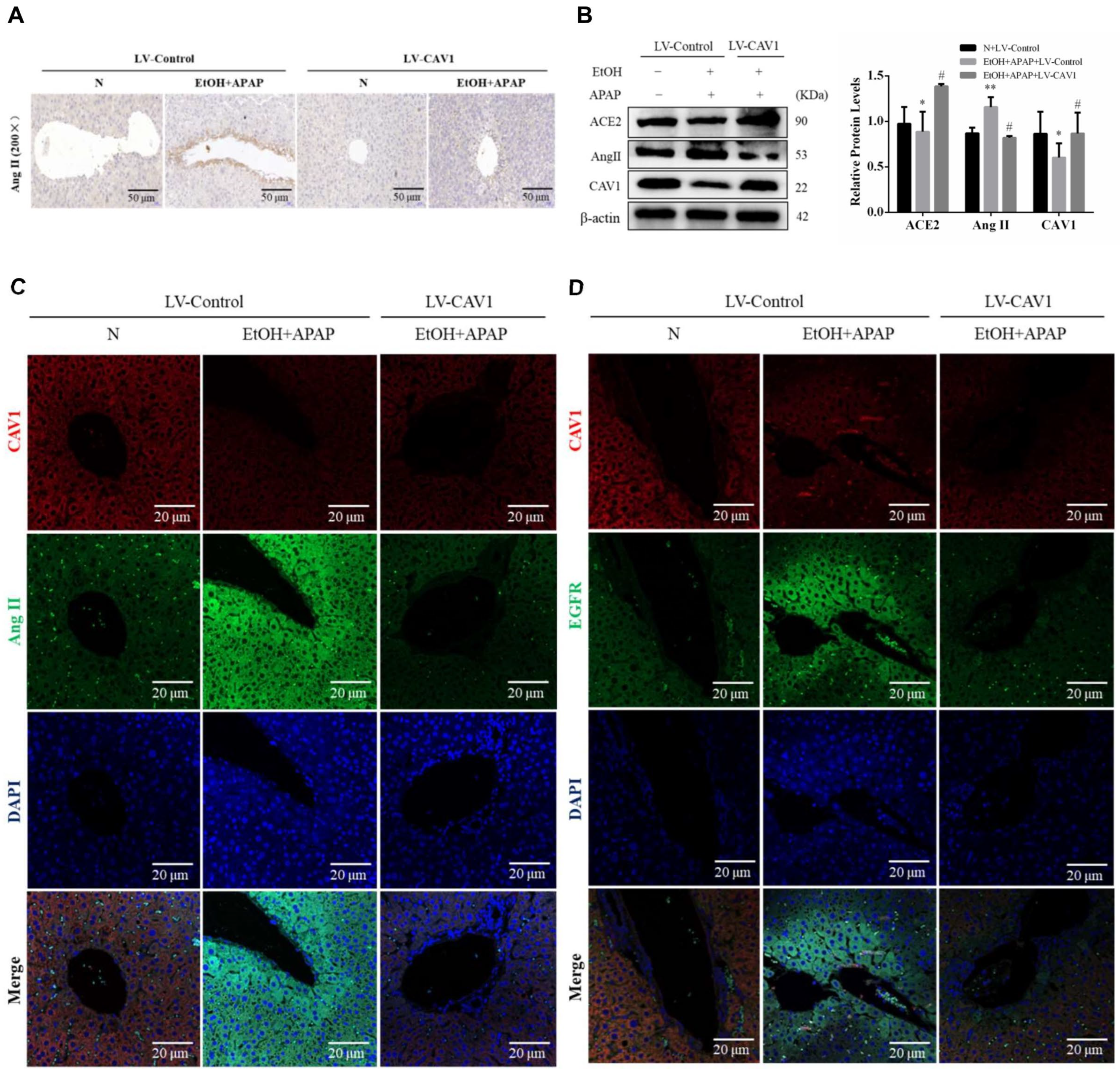
2.2. The Protein Expression of CAV1 and ACE2 Were Decreased While That of Ang II Was Increased in APAP-Treated AFLD Mice
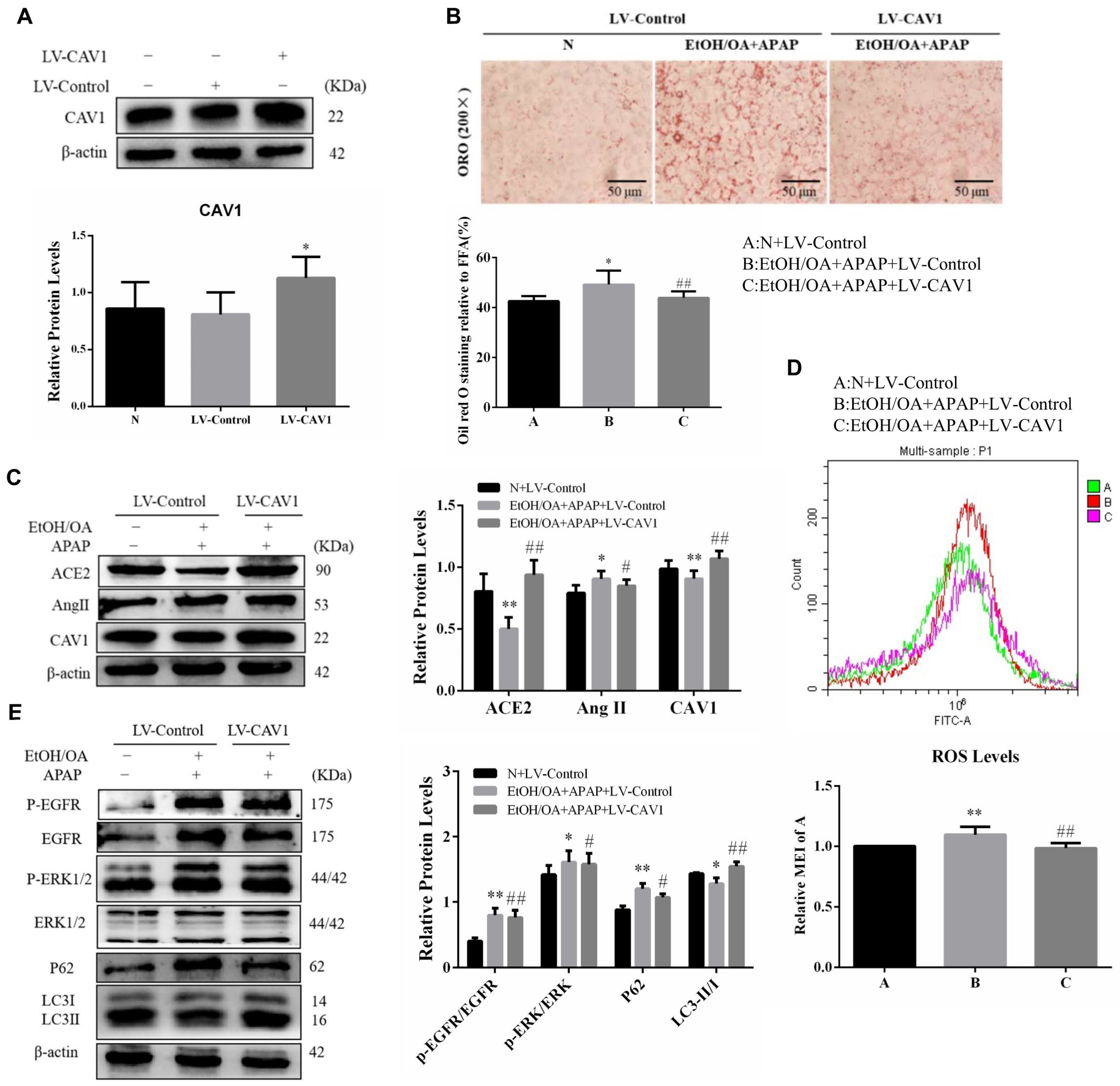
2.3. CAV1 Reduced Lipid Deposition and Liver Injury Caused by APAP in AFLD and Decreased the Level of Oxidative Stress
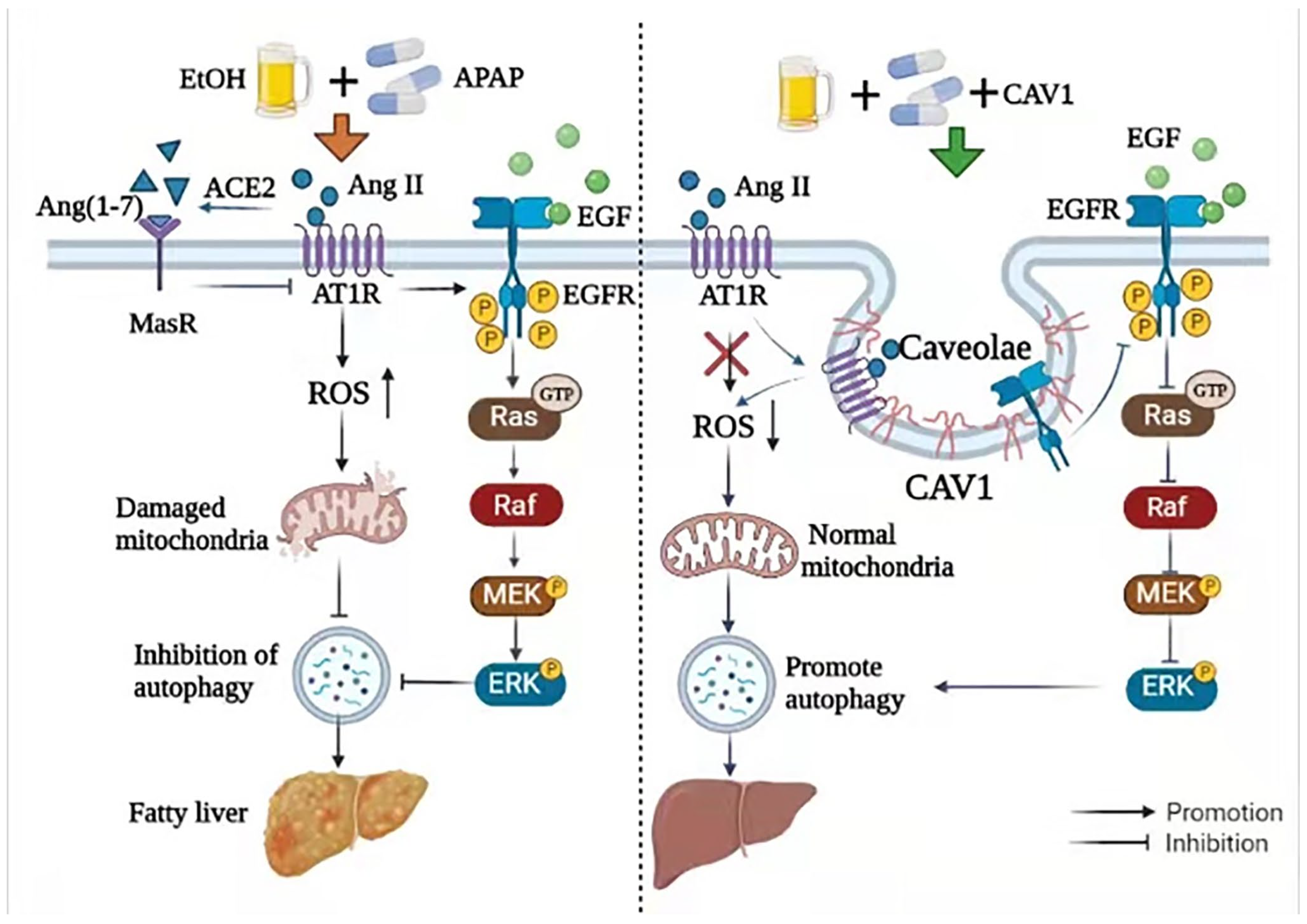
2.4. CAV1 May Alleviate APAP-Induced Lipid Accumulation in AFLD by Affecting the Ang II/EGFR/ERK Axis and Restoring Autophagic Flux
2.5. APAP Aggravated Lipid Deposition and Liver Injury Induced by Alcohol and Oleic Acid in L02 Cells
2.6. CAV1 Alleviated Lipid Accumulation and Oxidative Stress via Suppressing Ang II/EGFR/ERK Signaling and Restoring Autophagic Flux in L02 Cells
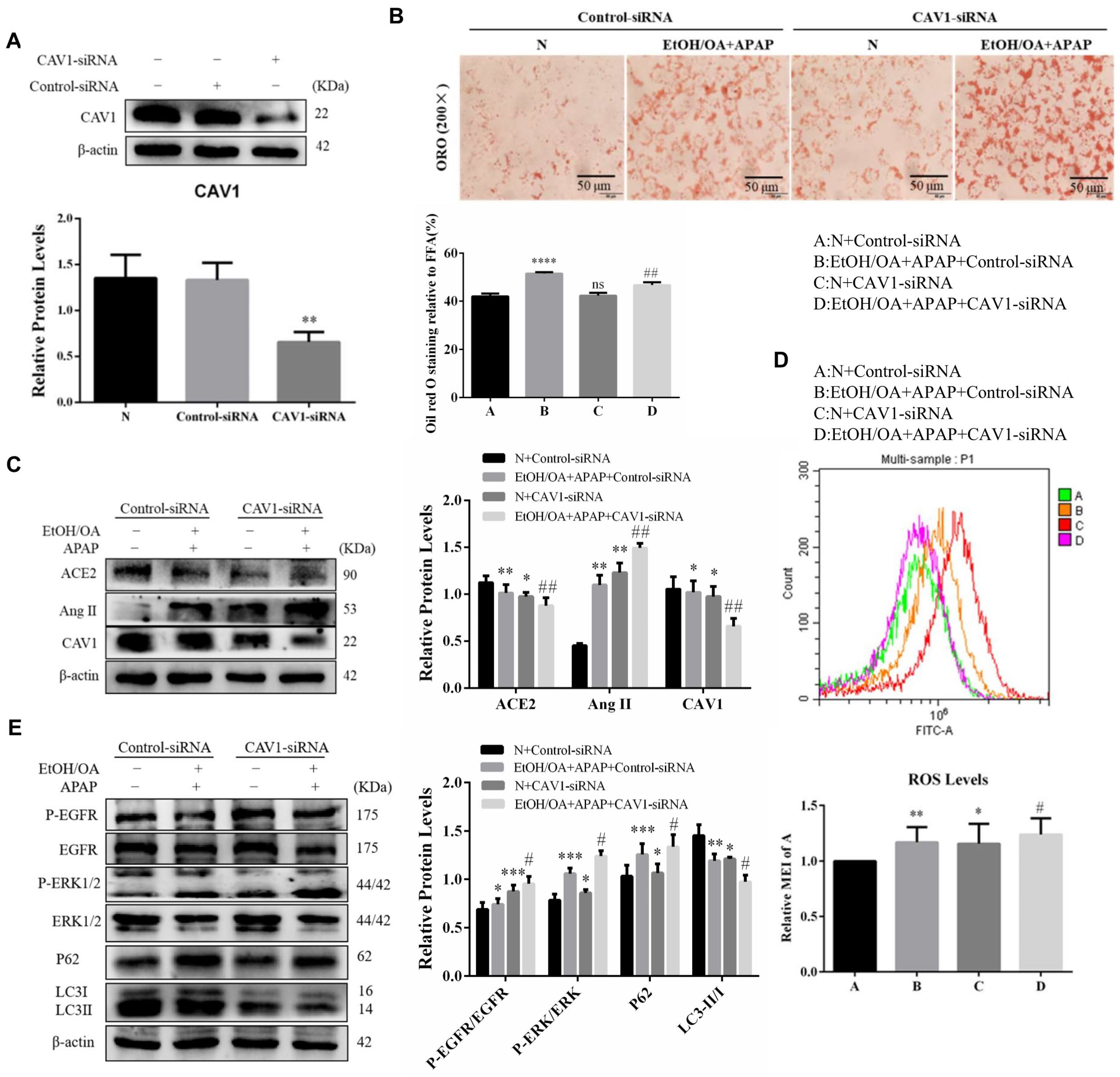
2.7. CAV1 Silencing Exacerbated Lipid Accumulation and Activated the Ang II/EGFR/ERK Signaling in L02 Cells Induced by EtOH/OA and APAP
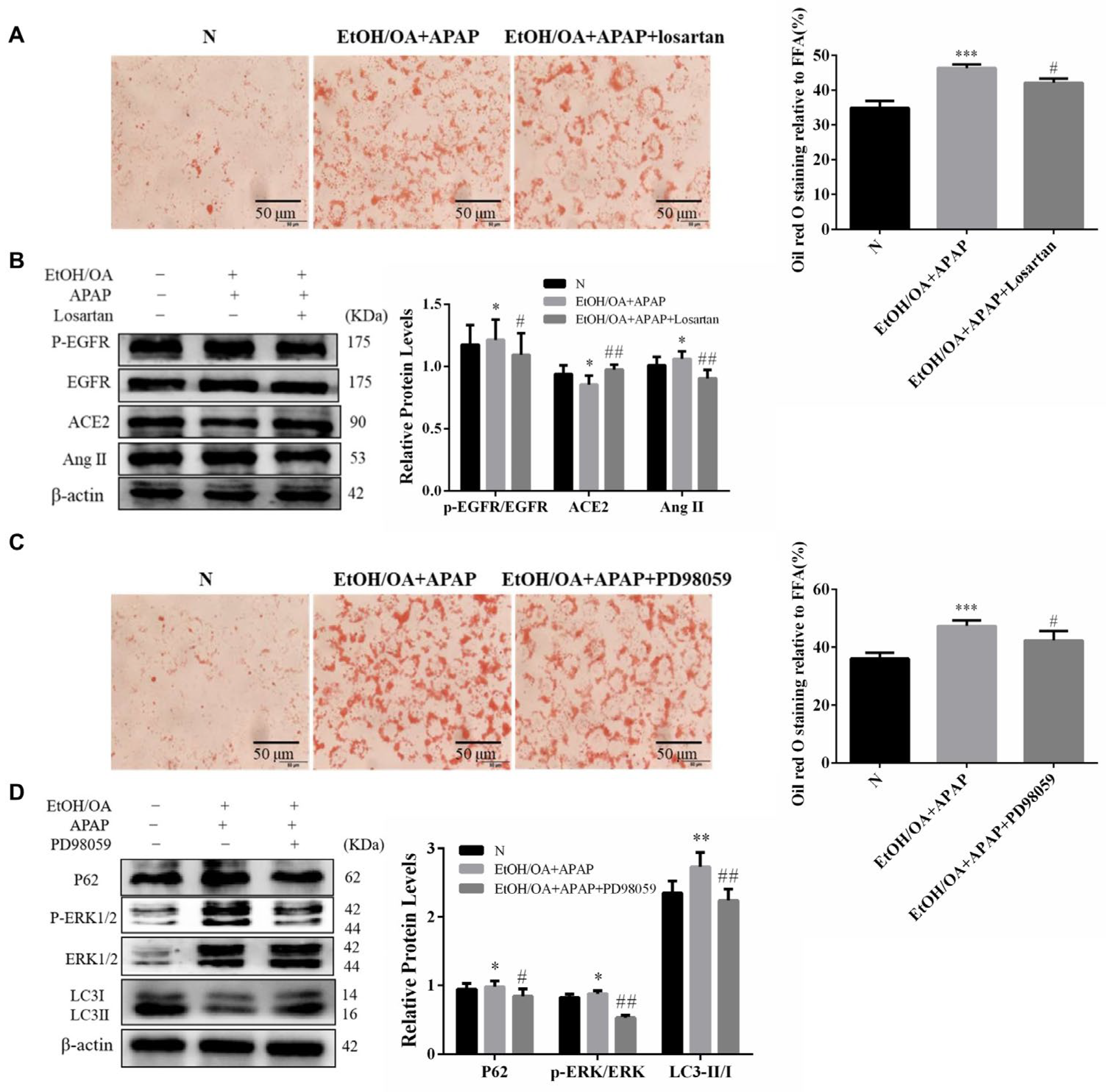
2.8. Inhibition of Ang II and ERK1/2 Alleviated Lipid Accumulation and Restored Autophagic Flux in L02 Cells
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Biochemical Measurements
4.3. Histopathologic Examination of Liver Tissue
4.4. Transmission Electron Microscopy (TEM)
4.5. Cell Culture and Treatment
4.6. Cell Transfection and Inhibitor Administration
4.7. ORO Staining and Measurement of Intracellular TG
4.8. Detection of Reactive Oxygen Species (ROS) Production
4.9. Western Blot Analysis
4.10. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Craig, D.G.N.; Bates, C.M.; Davidson, J.S.; Martin, K.G.; Hayes, P.C.; Simpson, K.J. Overdose pattern and outcome in paracetamol-induced acute severe hepatotoxicity. Br. J. Clin. Pharmacol. 2011, 71, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoine, D.J.; Dear, J.W.; Lewis, P.S.; Platt, V.; Coyle, J.; Masson, M.; Thanacoody, R.H.; Gray, A.J.; Webb, D.J.; Moggs, J.G.; et al. Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to hospital. Hepatology 2013, 58, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferner, R.E.; Dear, J.W.; Bateman, D.N. Management of paracetamol poisoning. BMJ 2011, 342, d2218. [Google Scholar] [CrossRef] [Green Version]
- Crabb, D.W.; Im, G.Y.; Szabo, G.; Mellinger, J.L.; Lucey, M.R. Diagnosis and Treatment of Alcohol-Associated Liver Diseases: 2019 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2020, 71, 306–333. [Google Scholar] [CrossRef] [Green Version]
- Avila, M.A.; Dufour, J.F.; Gerbes, A.L.; Zoulim, F.; Bataller, R.; Burra, P.; Cortez-Pinto, H.; Gao, B.; Gilmore, I.; Mathurin, P.; et al. Recent advances in alcohol-related liver disease (ALD): Summary of a Gut round table meeting. Gut 2020, 69, 764–780. [Google Scholar] [CrossRef] [PubMed]
- Michaut, A.; Moreau, C.; Robin, M.; Fromenty, B. Acetaminophen-induced liver injury in obesity and nonalcoholic fatty liver disease. Liver Int. 2014, 34, e171–e179. [Google Scholar] [CrossRef]
- Anderson, R.G. The caveolae membrane system. Annu. Rev. Biochem. 1998, 67, 199–225. [Google Scholar] [CrossRef] [Green Version]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Pike, L.J.; Han, X.; Chung, K.; Gross, R.W. Lipid Rafts Are Enriched in Arachidonic Acid and Plasmenylethanolamine and Their Composition Is Independent of Caveolin-1 Expression: A Quantitative Electrospray Ionization/Mass Spectrometric Analysis. Biochemistry 2002, 41, 2075–2088. [Google Scholar] [CrossRef]
- Orlandi, P.A.; Fishman, P.H. Filipin-dependent inhibition of cholera toxin: Evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 1998, 141, 905–915. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.; Wang, X.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.; Zhu, Y.; Li, B.; Huang, C.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parton, R.G.; Joggerst, B.; Simons, K. Regulated Internalization of Caveolae. J. Cell Biol. 1994, 127, 1199–1215. [Google Scholar] [CrossRef]
- Borem, L.; Neto, J.; Brandi, I.V.; Lelis, D.F.; Santos, S. The role of the angiotensin II type I receptor blocker telmisartan in the treatment of non-alcoholic fatty liver disease: A brief review. Hypertens. Res. 2018, 41, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiong, F.; Xu, W.; Liu, S. Increased Serum Angiotensin II Is a Risk Factor of Nonalcoholic Fatty Liver Disease: A Prospective Pilot Study. Gastroenterol. Res. Pract. 2019, 2019, 5647161. [Google Scholar] [CrossRef]
- Hirose, A.; Ono, M.; Saibara, T.; Nozaki, Y.; Masuda, K.; Yoshioka, A.; Takahashi, M.; Akisawa, N.; Iwasaki, S.; Oben, J.A.; et al. Angiotensin II type 1 receptor blocker inhibits fibrosis in rat nonalcoholic steatohepatitis. Hepatology 2007, 45, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Li, Y.; Ebihara, K.; Zhao, M.; Naowaboot, J.; Kusakabe, T.; Kuwahara, K.; Murray, M.; Nakao, K. Angiotensin II type 1 receptor-independent beneficial effects of telmisartan on dietary-induced obesity, insulin resistance and fatty liver in mice. Diabetologia 2010, 53, 1727–1731. [Google Scholar] [CrossRef] [Green Version]
- Paschos, P.; Tziomalos, K. Nonalcoholic fatty liver disease and the renin-angiotensin system: Implications for treatment. World J. Hepatol. 2012, 4, 327. [Google Scholar] [CrossRef]
- Hiroshi Kudo, Y.Y.T.T.; Takeshi Oya, S.M.M.S. Telmisartan attenuates progression of steatohepatitis in mice: Role of hepatic macrophage infiltration and e¡ects on adipose tissue. Liver Int. 2009, 29, 988–996. [Google Scholar] [CrossRef]
- Yokohama, S.; Yoneda, M.; Haneda, M.; Okamoto, S.; Okada, M.; Aso, K.; Hasegawa, T.; Tokusashi, Y.; Miyokawa, N.; Nakamura, K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 2004, 40, 1222–1225. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, W.; Xin, J.; Xue, W.; Shi, C.; Wen, J.; Huang, Y.; Hu, C. Caveolin-1 Alleviates Acetaminophen-Induced Fat Accumulation in Non-Alcoholic Fatty Liver Disease by Enhancing Hepatic Antioxidant Ability via Activating AMPK Pathway. Front. Pharmacol. 2021, 12, 717276. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wang, J.; Xue, W.; Xin, J.; Shi, C.; Wen, J.; Feng, X.; Huang, Y.; Hu, C. Caveolin-1 attenuates acetaminophen aggravated lipid accumulation in alcoholic fatty liver by activating mitophagy via the Pink-1/Parkin pathway. Eur. J. Pharmacol. 2021, 908, 174324. [Google Scholar] [CrossRef]
- Vallés, P.G.; Bocanegra, V.; Costantino, V.V.; Gil Lorenzo, A.F.; Benardon, M.E.; Cacciamani, V. The renal antioxidative effect of losartan involves heat shock protein 70 in proximal tubule cells. Cell Stress Chaperones 2020, 25, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Jin, R.; Konomi, J.V.; Cleeton, R.; Cruz, J.; Karpen, S.; Rodriguez, D.S.; Frediani, J.K.; McCracken, C.; Welsh, J. A randomized, controlled, crossover pilot study of losartan for pediatric nonalcoholic fatty liver disease. Pilot Feasibility Stud. 2018, 4, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.S.; Fan, J.G.; Zhang, Z.; Gao, B.; Wang, H.Y. The global burden of liver disease: The major impact of China. Hepatology 2014, 60, 2099–2108. [Google Scholar] [CrossRef]
- Cui, H.; Li, Y.; Cao, M.; Liao, J.; Liu, X.; Miao, J.; Fu, H.; Song, R.; Wen, W.; Zhang, Z.; et al. Untargeted Metabolomic Analysis of the Effects and Mechanism of Nuciferine Treatment on Rats with Nonalcoholic Fatty Liver Disease. Front. Pharmacol. 2020, 11, 858. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Abo El-Ela, F.I.; Alshahrani, F.K.; Bin-Jumah, M.; Al-Zharani, M.; Almutairi, B.; Alyousif, M.S.; Bungau, S.; Aleya, L.; Alkahtani, S. Protective effects of thymoquinone against acrylamide-induced liver, kidney and brain oxidative damage in rats. Environ. Sci. Pollut. Res. 2020, 27, 37709–37717. [Google Scholar] [CrossRef]
- Yang, R.; Song, C.; Chen, J.; Zhou, L.; Jiang, X.; Cao, X.; Sun, Y.; Zhang, Q. Limonin ameliorates acetaminophen-induced hepatotoxicity by activating Nrf2 antioxidative pathway and inhibiting NF-κB inflammatory response via upregulating Sirt1. Phytomedicine 2020, 69, 153211. [Google Scholar] [CrossRef]
- Pike, L.J. Rafts defined: A report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [Green Version]
- Michalopoulos, G.K.; Khan, Z. Liver regeneration, growth factors, and amphiregulin. Gastroenterology 2005, 128, 503–506. [Google Scholar] [CrossRef] [Green Version]
- Carver, R.S.; Stevenson, M.C.; Scheving, L.A.; Russell, W.E. Diverse expression of ErbB receptor proteins during rat liver development and regeneration. Gastroenterology 2002, 123, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Nishida, E.; Gotoh, Y. The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem. Sci. 1993, 18, 128–131. [Google Scholar] [CrossRef]
- Buday, L.; Downward, J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993, 73, 611–620. [Google Scholar] [CrossRef]
- Smart, E.J.; Ying, Y.S.; Mineo, C.; Anderson, R.G. A detergent-free method for purifying caveolae membrane from tissue culture cells. Proc. Natl. Acad. Sci. USA 1995, 92, 10104–10108. [Google Scholar] [CrossRef] [Green Version]
- Khan, E.M.; Heidinger, J.M.; Levy, M.; Lisanti, M.P.; Ravid, T.; Goldkorn, T. Epidermal Growth Factor Receptor Exposed to Oxidative Stress Undergoes Src- and Caveolin-1-dependent Perinuclear Trafficking. J. Biol. Chem. 2006, 281, 14486–14493. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Ren, D.; Wang, J.; Liu, X.; Zhang, H.; Wu, M.; Yang, G. Bruceine D induces lung cancer cell apoptosis and autophagy via the ROS/MAPK signaling pathway in vitro and in vivo. Cell Death Dis. 2020, 11, 126. [Google Scholar] [CrossRef] [Green Version]
- Mahli, A.; Saugspier, M.; Koch, A.; Sommer, J.; Dietrich, P.; Lee, S.; Thasler, R.; Schulze-Luehrmann, J.; Luehrmann, A.; Thasler, W.E.; et al. ERK activation and autophagy impairment are central mediators of irinotecan-induced steatohepatitis. Gut 2017, 64, 746–756. [Google Scholar] [CrossRef]
- Kim, K.; Park, K.; Kim, S.; Yu, S.; Park, S.; Kim, Y.; Seo, Y.; Ma, J.; Ahn, S. Inhibition of Autophagy Promotes Salinomycin-Induced Apoptosis via Reactive Oxygen Species-Mediated PI3K/AKT/mTOR and ERK/p38 MAPK-Dependent Signaling in Human Prostate Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1088. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Wang, J.; Jiang, W.; Shi, C.; Wang, X.; Huang, Y.; Hu, C. Caveolin-1 alleviates lipid accumulation in NAFLD associated with promoting autophagy by inhibiting the Akt/mTOR pathway. Eur. J. Pharmacol. 2020, 871, 172910. [Google Scholar] [CrossRef]
- Gao, L.; Zhou, Y.; Zhong, W.; Zhao, X.; Chen, C.; Chen, X.; Gu, Y.; Chen, J.; Lv, Z.; Shen, J. Caveolin-1 is essential for protecting against binge drinking-induced liver damage through inhibiting reactive nitrogen species. Hepatology 2014, 60, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic Liver Disease: Pathogenesis and New Therapeutic Targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, J.; You, T.; Jiang, X.; Fu, D.; Wang, J.; Jiang, W.; Feng, X.; Wen, J.; Huang, Y.; Hu, C. Caveolin-1 Alleviates Acetaminophen—Induced Hepatotoxicity in Alcoholic Fatty Liver Disease by Regulating the Ang II/EGFR/ERK Axis. Int. J. Mol. Sci. 2022, 23, 7587. https://doi.org/10.3390/ijms23147587
Xin J, You T, Jiang X, Fu D, Wang J, Jiang W, Feng X, Wen J, Huang Y, Hu C. Caveolin-1 Alleviates Acetaminophen—Induced Hepatotoxicity in Alcoholic Fatty Liver Disease by Regulating the Ang II/EGFR/ERK Axis. International Journal of Molecular Sciences. 2022; 23(14):7587. https://doi.org/10.3390/ijms23147587
Chicago/Turabian StyleXin, Jiao, Tingyu You, Xiangfu Jiang, Dongdong Fu, Jiarong Wang, Wei Jiang, Xiaowen Feng, Jiagen Wen, Yan Huang, and Chengmu Hu. 2022. "Caveolin-1 Alleviates Acetaminophen—Induced Hepatotoxicity in Alcoholic Fatty Liver Disease by Regulating the Ang II/EGFR/ERK Axis" International Journal of Molecular Sciences 23, no. 14: 7587. https://doi.org/10.3390/ijms23147587