Loss of Heterozygosity for KrasG12D Promotes Malignant Phenotype of Pancreatic Ductal Adenocarcinoma by Activating HIF-2α-c-Myc-Regulated Glutamine Metabolism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
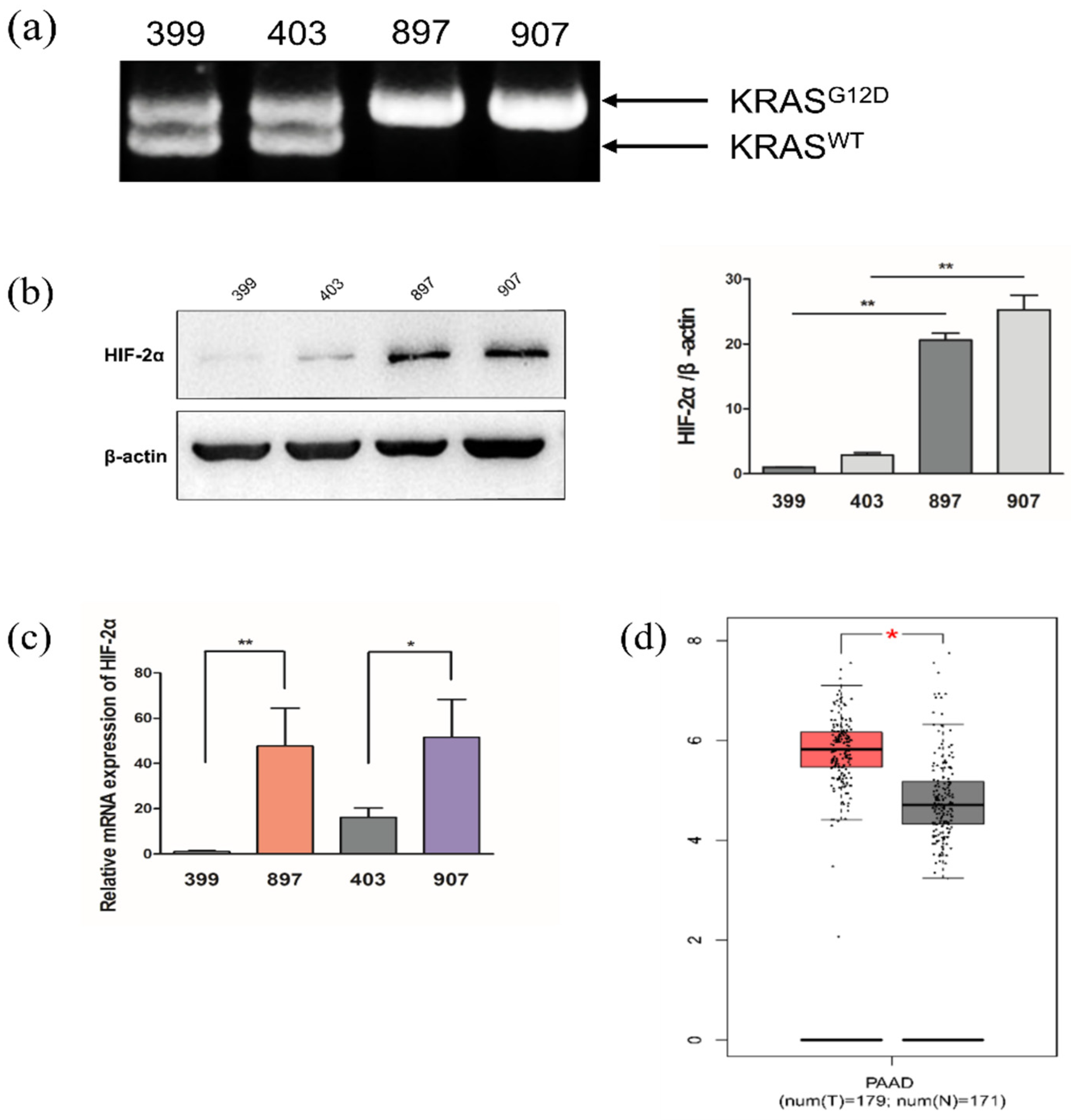
2.1. KrasG12D-LOH Elevated HIF-2α Expression in PDAC Cells at Both Protein and mRNA Levels
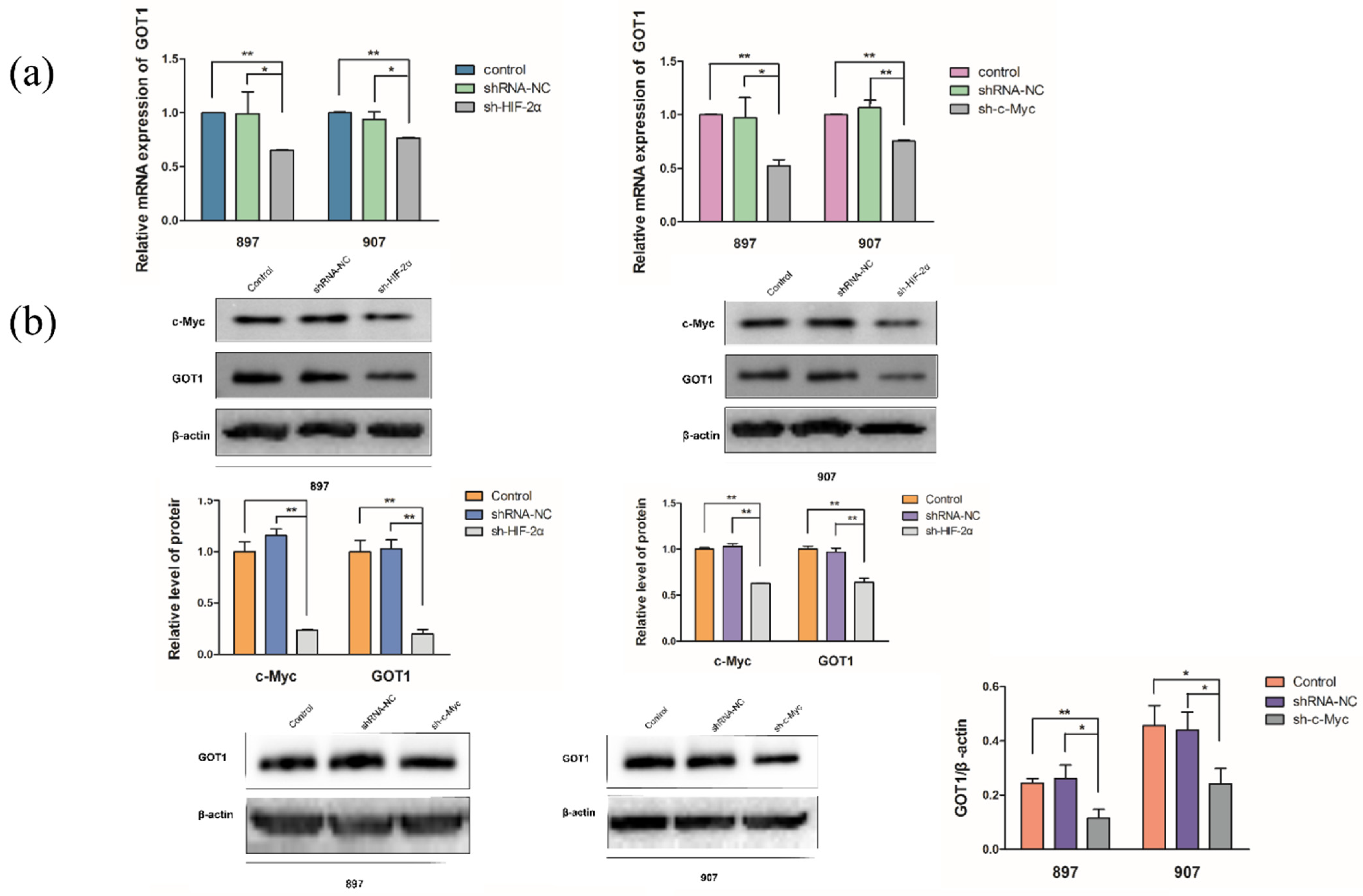
2.2. KrasG12D-LOH Triggered HIF-2α-Dependent c-Myc Activation
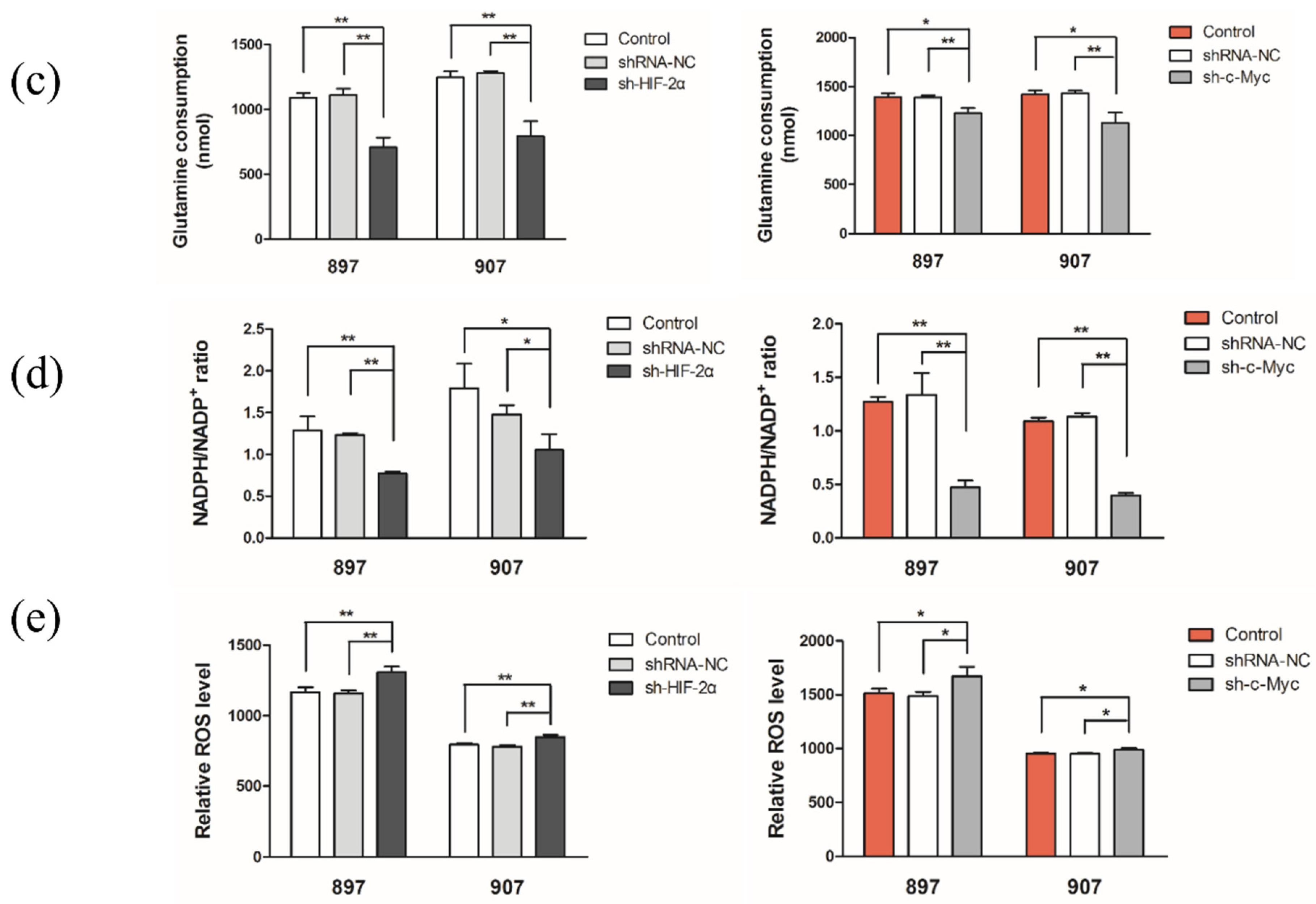
2.3. LOH-Activated HIF-2α-c-Myc Signaling Enhanced Noncanonical Gln Metabolism
2.4. HIF-2α Knockdown Impaired KrasG12D and KrasG12D-LOH PDAC Cell Growth
2.5. HIF-2α Knockdown Suppressed Invasion and Migration of KrasG12D and KrasG12D-LOH PDAC Cells
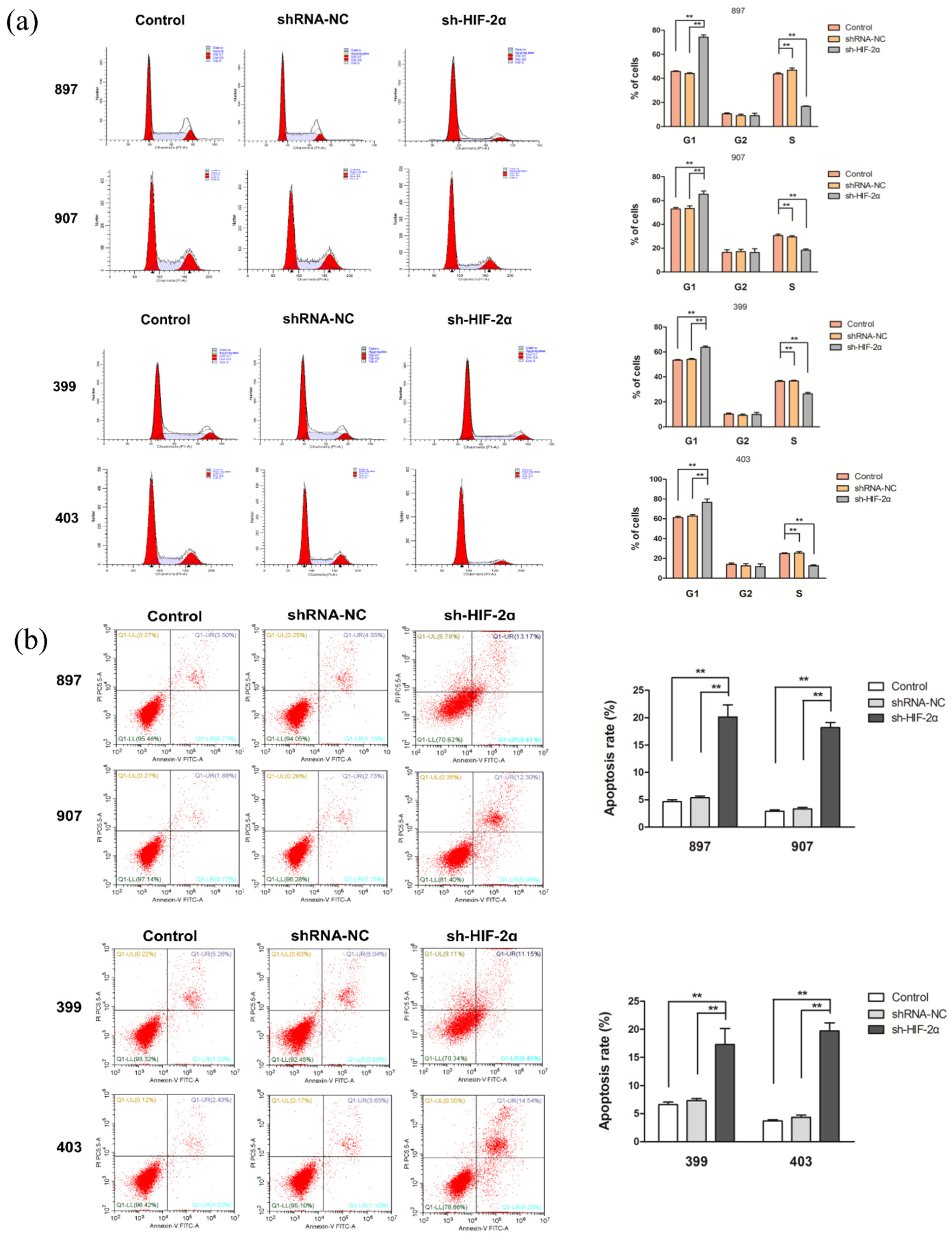
2.6. HIF-2α Knockdown Increased Cell Cycle Arrest and Apoptosis of KrasG12D and KrasG12D-LOH PDAC Cells
2.7. Role of HIF-2α in KrasG12D and KrasG12D-LOH PDAC Cells In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Polymerase Chain Reaction (PCR)
4.3. Western Blotting
4.4. Measurement of Gln Consumption, the NADPH:NADP+ Ratio, and Intracellular ROS Levels
4.5. Cell Viability, Colony Formation, Transwell, Cell Cycle, and Apoptosis Assays
4.6. Xenograft Mouse Model
4.7. Immunohistochemistry
4.8. GEPIA Database Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frappart, P.O.; Hofmann, T.G. Pancreatic ductal adenocarcinoma (PDAC) organoids: The shining light at the end of the tunnel for drug response prediction and personalized medicine. Cancers 2020, 12, 2750. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Gillen, S.; Schuster, T.; Büschenfelde, C.M.Z.; Friess, H.; Kleeff, J. Preoperative/neoadjuvant therapy in pancreatic cancer: A systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef]
- Grant, T.J.; Hua, K.; Singh, A. Molecular pathogenesis of pancreatic cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733.e9. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Hou, P.; Ma, X.; Yang, Z.; Zhang, Q.; Wu, C.J.; Li, J.; Tan, L.; Yao, W.; Yan, L.; Zhou, X.; et al. USP21 deubiquitinase elevates macropinocytosis to enable oncogenic KRAS bypass in pancreatic cancer. Genes Dev. 2021, 35, 1327–1332. [Google Scholar] [CrossRef]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Li, Q.K.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef]
- Velasco, A.; Pallares, J.; Santacana, M.; Yeramian, A.; Dolcet, X.; Eritja, N.; Puente, S.; Sorolla, A.; Llecha, N.; Matias-Guiu, X. Loss of heterozygosity in endometrial carcinoma. Int. J. Gynecol. Pathol. 2008, 27, 305–317. [Google Scholar] [CrossRef]
- Brown, J.; Bush, I.; Bozon, J.; Su, T.T. Cells with loss-of-heterozygosity after exposure to ionizing radiation in Drosophila are culled by p53-dependent and p53-independent mechanisms. PLoS Genet. 2020, 16, e1009056. [Google Scholar] [CrossRef]
- Shen, X.; Chang, L.G.; Hu, M.Y.; Yan, D.; Zhou, L.N.; Ma, Y.; Ling, S.K.; Fu, Y.Q.; Zhang, S.Y.; Kong, B.; et al. KrasG12D-LOH promotes malignant biological behavior and energy metabolism of pancreatic ductal adenocarcinoma cells through the mTOR signaling pathway. Neoplasma 2018, 65, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Chen, X.; Ma, L.; Ma, Y.; Li, Y.; Song, H.; Xu, J.; Zhou, L.; Li, X.; Jiang, Y.; et al. AMPK inhibition suppresses the malignant phenotype of pancreatic cancer cells in part by attenuating aerobic glycolysis. J. Cancer 2019, 10, 1870–1878. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Li, Y.; Ling, S.; Li, X.; Kong, B.; Hu, M.; Huang, P. Loss of heterozygosity for KrasG12D promotes REDD1-dependent, non-canonical glutamine metabolism in pancreatic ductal adenocarcinoma. Biochem. Biophys. Res. Commun. 2020, 526, 880–888. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Xu, P.; Oosterveer, M.H.; Stein, S.; Demagny, H.; Ryu, D.; Moullan, N.; Wang, X.; Can, E.; Zamboni, N.; Comment, A.; et al. LRH-1 dependent programming of mitochondrial glutamine processing drives liver cancer. Genes Dev. 2016, 30, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Rubin, H. Deprivation of glutamine in cell culture reveals its potential for treating cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6964–6968. [Google Scholar] [CrossRef] [Green Version]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recouvreux, M.V.; Moldenhauer, M.R.; Galenkamp, K.M.O.; Jung, M.; James, B.; Zhang, Y.; Lowy, A.; Bagchi, A.; Commisso, C. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J. Exp. Med. 2020, 217, e20200388. [Google Scholar] [CrossRef]
- Reyes-Castellanos, G.; Masoud, R.; Carrier, A. Mitochondrial metabolism in PDAC: From better knowledge to new targeting strategies. Biomedicines 2020, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Kim, Y.E.; Lee, M.; Gu, H.; Kim, J.; Jeong, S.; Yeo, S.; Lee, Y.J.; Im, S.H.; Sung, Y.C.; Kim, H.J.; et al. HIF-1α activation in myeloid cells accelerates dextran sodium sulfate-induced colitis progression in mice. Dis. Models Mech. 2018, 11, dmm033241. [Google Scholar] [CrossRef] [Green Version]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Gastelum, G.; Poteshkina, A.; Veena, M.; Artiga, E.; Weckstein, G.; Frost, P. Restoration of the prolyl-hydroxylase domain protein-3 oxygen-sensing mechanism is responsible for regulation of HIF-2α expression and induction of sensitivity of myeloma cells to hypoxia-mediated apoptosis. PLoS ONE 2017, 12, e0188438. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.J.; Bao, Y.Y.; Zhou, S.H. The role of hypoxia inducible factor in nasal inflammations. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 5067–5076. [Google Scholar] [PubMed]
- Huang, L.; Liu, C.; Deng, Y.; Liu, Y.; Zhao, J.; Huang, X.; Tang, W.; Sun, Y.; Qin, X.; Li, S. Association of hypoxia-inducible factor-2 alpha gene polymorphisms with the risk of hepatitis B virus-related liver disease in Guangxi Chinese: A case-control study. PLoS ONE 2016, 11, e0158241. [Google Scholar] [CrossRef]
- Pérez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hée, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Chen, C.; Zhao, X.; Ye, H.; Zhao, Y.; Fu, Z.; Pan, W.; Zheng, S.; Wei, L.; Nong, T.; et al. HIF-2α regulates non-canonical glutamine metabolism via activation of PI3K/mTORC2 pathway in human pancreatic ductal adenocarcinoma. J. Cell. Mol. Med. 2017, 21, 2896–2908. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mennerich, D.; Kubaichuk, K.; Kietzmann, T. DUBs, hypoxia, and cancer. Trends Cancer 2019, 5, 632–653. [Google Scholar] [CrossRef] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Gradin, K.; Poellinger, L.; Yamamoto, M. Regulation of hypoxia-inducible gene expression after HIF activation. Exp. Cell Res. 2017, 356, 182–186. [Google Scholar] [CrossRef]
- Gkagkalidis, K.; Kampantais, S.; Dimitriadis, G.; Gourvas, V.; Kapoukranidou, D.; Mironidou-Tzouveleki, M. Expression of HIF-2a in clear-cell renal cell carcinoma independently predicts overall survival. Med. Mol. Morphol. 2020, 53, 229–237. [Google Scholar] [CrossRef]
- Kaelin, W.G. Proline hydroxylation and gene expression. Annu. Rev. Biochem. 2005, 74, 115–128. [Google Scholar] [CrossRef]
- Mohlin, S.; Wigerup, C.; Jögi, A.; Påhlman, S. Hypoxia, pseudohypoxia and cellular differentiation. Exp. Cell Res. 2017, 356, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, B.; Wu, W.; Cheng, T.; Schlitter, A.M.; Qian, C.; Bruns, P.; Jian, Z.; Jäger, C.; Regel, I.; Raulefs, S.; et al. A subset of metastatic pancreatic ductal adenocarcinomas depends quantitatively on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut 2016, 654, 647–657. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Ling, S.; Li, Y.; Hu, M.; Kong, B.; Huang, P.; Liu, H. Loss of Heterozygosity for KrasG12D Promotes Malignant Phenotype of Pancreatic Ductal Adenocarcinoma by Activating HIF-2α-c-Myc-Regulated Glutamine Metabolism. Int. J. Mol. Sci. 2022, 23, 6697. https://doi.org/10.3390/ijms23126697
Ma Y, Ling S, Li Y, Hu M, Kong B, Huang P, Liu H. Loss of Heterozygosity for KrasG12D Promotes Malignant Phenotype of Pancreatic Ductal Adenocarcinoma by Activating HIF-2α-c-Myc-Regulated Glutamine Metabolism. International Journal of Molecular Sciences. 2022; 23(12):6697. https://doi.org/10.3390/ijms23126697
Chicago/Turabian StyleMa, Yu, Sunkai Ling, Yuan Li, Mingyue Hu, Bo Kong, Peilin Huang, and Hui Liu. 2022. "Loss of Heterozygosity for KrasG12D Promotes Malignant Phenotype of Pancreatic Ductal Adenocarcinoma by Activating HIF-2α-c-Myc-Regulated Glutamine Metabolism" International Journal of Molecular Sciences 23, no. 12: 6697. https://doi.org/10.3390/ijms23126697