
Antithrombotic Effects of Fostamatinib in Combination with Conventional Antiplatelet Drugs

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
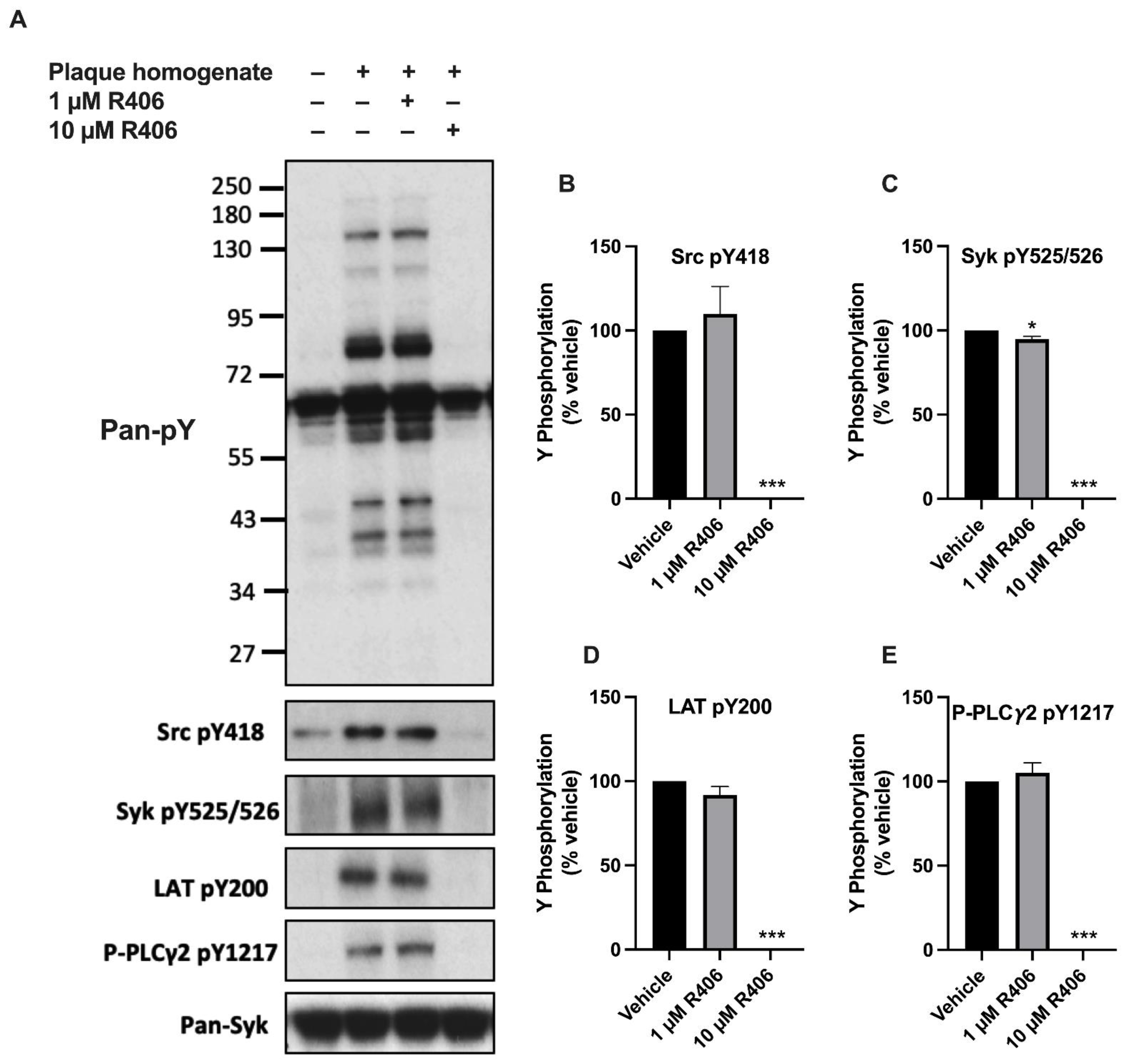
2.1. R406 Inhibits Atherosclerotic Plaque-Induced Platelet Signalling
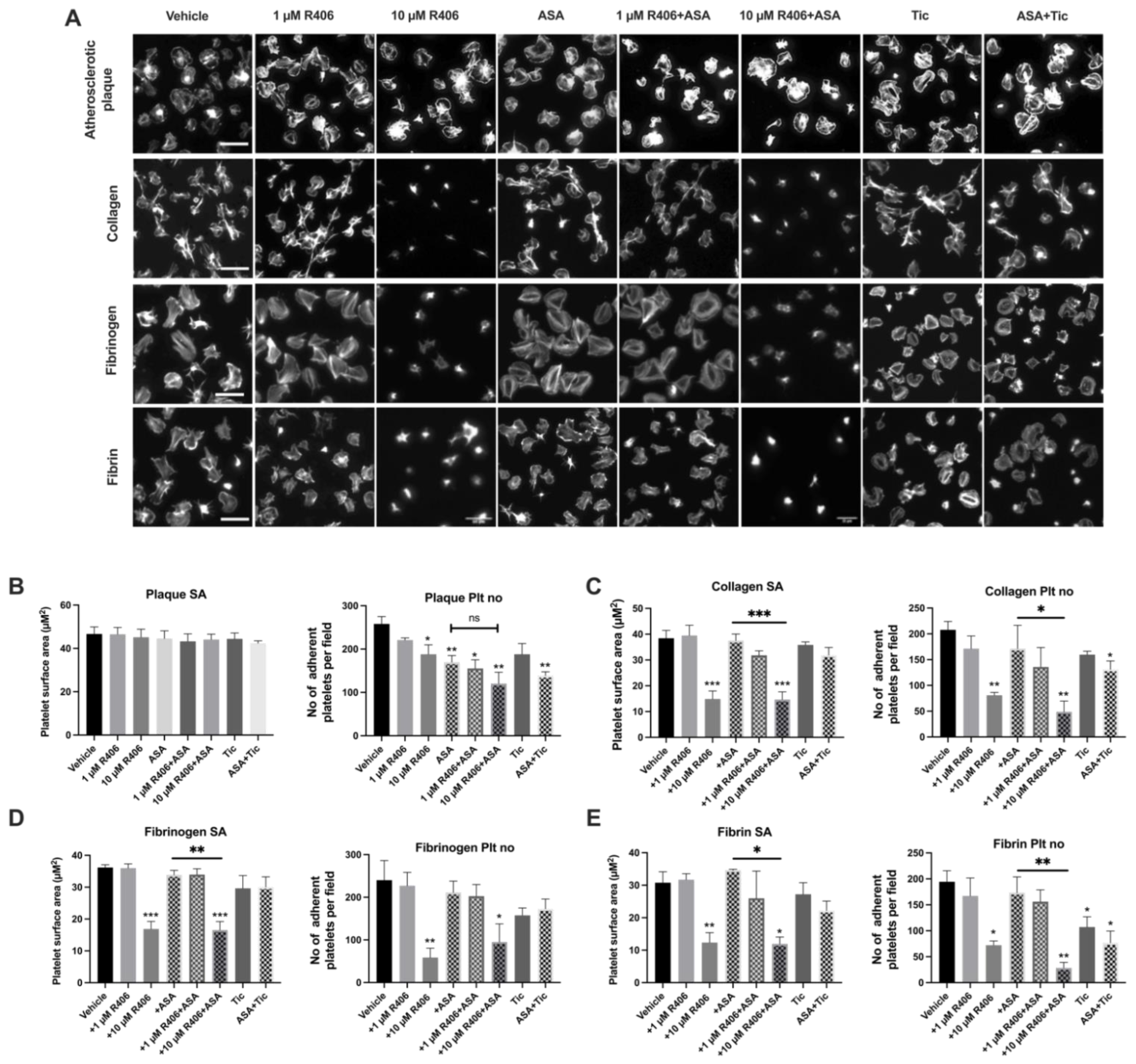
2.2. R406 Reduces Platelet Adhesion on Atherosclerotic Plaque under Static Conditions
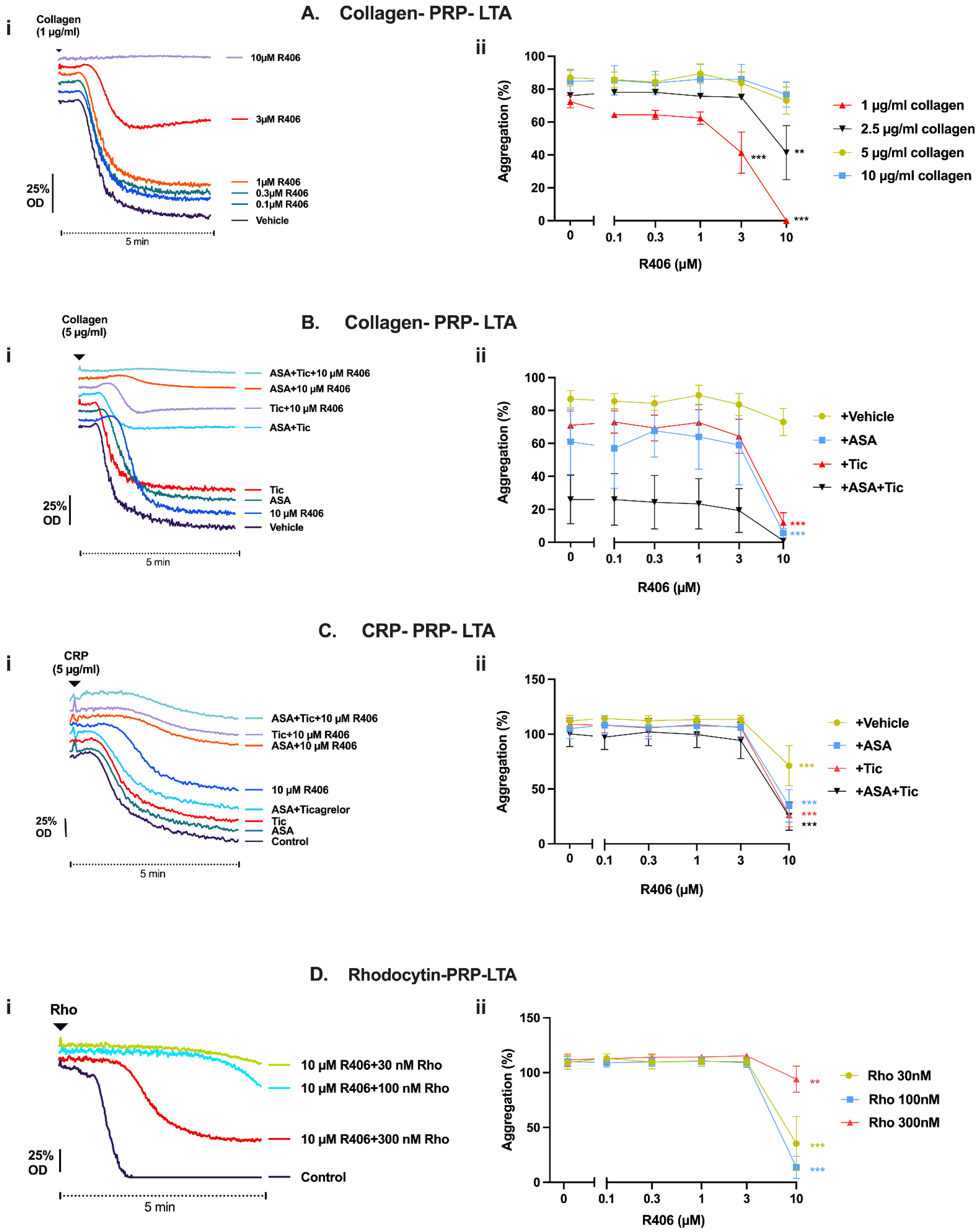
2.3. R406 Inhibits Atherosclerotic Plaque-Induced Platelet Aggregation
2.4. When Combined with Aspirin and Ticagrelor, R406 Provides Mild Additional Inhibition of Platelet Adhesion and Thrombus Formation over Atherosclerotic Plaque under Flow Conditions
2.5. Effect of R406 on GPVI-Mediated Platelet Function Is Amplified by Aspirin and Ticagrelor
2.6. R406 Inhibits Platelet Aggregate Formation and Platelet Adhesion on Collagen under Flow Conditions When Added Ex Vivo to the Blood of Patients Treated with Aspirin and Ticagrelor
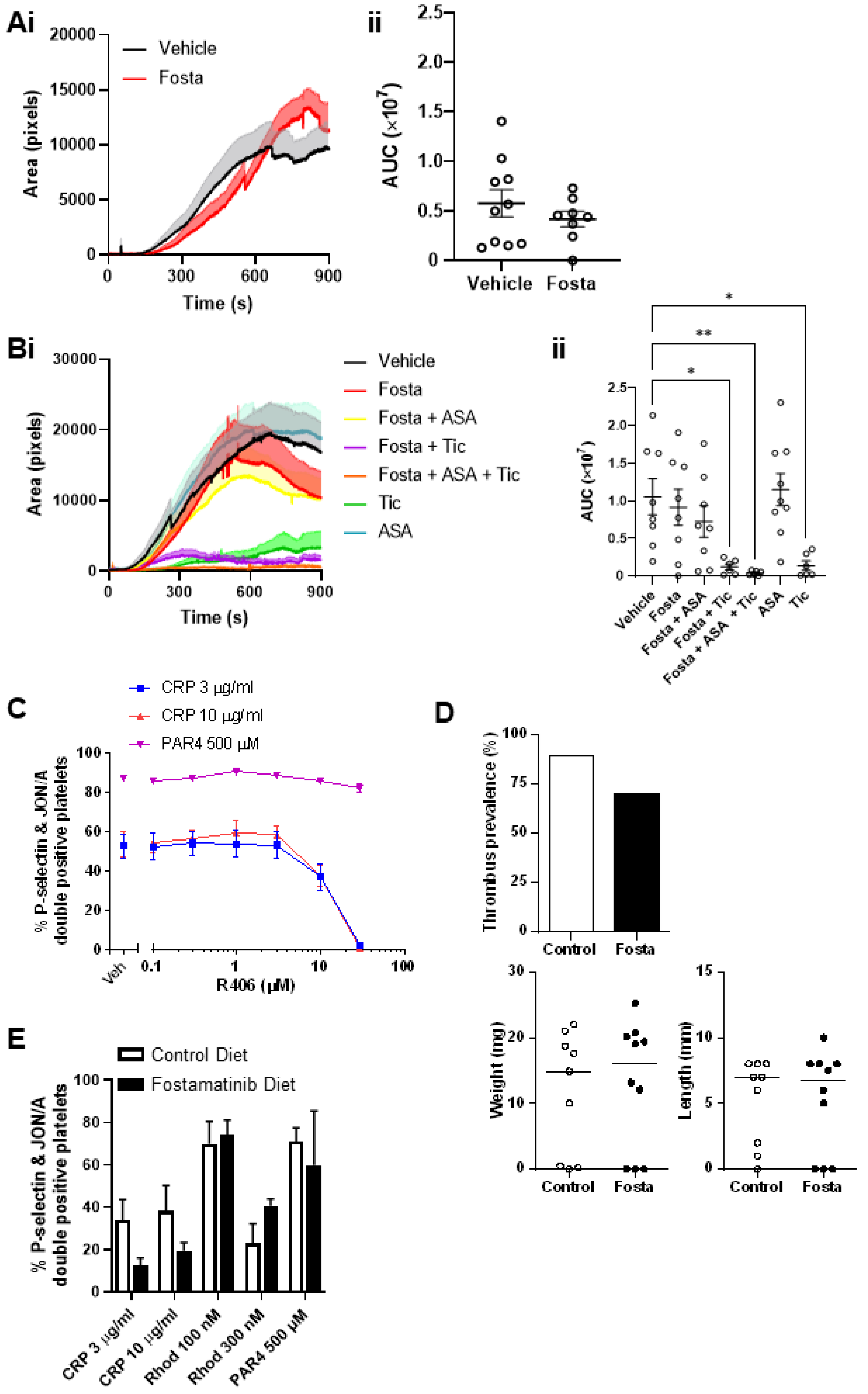
2.7. Fostamatinib Does Not Affect Arterial or Venous Thrombus Formation in Mice
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Platelet Function Tests
4.3. Blood Collection and Ethical Approval
4.4. Atherosclerotic Plaque Preparation
4.5. Animals
4.6. In Vivo Thrombosis
4.7. Platelet Preparation
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonaca, M.P.; Bhatt, D.L.; Cohen, M.; Steg, P.G.; Storey, R.F.; Jensen, E.C.; Magnani, G.; Bansilal, S.; Fish, M.P.; Im, K.; et al. Long-Term Use of Ticagrelor in Patients with Prior Myocardial Infarction. N. Engl. J. Med. 2015, 372, 1791–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiviott, S.D.; Braunwald, E.; McCabe, C.H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.-J.; Ardissino, D.; De Servi, S.; Murphy, S.A.; et al. Prasugrel versus Clopidogrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2007, 357, 2001–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H.; et al. Ticagrelor versus Clopidogrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef]
- Fox, K.A.; Dabbous, O.H.; Goldberg, R.J.; Pieper, K.S.; Eagle, K.A.; Van de Werf, F.; Avezum, Á.; Goodman, S.G.; Flather, M.D.; Anderson, F. A Prediction of risk of death and myocardial infarction in the six months after presentation with acute coronary syndrome: Prospective multinational observational study (GRACE). BMJ 2006, 333, 1091. [Google Scholar] [CrossRef] [Green Version]
- Lockyer, S.; Okuyama, K.; Begum, S.; Le, S.; Sun, B.; Watanabe, T.; Matsumoto, Y.; Yoshitake, M.; Kambayashi, J.; Tandon, N.N. GPVI-deficient mice lack collagen responses and are protected against experimentally induced pulmonary thromboembolism. Thromb. Res. 2006, 118, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Schulte, V.; Bergmeier, W.; Mokhtari-Nejad, R.; Rackebrandt, K.; Cazenave, J.-P.; Ohlmann, P.; Gachet, C.; Zirngibl, H. Long-Term Antithrombotic Protection by in vivo Depletion of Platelet Glycoprotein VI in Mice. J. Exp. Med. 2001, 193, 459–470. [Google Scholar] [CrossRef]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef]
- Alshehri, O.M.; Hughes, C.E.; Montague, S.; Watson, S.K.; Frampton, J.; Bender, M.; Watson, S.P. Fibrin activates GPVI in human and mouse platelets. Blood 2015, 126, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Mammadova-Bach, E.; Ollivier, V.; Loyau, S.; Schaff, M.; Dumont, B.; Favier, R.; Freyburger, G.; Latger-Cannard, V.; Nieswandt, B.; Gachet, C.; et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood 2015, 126, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Quek, L.S.; Bolen, J.; Watson, S.P. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr. Biol. 1998, 8, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Onselaer, M.-B.; Hardy, A.T.; Wilson, C.; Sanchez, X.; Babar, A.K.; Miller, J.L.C.; Watson, C.N.; Watson, S.K.; Bonna, A.; Philippou, H.; et al. Fibrin and D-dimer bind to monomeric GPVI. Blood Adv. 2017, 1, 1495–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbins, J.; Asselin, J.; Farndale, R.; Barnes, M.; Law, C.L.; Watson, S.P. Tyrosine phosphorylation of the Fc receptor gamma-chain in collagen-stimulated platelets. J. Biol. Chem. 1996, 271, 18095–18099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiefer, F.; Brumell, J.; Al-Alawi, N.; Latour, S.; Cheng, A.; Veillette, A.; Grinstein, S.; Pawson, T. The Syk protein tyrosine kinase is essential for Fcγ receptor signaling in macrophages and neutrophils. Mol. Cell. Biol. 1998, 18, 4209–4220. [Google Scholar] [CrossRef] [Green Version]
- Mócsai, A.; Ruland, J.; Tybulewicz, V. The SYK tyrosine kinase: A crucial player in diverse biological functions. Nat. Rev. Immunol. 2010, 10, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.; Gibbins, J.M.; Turner, M.; Van Vugt, M.J.; Van de Winkel JG, J.; Saito, T. The Fc receptor γ-chain and the tyrosine kinase Syk are essential for activation of mouse platelets by collagen. EMBO J. 1997, 16, 2333–2341. [Google Scholar] [CrossRef] [Green Version]
- Andre, P.; Morooka, T.; Sim, D.; Abe, K.; Lowell, C.; Nanda, N.; Delaney, S.; Siu, G.; Yan, Y.; Hollenbach, S.; et al. Critical role for Syk in responses to vascular injury. Blood 2011, 118, 5000–5010. [Google Scholar] [CrossRef] [Green Version]
- Hess, P.R.; Rawnsley, D.R.; Jakus, Z.; Yang, Y.; Sweet, D.T.; Fu, J.; Herzog, B.; Lu, M.; Nieswandt, B.; Oliver, G.; et al. Platelets mediate lymphovenous hemostasis to maintain blood-lymphatic separation throughout life. J. Clin. Investig. 2013, 124, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Reilly, M.P.; Sinha, U.; André, P.; Taylor, S.M.; Pak, Y.; DeGuzman, F.R.; Nanda, N.; Pandey, A.; Stolla, M.; Bergmeier, W.; et al. PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood 2011, 117, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Braselmann, S.; Taylor, V.; Zhao, H.; Wang, S.; Sylvain, C.; Baluom, M.; Qu, K.; Herlaar, E.; Lau, A.; Young, C.; et al. R406, an Orally Available Spleen Tyrosine Kinase Inhibitor Blocks Fc Receptor Signaling and Reduces Immune Complex-Mediated Inflammation. J. Pharmacol. Exp. Ther. 2006, 319, 998–1008. [Google Scholar] [CrossRef]
- van Eeuwijk, J.M.; Stegner, D.; Lamb, D.J.; Kraft, P.; Beck, S.; Thielmann, I. The Novel Oral Syk Inhibitor, Bl1002494, Protects Mice from Arterial Thrombosis and Thromboinflammatory Brain Infarction. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Bussel, J.; Arnold, D.M.; Grossbard, E.; Mayer, J.; Treliński, J.; Homenda, W.; Hellmann, A.; Windyga, J.; Sivcheva, L.; Khalafallah, A.; et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am. J. Hematol. 2018, 93, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Newland, A.; McDonald, V. Fostamatinib: A review of its clinical efficacy and safety in the management of chronic adult immune thrombocytopenia. Immunotherapy 2020, 12, 1325–1340. [Google Scholar] [CrossRef] [PubMed]
- Wonerow, P.; Pearce, A.C.; Vaux, D.J.; Watson, S.P. A critical role for phospholipase Cγ2 in αIIbβ3-mediated platelet spreading. J. Biol. Chem. 2003, 278, 37520–37529. [Google Scholar] [CrossRef] [Green Version]
- Penz, S.; Reininger, A.J.; Brandl, R.; Goyal, P.; Rabie, T.; Bernlochner, I.; Rother, E.; Goetz, C.; Engelmann, B.; Smethurst, P.A.; et al. Human atheromatous plaques stimulate thrombus formation by activating platelet glycoprotein VI. FASEB J. 2005, 19, 898–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, C.; Penz, S.; Hoffmann, C.; Langer, H.; Gillitzer, A.; Schneider, S.; Brandl, R.; Seidl, S.; Massberg, S.; Pichler, B.; et al. Platelet GPVI binds to collagenous structures in the core region of human atheromatous plaque and is critical for atheroprogression in vivo. Basic Res. Cardiol. 2008, 103, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Altomare, I.; Markovtsov, V.V.; Todd, L.; Weerasinghe, D.; Numerof, R.P.; Tong, S.; Masuda, E.; Bussel, J.B. Potential Anti-Thrombotic Effect without Accompanying Hemorrhage with Fostamatinib Use in Patients with Immune Thrombocytopenia. Blood 2019, 134 (Suppl. 1), 4889. [Google Scholar] [CrossRef]
- Cooper, N.; Altomare, I.; Thomas, M.R.; Nicolson, P.L.R.; Watson, S.P.; Markovtsov, V.; Todd, L.K.; Masuda, E.; Bussel, J.B. Assessment of thrombotic risk during long-term treatment of immune thrombocytopenia with fostamatinib. Ther. Adv. Hematol. 2021, 12. [Google Scholar] [CrossRef]
- Payne, H.; Ponomaryov, T.; Watson, S.P.; Brill, A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood 2017, 129, 2013–2020. [Google Scholar] [CrossRef] [Green Version]
- Nicolson, P.L.; Welsh, J.D.; Chauhan, A.; Thomas, M.; Kahn, M.L.; Watson, S.P. A rationale for blocking thromboinflammation in COVID-19 with Btk inhibitors. Platelets 2020, 31, 685–690. [Google Scholar] [CrossRef]
- Spalton, J.C.; Mori, J.; Pollitt, A.; Hughes, C.; Eble, J.A.; Watson, S.P. The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. J. Thromb. Haemost. 2009, 7, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Zheng, T.J.; Lofurno, E.R.; Melrose, A.R.; Lakshmanan, H.H.S.; Pang, J.; Phillips, K.G.; Fallon, M.E.; Kohs, T.C.L.; Ngo, A.T.P.; Shatzel, J.J.; et al. Assessment of the effects of Syk and BTK inhibitors on GPVI-mediated platelet signaling and function. Am. J. Physiol. Physiol. 2021, 320, C902–C915. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, A.-K.M.; Jamasbi, J.; Uhland, K.; Degen, H.; Münch, G.; Ungerer, M.; Brandl, R.; Megens, R.; Weber, C.; Lorenz, R.; et al. Recombinant GPVI-Fc added to single or dual antiplatelet therapy in vitro prevents plaque-induced platelet thrombus formation. Thromb. Haemost. 2017, 117, 1651–1659. [Google Scholar] [CrossRef]
- Mangoni, A.A.; Jackson, S.H.D. Age-related changes in pharmacokinetics and pharmacodynamics: Basic principles and practical applications. Br. J. Clin. Pharmacol. 2004, 57, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tullemans, B.M.; Karel, M.F.; Léopold, V.; Brink, M.S.T.; Baaten, C.C.; Maas, S.L.; de Vos, A.F.; Eble, J.A.; Nijziel, M.R.; van der Vorst, E.P.; et al. Comparison of inhibitory effects of irreversible and reversible Btk inhibitors on platelet function. eJHaem 2021, 2, 685–699. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Rolf, M.G.; Curwen, J.O.; Veldmanjones, M.H.; Eberlein, C.; Wang, J.; Harmer, A.; Hellawell, C.J.; Braddock, M. In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacol. Res. Perspect. 2015, 3, e00175. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Han, X.; Kantarjian, H.; Cortes, J. Tyrosine kinase inhibitor–induced platelet dysfunction in patients with chronic myeloid leukemia. Blood 2009, 114, 261–263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Diamond, S.L. Src family kinases inhibition by dasatinib blocks initial and subsequent platelet deposition on collagen under flow, but lacks efficacy with thrombin generation. Thromb. Res. 2020, 192, 141–151. [Google Scholar] [CrossRef]
- Bender, M.; May, F.; Lorenz, V.; Thielmann, I.; Hagedorn, I.; Finney, B.A.; Vögtle, T.; Remer, K.; Braun, A.; Bösl, M.; et al. Combined In Vivo Depletion of Glycoprotein VI and C-Type Lectin-Like Receptor 2 Severely Compromises Hemostasis and Abrogates Arterial Thrombosis in Mice. Arter. Thromb. Vasc. Biol. 2013, 33, 926–934. [Google Scholar] [CrossRef] [Green Version]
- Law, D.A.; Nannizzi-Alaimo, L.; Ministri, K.; Hughes, P.E.; Forsyth, J.; Turner, M.; Shattil, S.J.; Ginsberg, M.H.; Tybulewicz, V.L.; Phillips, D.R. Genetic and pharmacological analyses of Syk function in αIIbβ3 signaling in platelets. Blood 1999, 93, 2645–2652. [Google Scholar] [CrossRef]
- Liu, J.; Joglekar, M.; Ware, J.; Fitzgerald, M.E.C.; Lowell, C.A.; Berndt, M.C.; Gartner, T.K. Evaluation of the physiological significance of botrocetin/von Willebrand factor in vitro signaling. J. Thromb. Haemost. 2008, 6, 1915–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, S.P.; Mackman, N. How useful are ferric chloride models of arterial thrombosis? Platelets 2020, 31, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Hardy, I.R.; Clarke, R.L.; Lundy, N.; Pine, P.; Turner, B.C.; Potter, T.A.; Refaeli, Y. Mouse models of non-Hodgkin lymphoma reveal Syk as an important therapeutic target. Blood 2009, 113, 2508–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, M.; Hagedorn, I.; Nieswandt, B. Genetic and antibody-induced glycoprotein VI deficiency equally protects mice from mechanically and FeCl3-induced thrombosis. J. Thromb. Haemost. 2011, 9, 1423–1426. [Google Scholar] [CrossRef] [PubMed]
- Haining, E.J.; Cherpokova, D.; Wolf, K.; Becker, I.C.; Beck, S.; Eble, J.A.; Stegner, D.; Watson, S.P.; Nieswandt, B. CLEC-2 contributes to hemostasis independently of classical hemITAM signaling in mice. Blood 2017, 130, 2224–2228. [Google Scholar] [CrossRef]
- Podolanczuk, A.; Lazarus, A.H.; Crow, A.R.; Grossbard, E.; Bussel, J.B. Of mice and men: An open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood 2009, 113, 3154–3160. [Google Scholar] [CrossRef] [Green Version]
- Boccia, R.; Cooper, N.; Ghanima, W.; A Boxer, M.; Hill, Q.A.; Sholzberg, M.; Tarantino, M.D.; Todd, L.K.; Tong, S.; Bussel, J.B.; et al. Fostamatinib is an effective second-line therapy in patients with immune thrombocytopenia. Br. J. Haematol. 2020, 190, 933–938. [Google Scholar] [CrossRef]
- Bussel, J.B.; Arnold, D.M.; Boxer, M.A.; Cooper, N.; Mayer, J.; Zayed, H.; Tong, S.; Duliege, A. Long-term fostamatinib treatment of adults with immune thrombocytopenia during the phase 3 clinical trial program. Am. J. Hematol. 2019, 94, 546–553. [Google Scholar] [CrossRef]
- Lhermusier, T.; VAN Rottem, J.; Garcia, C.; Xuereb, J.-M.; Ragab, A.; Martin, V.; Gratacap, M.-P.; Sié, P.; Payrastre, B. The Syk-kinase inhibitor R406 impairs platelet activation and monocyte tissue factor expression triggered by heparin-PF4 complex directed antibodies. J. Thromb. Haemost. 2011, 9, 2067–2076. [Google Scholar] [CrossRef]
- Woulfe, D.S. Platelet G protein-coupled receptors in hemostasis and thrombosis. J. Thromb. Haemost. 2005, 3, 2193–2200. [Google Scholar] [CrossRef]
- Chan, M.V.; Warner, T.D. Standardised optical multichannel (optimul) platelet aggregometry using high-speed shaking and fixed time point readings. Platelets 2011, 23, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Pike, J.A.; Simms, V.A.; Smith, C.W.; Morgan, N.V.; Khan, A.O.; Poulter, N.S.; Styles, I.B.; Thomas, S.G. An adaptable analysis workflow for characterization of platelet spreading and morphology. Platelets 2020, 32, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki-Inoue, K.; Fuller, G.L.J.; García, Á.; Eble, J.A.; Pöhlmann, S.; Inoue, O.; Gartner, T.K.; Hughan, S.C.; Pearce, A.C.; Laing, G.D.; et al. A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood 2006, 107, 542–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluom, M.; Grossbard, E.B.; Mant, T.; Lau, D.T.W. Pharmacokinetics of fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, in healthy human subjects following single and multiple oral dosing in three phase I studies. Br. J. Clin. Pharmacol. 2013, 76, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolson, P.L.; Nock, S.H.; Hinds, J.; Garcia-Quintanilla, L.; Smith, C.W.; Campos, J.; Brill, A.; Pike, J.A.; Khan, A.O.; Poulter, N.S.; et al. Low-dose Btk inhibitors selectively block platelet activation by CLEC-2. Haematologica 2021, 106, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.W.; Thomas, S.G.; Raslan, Z.; Patel, P.; Byrne, M.; Lordkipanidzé, M.; Bem, D.; Meyaard, L.; Senis, Y.A.; Watson, S.P.; et al. Mice Lacking the Inhibitory Collagen Receptor LAIR-1 Exhibit a Mild Thrombocytosis and Hyperactive Platelets. Arter. Thromb. Vasc. Biol. 2017, 37, 823–835. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harbi, M.H.; Smith, C.W.; Alenazy, F.O.; Nicolson, P.L.R.; Tiwari, A.; Watson, S.P.; Thomas, M.R. Antithrombotic Effects of Fostamatinib in Combination with Conventional Antiplatelet Drugs. Int. J. Mol. Sci. 2022, 23, 6982. https://doi.org/10.3390/ijms23136982
Harbi MH, Smith CW, Alenazy FO, Nicolson PLR, Tiwari A, Watson SP, Thomas MR. Antithrombotic Effects of Fostamatinib in Combination with Conventional Antiplatelet Drugs. International Journal of Molecular Sciences. 2022; 23(13):6982. https://doi.org/10.3390/ijms23136982
Chicago/Turabian StyleHarbi, Maan H., Christopher W. Smith, Fawaz O. Alenazy, Phillip L. R. Nicolson, Alok Tiwari, Steve P. Watson, and Mark R. Thomas. 2022. "Antithrombotic Effects of Fostamatinib in Combination with Conventional Antiplatelet Drugs" International Journal of Molecular Sciences 23, no. 13: 6982. https://doi.org/10.3390/ijms23136982