
ATR Contributes More Than ATM in Intra-S-Phase Checkpoint Activation after IR, and DNA-PKcs Facilitates Recovery: Evidence for Modular Integration of ATM/ATR/DNA-PKcs Functions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
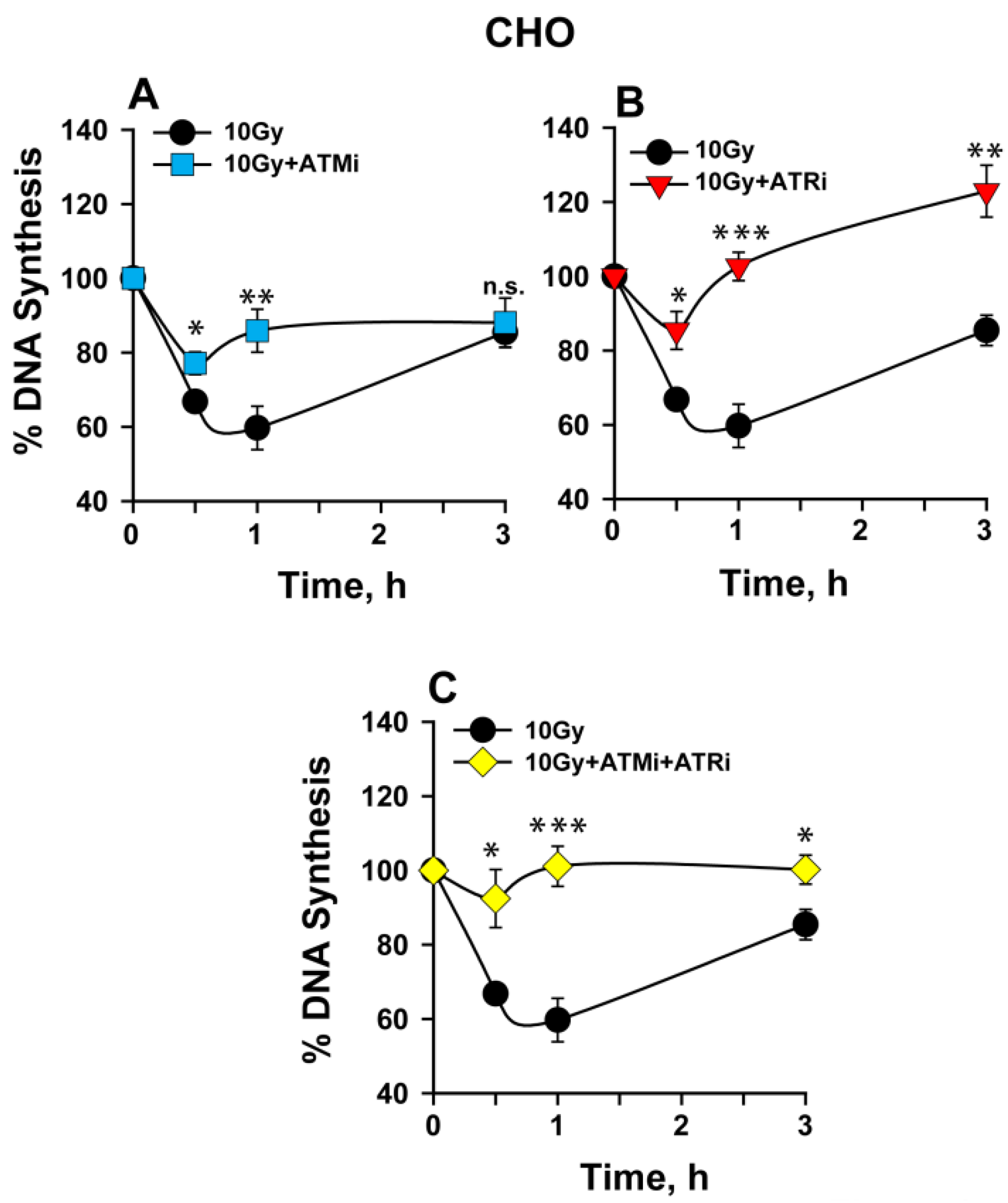
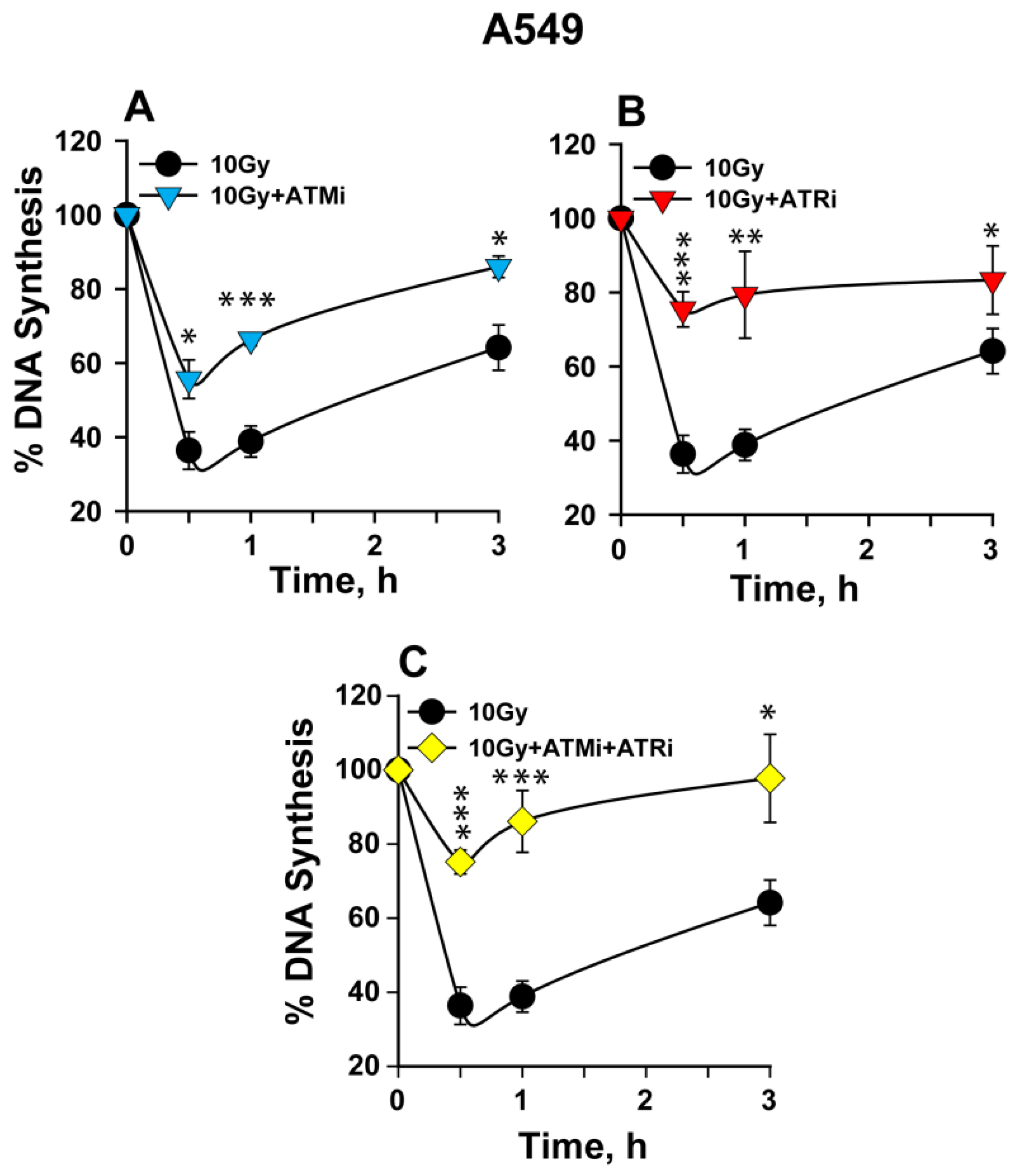
2.1. The Activation of the Intra-S-Phase Checkpoint Is Strongly Dependent on the Functions of Both ATM and ATR
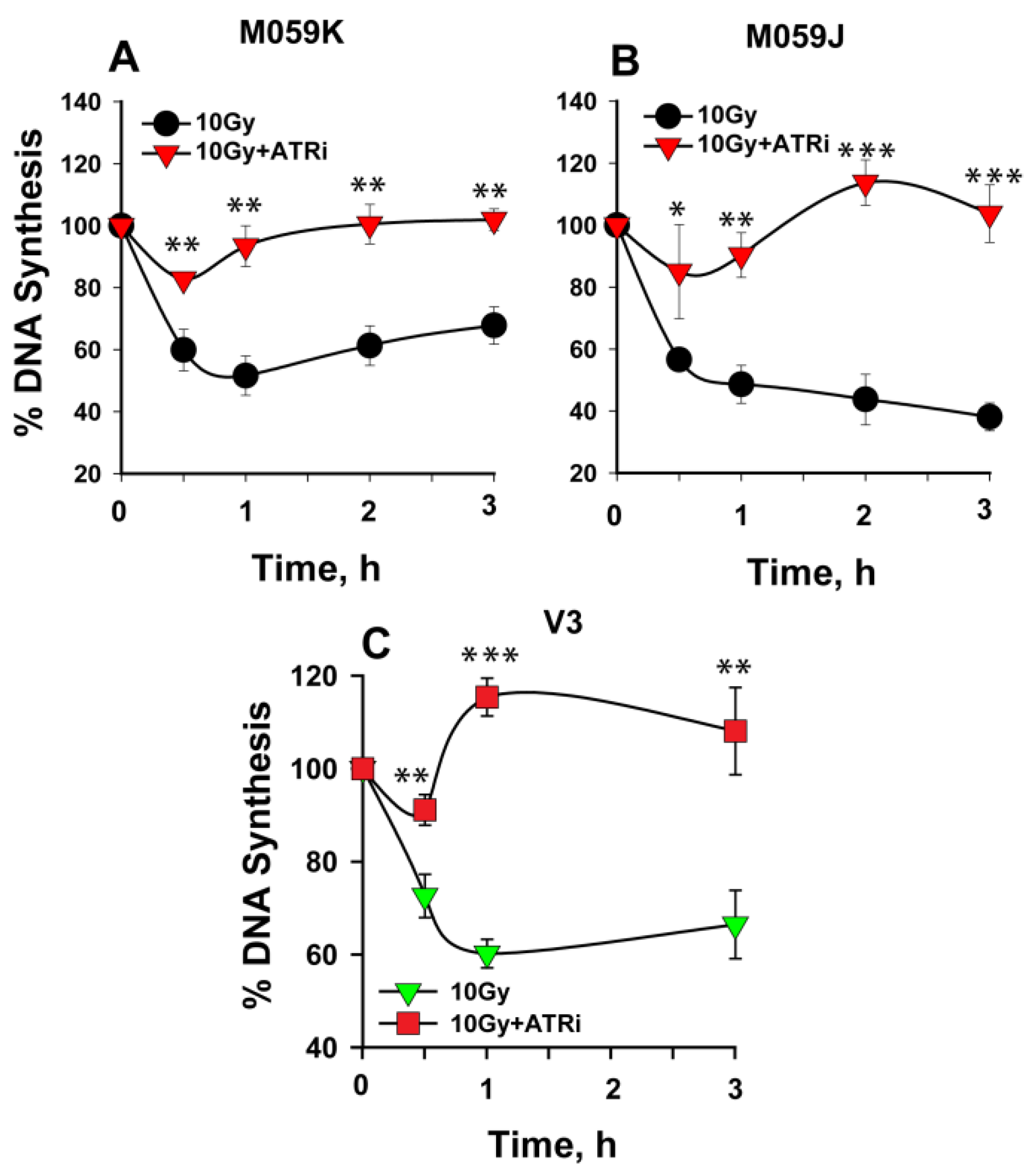
2.2. DNA-PKcs Contributes to the Recovery from IR-Induced Intra-S-Phase Checkpoint
2.3. Persistent Intra-S-Phase Checkpoint in DNA-PKcs Mutants Is Independent of ATM Activity
2.4. ATR Is the Key Activator of the Persistent Intra-S-Phase Checkpoint in DNA-PKcs Mutants
2.5. The Degree of DNA End-Resection at DSBs in the S-Phase Correlates with the Strength of the Intra-S-Phase Checkpoint
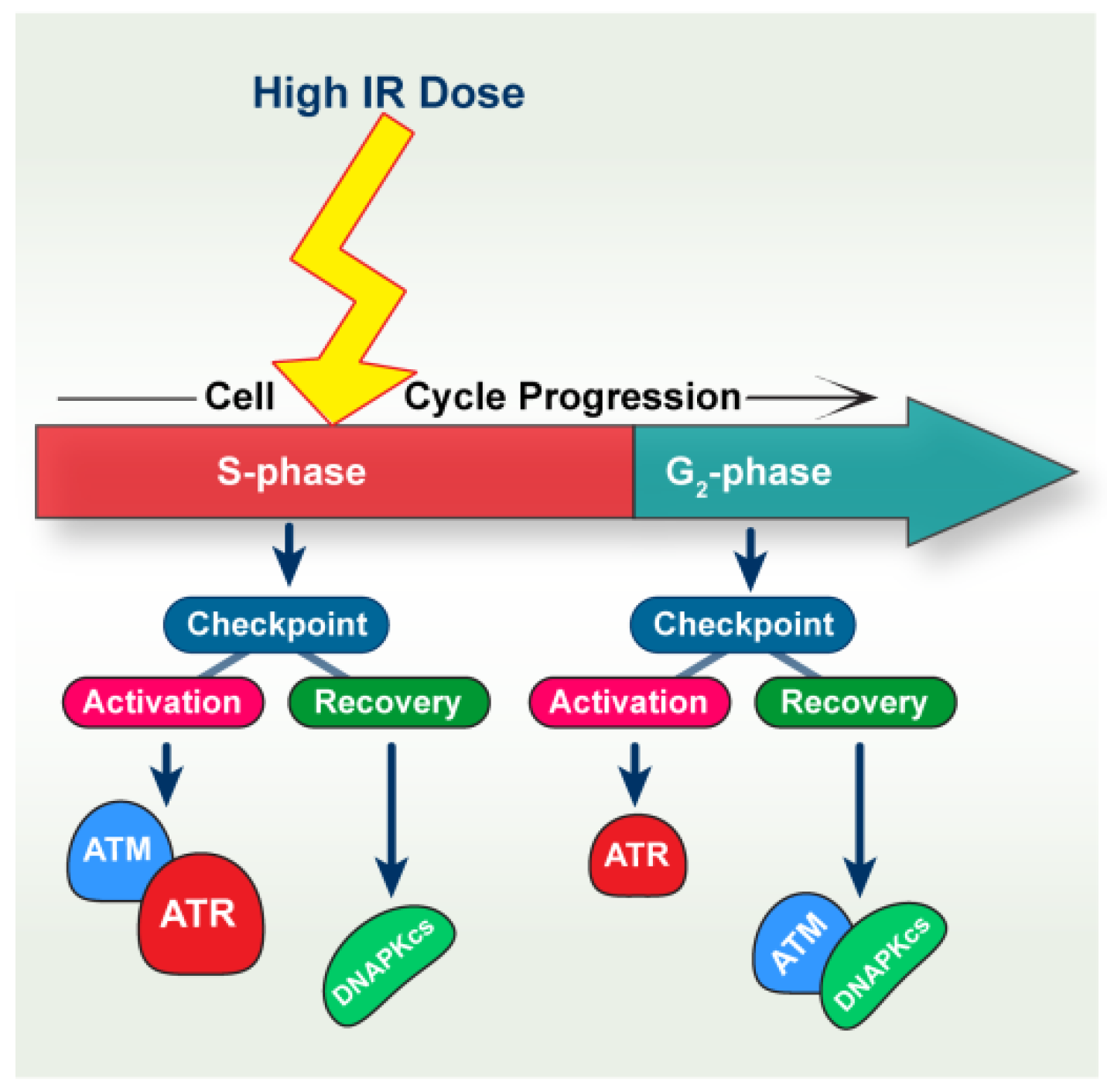
3. Discussion
3.1. ATM and ATR Contribute to the Intra-S-Phase Checkpoint
3.2. DNA-PKcs Defects Cause Mainly ATR-Dependent Potentiation of Intra-S-Phase Checkpoint
4. Methods and Materials
4.1. Cell Culture and Growth Conditions
4.2. Chemicals and Inhibitors
4.3. Irradiation
4.4. Measurement of Radio-Resistant DNA Synthesis
4.5. Flow Cytometry-Based DSB end Resection Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartwell, L.H.; Kastan, M.B. Cell cycle control and cancer. Science 1994, 266, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Wang, Y.; Guan, J.; Wang, H. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 2003, 22, 5834–5847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krempler, A.; Deckbar, D.; Jeggo, P.A.; Löbrich, M. An Imperfect G2/M Checkpoint Contributes to Chromosome Instability Following Irradiation of S and G2 Phase Cells. Cell Cycle 2007, 6, 1682–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, A.; Mladenov, E.; Iliakis, G. Proficiency in homologous recombination repair is prerequisite for activation of G2-checkpoint at low radiation doses. DNA Repair 2021, 101, 103076. [Google Scholar] [CrossRef]
- Terzoudi, G.I.; Manola, K.N.; Pantelias, G.E.; Iliakis, G. Checkpoint Abrogation in G2 Compromises Repair of Chromosomal Breaks in Ataxia Telangiectasia Cells. Cancer Res. 2005, 65, 11292–11296. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Parplys, A.C.; Petermann, E.; Petersen, C.; Dikomey, E.; Borgmann, K. DNA damage by X-rays and their impact on replication processes. Radiother. Oncol. 2012, 102, 466–471. [Google Scholar] [CrossRef]
- Kinner, A.; Wu, W.Q.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res./Genet. Toxicol. Environ. Mutagenesis 2015, 793, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar]
- Kastan, M.B.; Lim, D.-S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000, 1, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [Green Version]
- Adams, K.E.; Medhurst, A.L.; Dart, D.A.; Lakin, N.D. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene 2006, 25, 3894–3904. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Myers, J.S.; Cortez, D. Rapid Activation of ATR by Ionizing Radiation Requires ATM and Mre11. J. Biol. Chem. 2006, 281, 9346–9350. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.-Y.; Wang, X.; Hu, B.; Guan, J.; Iliakis, G.; Wang, Y. An ATM-independent S-Phase Checkpoint Response Involves CHK1 Pathway. Cancer Res. 2002, 62, 1598–1603. Available online: http://cancerres.aacrjournals.org/content/62/6/1598 (accessed on 1 July 2022).
- Dueva, R.; Iliakis, G. Replication protein A: A multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2015, 26, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothkamm, K.; Krüger, I.; Thompson, L.H.; Löbrich, M. Pathways of DNA Double-Strand Break Repair during the Mammalian Cell Cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [Green Version]
- Rathmell, W.K.; Kaufmann, W.K.; Hurt, J.C.; Byrd, L.L.; Chu, G. DNA-dependent protein kinase is not required for accumulation of p53 or cell cycle arrest after DNA damage. Cancer Res. 1997, 1997, 68–74. [Google Scholar]
- Burma, S.; Kurimasa, A.; Xie, G.; Taya, Y.; Araki, R.; Abe, M.; Crissman, H.A.; Ouyang, H.; Li, G.C.; Chen, D.J. DNA-dependent protein kinase-independent activation of p53 in response to DNA damage. J. Biol. Chem. 1999, 274, 17139–17143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhappan, C.; Yusufzai, T.M.; Anderson, S.; Anver, M.R.; Merlino, G. The p53 response to DNA damage in vivo is independent of DNA-dependent protein kinase. Mol. Cell. Biol. 2000, 20, 4075–4083. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM epistatically suppress DNA end resection and hyperactivation of ATR-dependent G2-checkpoint in S-phase irradiated cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef]
- Wang, X.; Khadpe, J.; Hu, B.; Iliakis, G.; Wang, Y. An Over-activated ATR/CHK1 Pathway is Responsible for the Prolonged G2 Accumulation in Irradiated AT Cells. J. Biol. Chem. 2003, 278, 30869–30874. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Mladenov, E.; Dueva, R.; Stuschke, M.; Timmermann, B.; Iliakis, G. Shift in G1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation. Cells 2022, 11, 63. [Google Scholar] [CrossRef]
- Painter, R.B.; Young, B.R. Radiosensitivity in ataxia-telangiectasia: A new explanation. Proc. Natl. Acad. Sci. USA 1980, 77, 7315–7317. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, C.S.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.-B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; DiBiase, S.; Iliakis, G. The catalytic subunit DNA-dependent protein kinase (DNA-PKcs) facilitates recovery from radiation-induced inhibition of DNA replication. Nucleic Acids Res. 2000, 28, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, T.N.; Yin, Y.; Calderon, M.J.; Qian, C.; Schamus-Haynes, S.; Sugitani, N.; Osmanbeyoglu, H.U.; Rothenberg, E.; Watkins, S.C.; Bakkenist, C.J. An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc. Natl. Acad. Sci. USA 2019, 116, 13374–13383. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, T.; Hood, B.; Schamus, S.; O’Connor, M.J.; Conrads, T.P.; Bakkenist, C.J. ATR kinase inhibition induces unscheduled origin firing through a Cdc7-dependent association between GINS and And-1. Nat. Commun. 2017, 8, 1392. [Google Scholar] [CrossRef] [Green Version]
- Parplys, A.C.; Seelbach, J.I.; Becker, S.; Behr, M.; Wrona, A.; Jend, C.; Mansour, W.Y.; Joosse, S.A.; Stuerzbecher, H.-W.; Pospiech, H.; et al. High levels of RAD51 perturb DNA replication elongation and cause unscheduled origin firing due to impaired CHK1 activation. Cell Cycle 2015, 14, 3190–3202. [Google Scholar] [CrossRef]
- Sokka, M.; Koalick, D.; Hemmerich, P.; Syvaoja, J.E.; Pospiech, H. The ATR-Activation Domain of TopBP1 Is Required for the Suppression of Origin Firing during the S Phase. Int. J. Mol. Sci. 2018, 19, 2376. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Li, F.; Mladenov, E.; Soni, A.; Mladenova, V.; Bing, P.; Dueva, R.; Stuschke, M.; Timmermann, B.; Iliakis, G. Increased resection at DSBs in G2-phase is a unique phenotype associated with DNA-PKcs defects that is not shared by other factors of c-NHEJ. Cells 2022, 14, 197. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 1211–1228. [Google Scholar] [CrossRef]
- Jeggo, P.; Löbrich, M. Radiation-induced DNA damage responses. Radiat. Prot. Dosim. 2007, 122, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, J.; Lukas, C.; Bartek, J. Mammalian cell cycle checkpoints: Signalling pathways and their organization in space and time. DNA Repair 2004, 3, 997–1007. [Google Scholar] [CrossRef]
- Falck, J.; Mailand, N.; Syljuasen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, V.; Robertson, K.; Ying, C.Y.; Kim, E.; Avvedimento, E.; Gottesman, M.; Grieco, D.; Gautier, J. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol. Cell 2000, 6, 649–659. [Google Scholar] [CrossRef]
- Falck, J.; Petrini, J.H.J.; Williams, B.R.; Lukas, J.; Bartek, J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat. Genet. 2002, 30, 290–294. [Google Scholar] [CrossRef]
- Mailand, N.; Falck, J.; Lukas, C.; Syljuasen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid destruction of human Cdc25A in response to DNA damage [In Process Citation]. Science 2000, 288, 1425–1429. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef]
- Deckbar, D.; Jeggo, P.A.; Lobrich, M. Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Pantelias, G.E.; Iliakis, G. Chromosome breaks generated by low doses of ionizing radiation in G2-phase are processed exclusively by gene conversion. DNA Repair 2020, 89, 102828. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar]
- Shrivastav, M.; Miller, C.A.; De Haro, L.P.; Durant, S.T.; Chen, B.P.C.; Chen, D.J.; Nickoloff, J.A. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair 2009, 8, 920–929. [Google Scholar] [CrossRef] [Green Version]
- Tomimatsu, N.; Mukherjee, B.; Burma, S. Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep. 2009, 10, 629–635. [Google Scholar]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Peng, A.; Chen, P.-L. NFBD1/Mdc1 Mediates ATR-Dependent DNA Damage Response. Cancer Res. 2005, 65, 1158–1163. [Google Scholar]
- Zhou, Y.; Lee, J.H.; Jiang, W.; Crowe, J.L.; Zha, S.; Paull, T.T. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol. Cell 2017, 65, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.P.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Lobrich, M.; Shiloh, Y.; Chen, D.J. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong suppression of gene conversion with increasing DNA double-strand break load delimited by 53BP1 and RAD52. Nucleic Acids Res. 2020, 48, 1905–1924. [Google Scholar] [CrossRef] [Green Version]
- Kenny, M.K.; Schlegel, U.; Furneaux, H.; Hurwitz, J. The role of human single-stranded DNA binding protein and its individual subunits in simian virus 40 DNA replication. J. Biol. Chem. 1990, 265, 7693–7700. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soni, A.; Duan, X.; Stuschke, M.; Iliakis, G. ATR Contributes More Than ATM in Intra-S-Phase Checkpoint Activation after IR, and DNA-PKcs Facilitates Recovery: Evidence for Modular Integration of ATM/ATR/DNA-PKcs Functions. Int. J. Mol. Sci. 2022, 23, 7506. https://doi.org/10.3390/ijms23147506
Soni A, Duan X, Stuschke M, Iliakis G. ATR Contributes More Than ATM in Intra-S-Phase Checkpoint Activation after IR, and DNA-PKcs Facilitates Recovery: Evidence for Modular Integration of ATM/ATR/DNA-PKcs Functions. International Journal of Molecular Sciences. 2022; 23(14):7506. https://doi.org/10.3390/ijms23147506
Chicago/Turabian StyleSoni, Aashish, Xiaolu Duan, Martin Stuschke, and George Iliakis. 2022. "ATR Contributes More Than ATM in Intra-S-Phase Checkpoint Activation after IR, and DNA-PKcs Facilitates Recovery: Evidence for Modular Integration of ATM/ATR/DNA-PKcs Functions" International Journal of Molecular Sciences 23, no. 14: 7506. https://doi.org/10.3390/ijms23147506