
Characterization of Phytohormones and Transcriptomic Profiling of the Female and Male Inflorescence Development in Manchurian Walnut (Juglans mandshurica Maxim.)




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
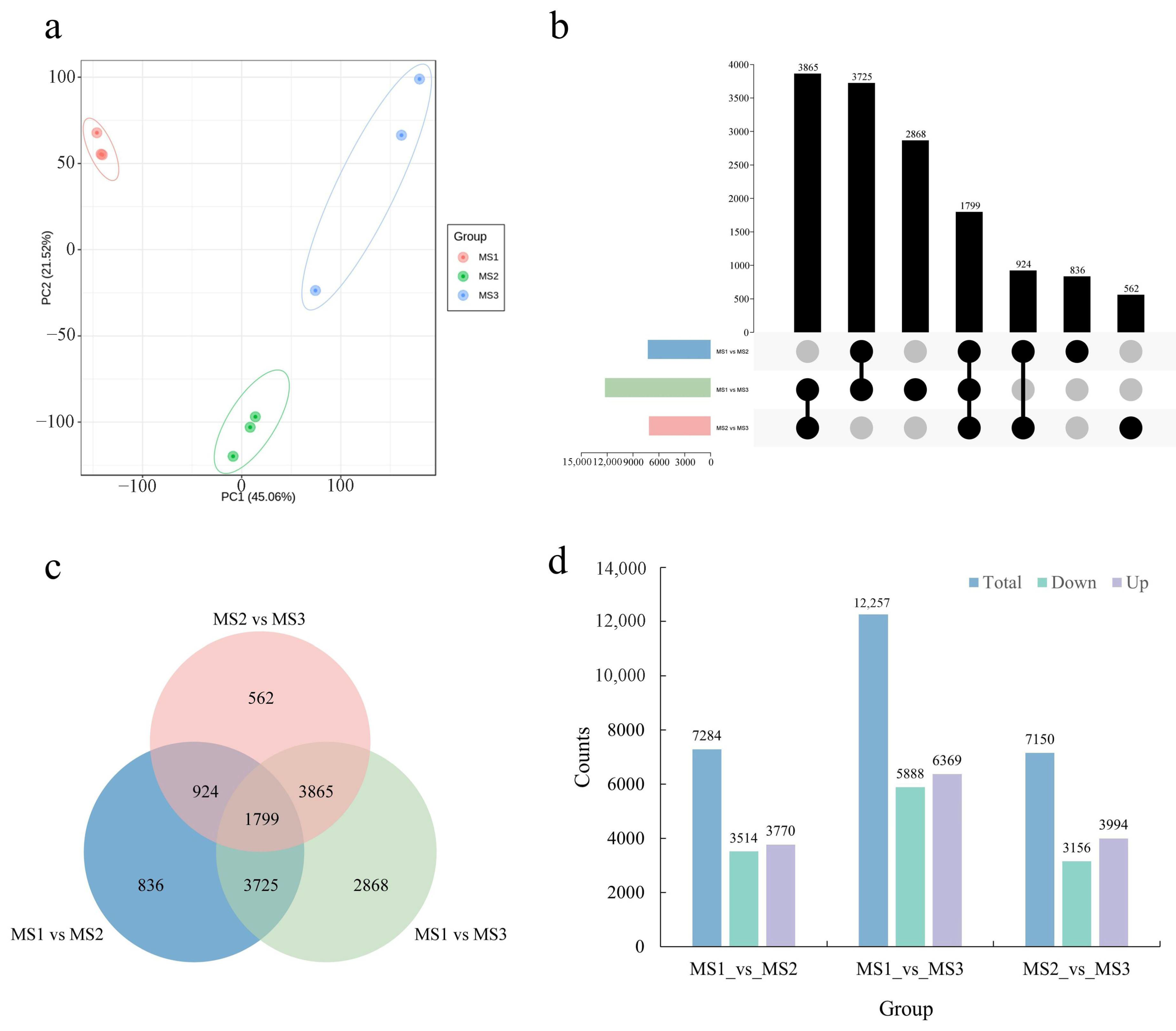
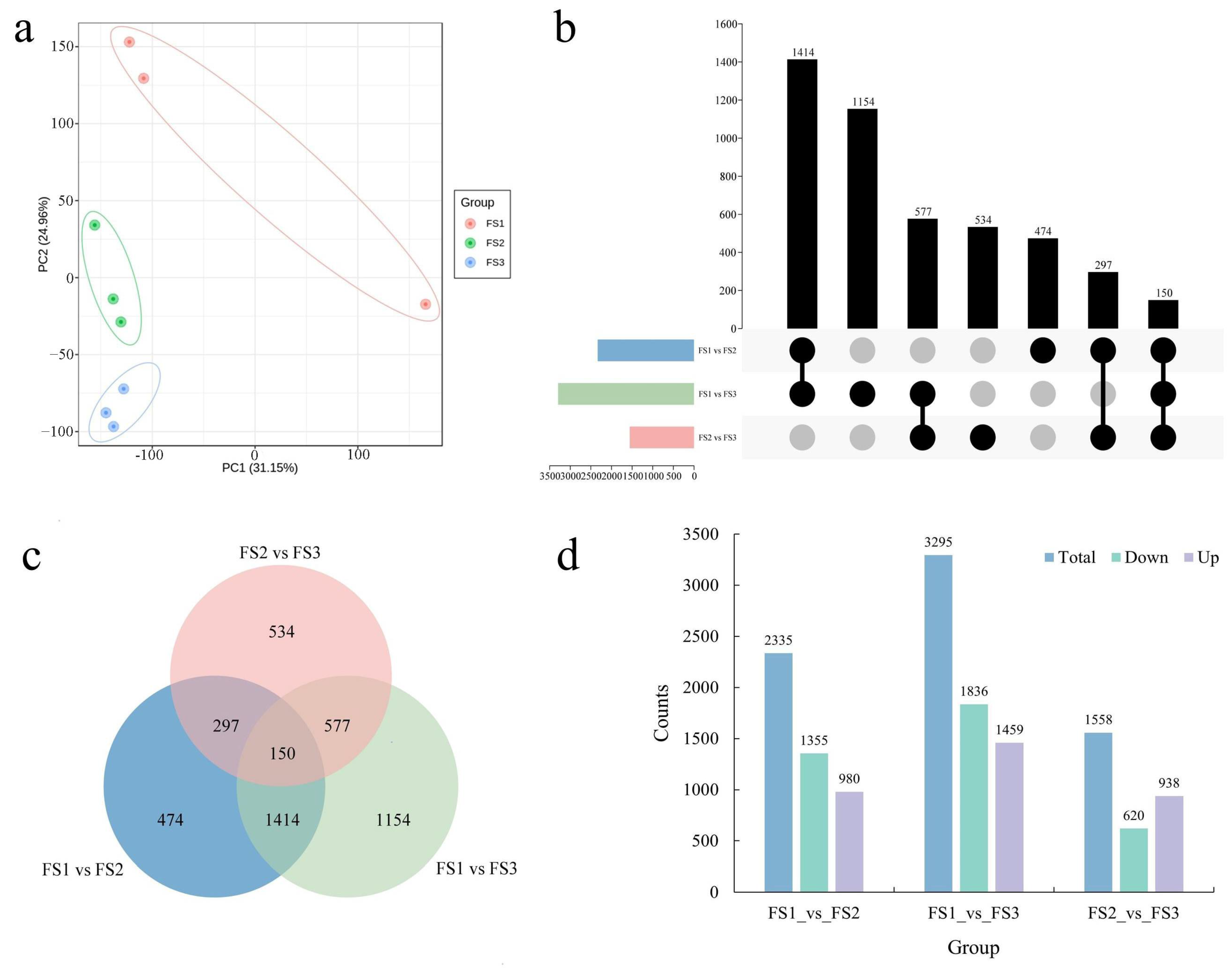
2. Results
2.1. Morphological Characteristics of Female/Male Flowering Transitions
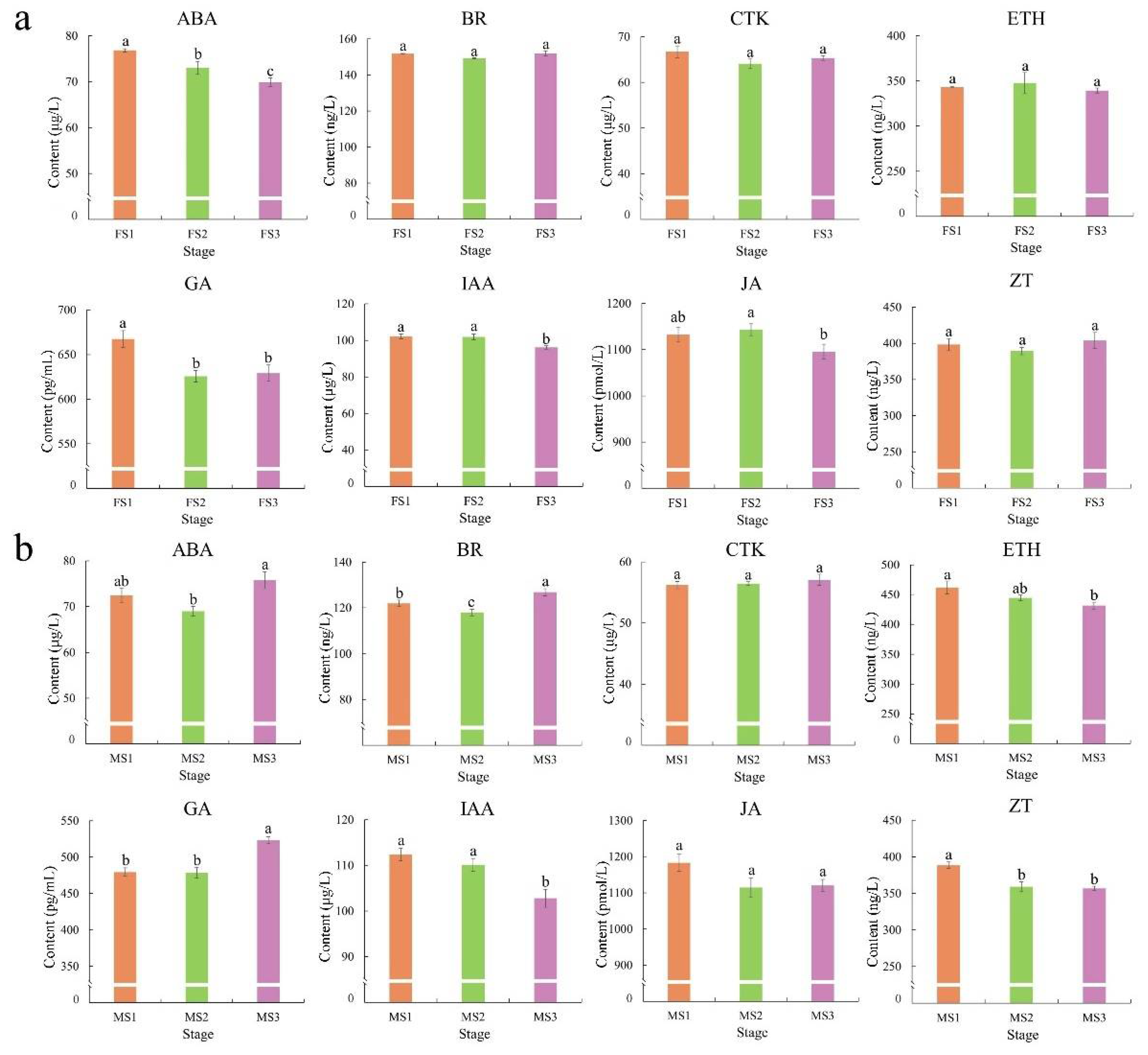
2.2. Changes in Phytohormones during the Flowering Transition Process
2.3. Identification and Functional Enrichment of DEGs during the Development of Male Inflorescence
2.4. Identification and Functional Enrichment of DEGs during Female Inflorescence Development
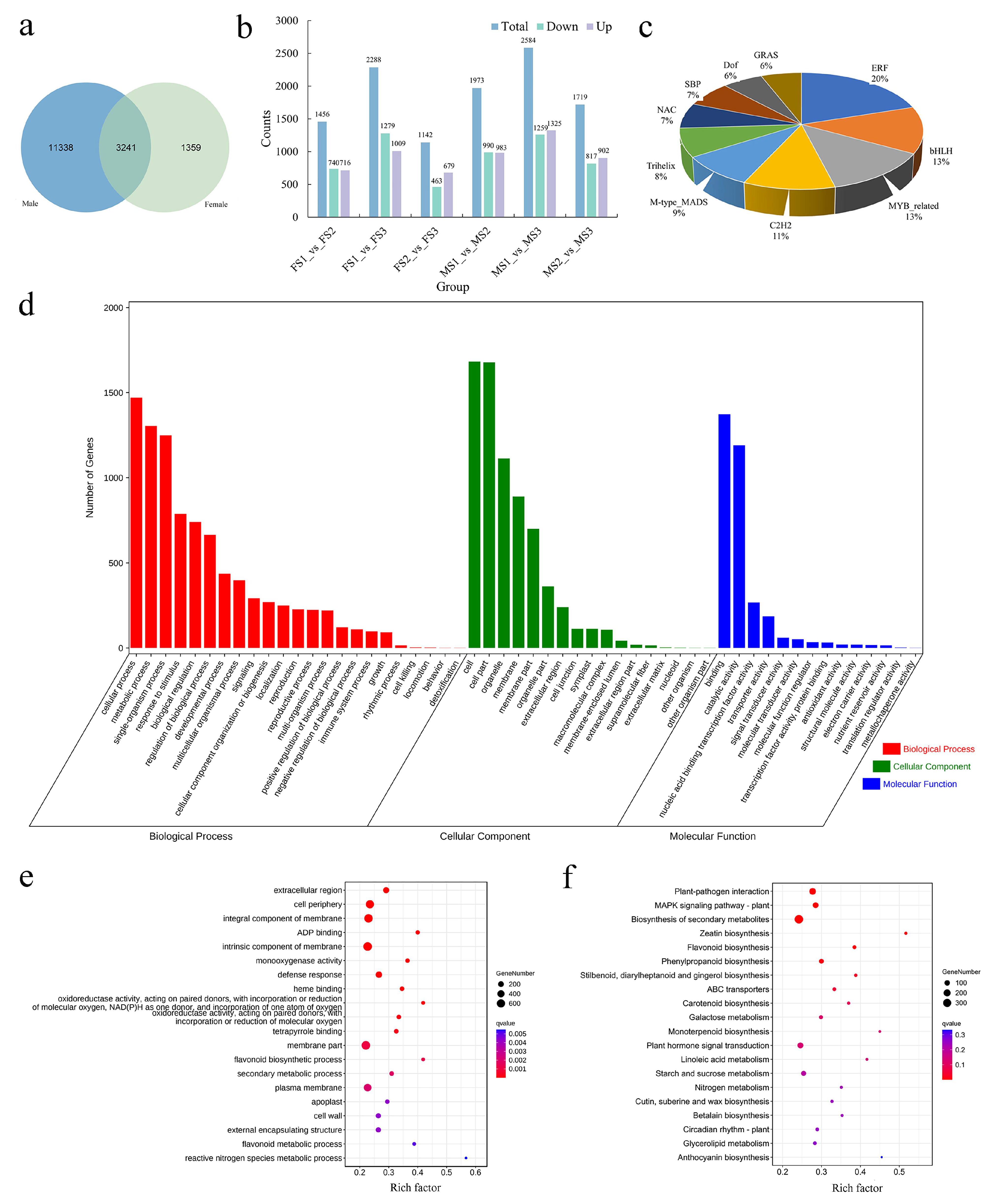
2.5. Differentially Expressed Transcription Factors during the Transition to Flowering
2.6. Functional Analysis of the Common DEGs between Male and Female Inflorescence during the Transition to Flowering
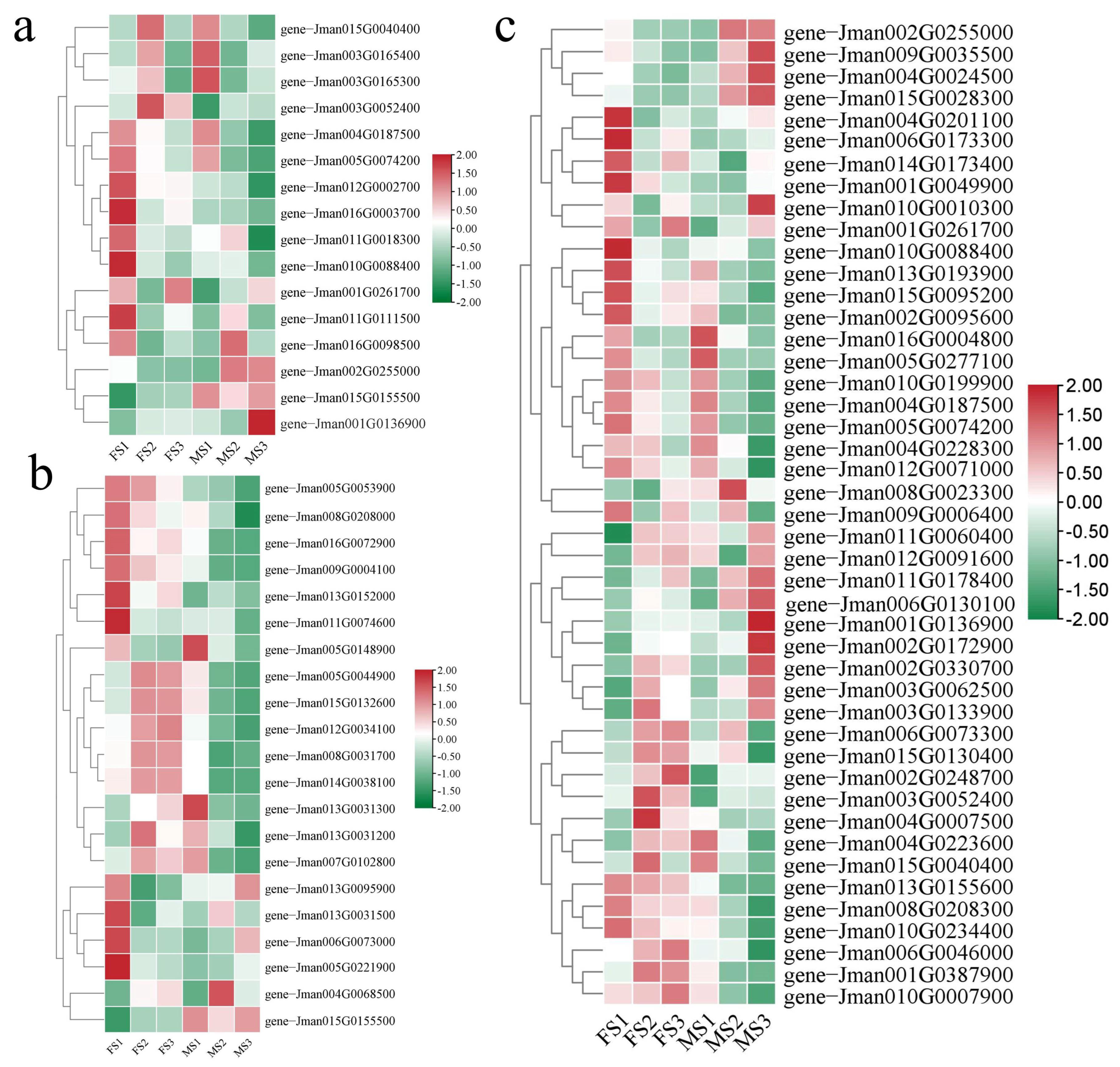
2.7. Recognizing DEGs Related to Floral Transition and Flower Development
2.8. Identification and Expression Analysis of Genes Involved in Biosynthesis and Reception of Phytohormones
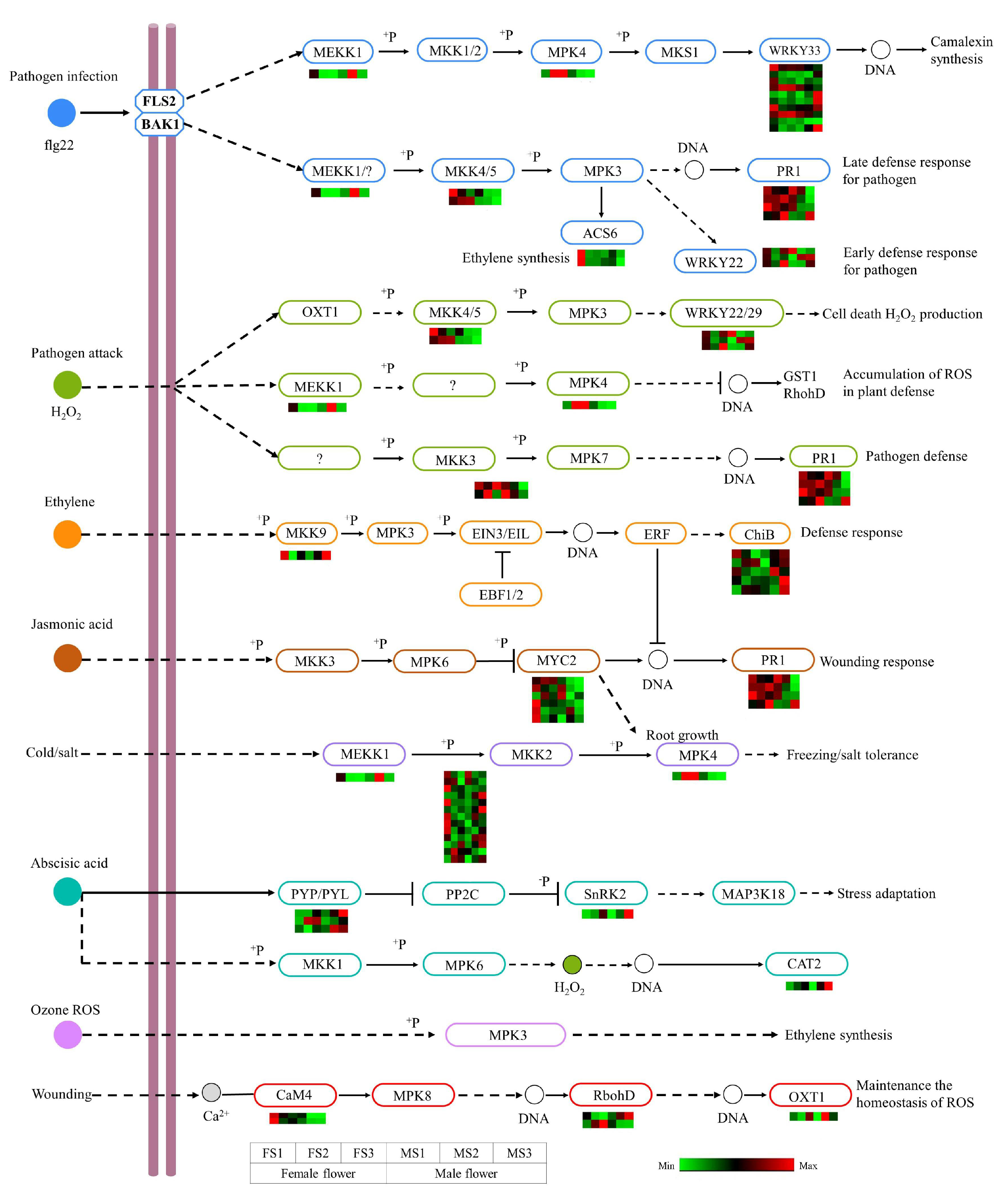
2.9. DEGs Involved in MAPK Signaling Pathway
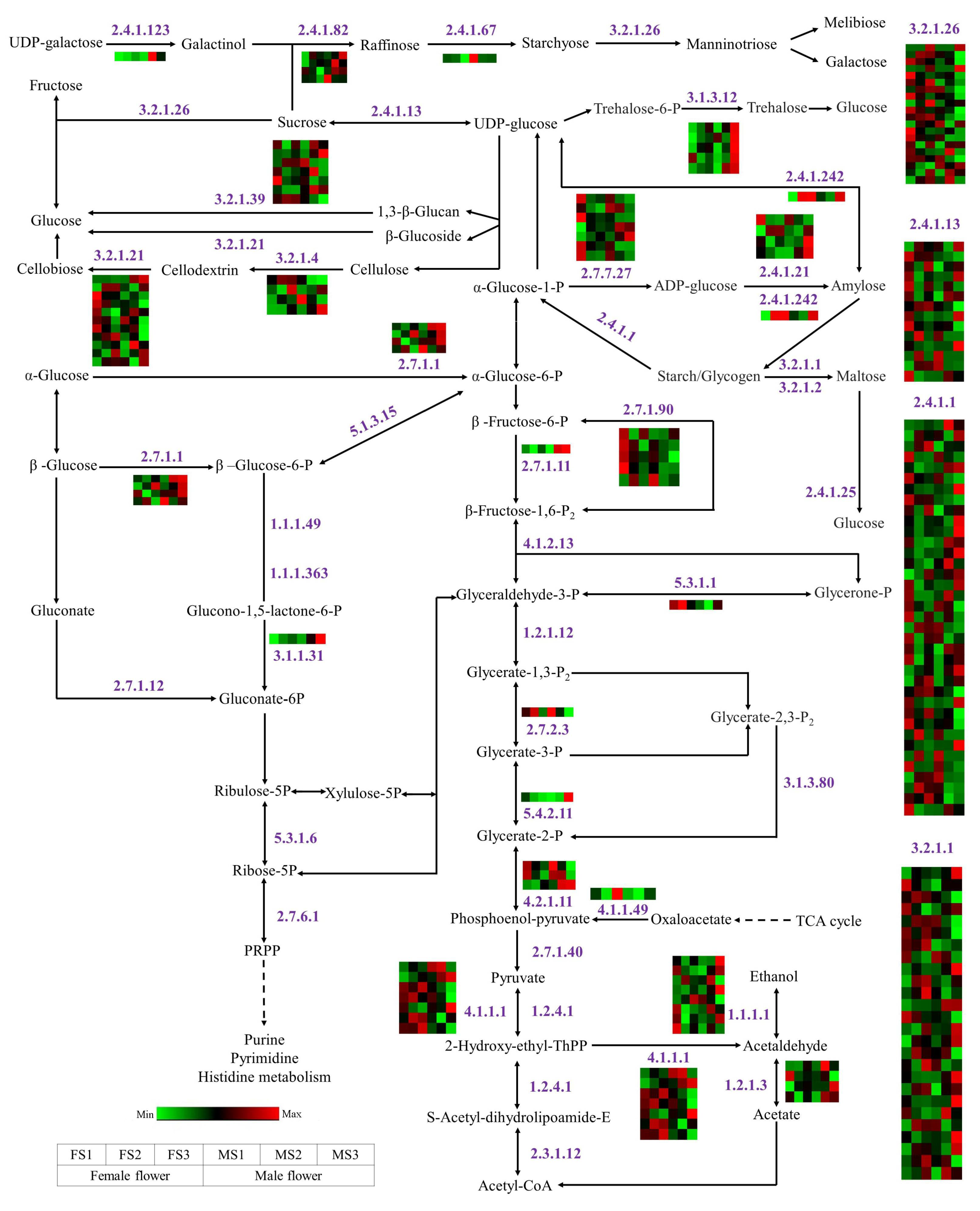
2.10. DEGs Involved in Energy Metabolism during Flowering Process
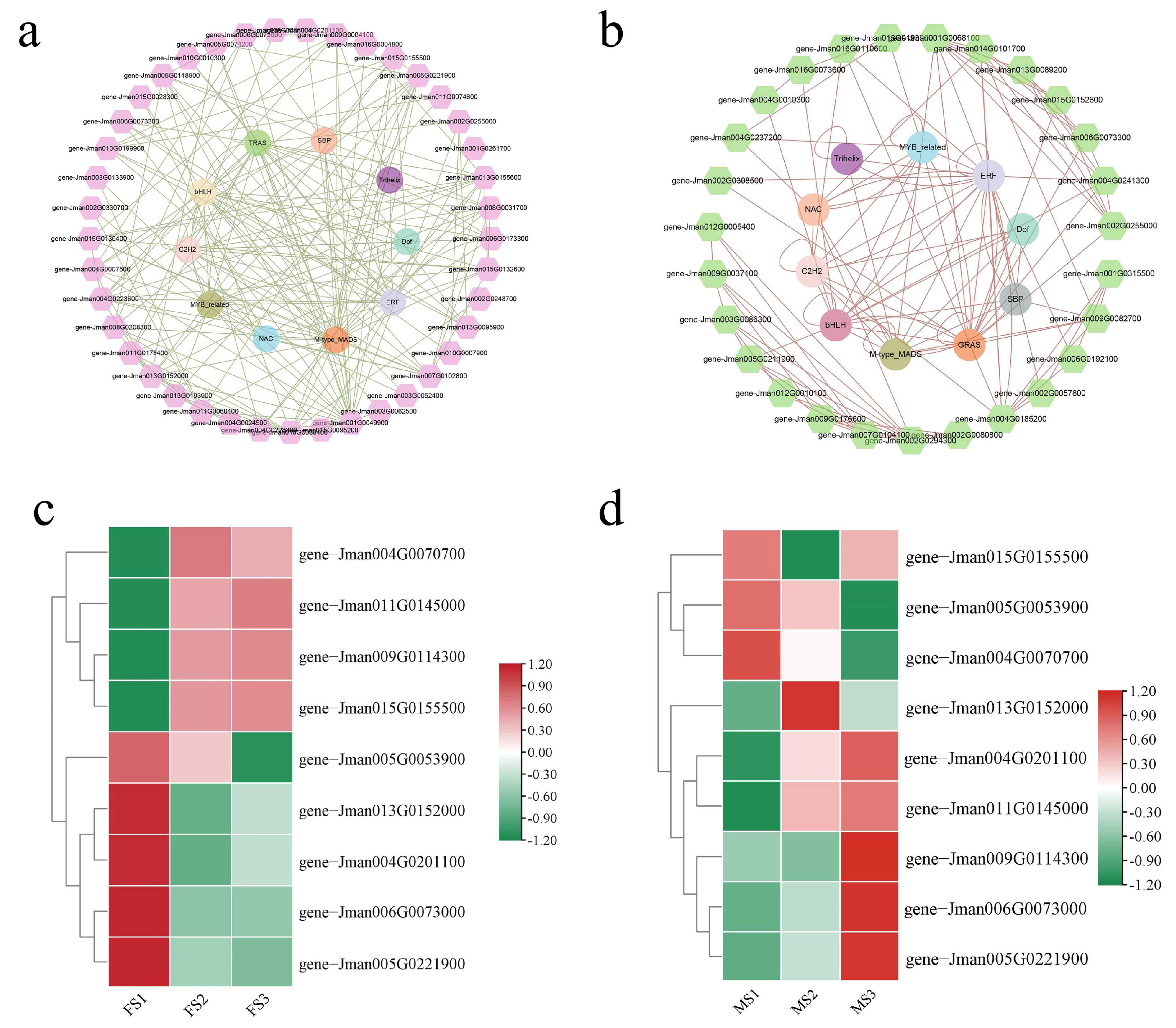
2.11. Co-Expression Network Analysis and Identification of MADS Genes
2.12. Validation of RNA-Seq Data by qRT-PCR
3. Discussion
3.1. Identification of DEGs Involved in Flower Development in J. mandshurcia
3.2. Underlying Functions of Phytohormone in Flower Development in J. mandshurcia
3.3. Energy Metabolism during Flower Development
3.4. MADS-Box Genes Contribute to the Flowering of J. mandshurica
4. Materials and Methods
4.1. Plant Materials and RNA Preparation
4.2. Measurement of Hormone Content
4.3. RNA-Seq Library Construction and Sequencing
4.4. Bioinformatics Analysis of RNA-Seq Data
4.5. Differential Gene Expression Analysis and GO Enrichment Analysis
4.6. Interaction Network Construction
4.7. Verifying Gene Expression Levels by qRT-PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, C.M.; Liu, X.C.; Silva, J.A.T.D.; Liu, N.; Duan, J. Transcriptome sequencing and metabolite profiling analyses provide comprehensive insight into molecular mechanisms of flower development in Dendrobium officinale (Orchidaceae). Plant Mol. Biol. 2020, 104, 529–548. [Google Scholar] [CrossRef]
- Wang, M.; Xi, D.; Chen, Y.; Zhu, C.C.; Zhao, Y.Q.; Geng, G.M. Morphological characterization and transcriptome analysis of pistillate flowering in pecan (Carya illinoinensis). Sci. Hortic. 2019, 257, 108674. [Google Scholar] [CrossRef]
- Wang, H.X.; Yang, Y.H.; Zhang, Y.Y.; Zhao, T.T.; Jiang, J.B.; Li, J.F.; Xu, X.Y.; Yang, H.H. Transcriptome analysis of flower development and mining of genes related to flowering time in Tomato (Solanum lycopersicum). Int. J. Mol. Sci. 2021, 22, 8128. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.L.; Yu, C.; Luo, L.; Wan, H.H.; Zhen, N.; Xu, T.L.; Tan, J.R.; Pan, H.T.; Zhang, Q.X. Transcriptome of the floral transition in Rosa chinensis ‘Old Blush’. BMC Genom. 2017, 18, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, D.L.; Chen, W.W.; Hu, R.Q.; Zhang, Y.C.; Xia, Y.; Wang, S.M.; He, Q.; Guo, Q.G.; Liang, G.L. An integrative analysis of transcriptome, proteome and hormones reveals key differentially expressed genes and metabolic pathways involved in flower development in Loquat. Int. J. Mol. Sci. 2020, 21, 5107. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.Z.; Hu, H.M.; Ren, H.M.; Yang, Z.L.; Qiu, Q.; Qi, W.W.; Liu, X.Y.; Chen, X.M.; Cui, X.K.; Li, S.S.; et al. The Arabidopsis H3K27me3 demethylase JUMONJI 13 is a temperature and photoperiod dependent flowering repressor. Nat. Commun. 2019, 10, 1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.L.; An, H.R.; Tong, C.G.; Jang, S. Flowering and flowering genes: From model plants to orchids. Hortic. Environ. Biotechnol. 2021, 62, 135–148. [Google Scholar] [CrossRef]
- Liu, Y.P.; Yang, J.; Yang, M.F. Pathways of flowering regulation in plants. EMBO Rep. 2015, 31, 1553–1566. [Google Scholar]
- Song, Y.H.; Ito, S.; Imaizumi, T. Flowering time regulation: Photoperiod- and temperature-sensing in leaves. Trends Plant Sci. 2013, 18, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Blázquez, M.; Green, R.; Nilsson, O.; Weigel, S.D. Gibberellins promote flowering of Arabidopsis by activating the LEAFY promoter. Plant Cell 1998, 10, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Samach, A.; Onouchi, H.; Gold, S.E.; Ditta, G.S.; Schwarz-Sommer, Z.; Yanofsky, M.F.; Coupland, G. Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science 2000, 288, 1613–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komeda, Y. Genetic regulation of time to flower in Arabidopsis thaliana. Annu. Rev. Plant Biol. 2004, 55, 521–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.H.; Luo, X.M.; Xu, D.G.; Tian, X.L.; Song, J.; Xia, X.C.; Chu, C.C.; He, Z.H. Genetic architecture underlying light and temperature mediated flowering in Arabidopsis, rice and temperate cereals. New Phytol. 2021, 230, 1731–1745. [Google Scholar] [CrossRef]
- Cerdan, P.D.; Chory, J. Regulation of flowering time by light quality. Nature 2003, 423, 881–885. [Google Scholar] [CrossRef]
- Kinmonth-Schultz, H.A.; Tong, X.; Lee, J.; Song, Y.H.; Ito, S.; Kim, S.H.; Imaizumi, T. Cool night-time temperatures induce the expression of CONSTANS and FLOWERING LOCUS T to regulate flowering in Arabidopsis. New Phytol. 2016, 211, 208–224. [Google Scholar] [CrossRef] [Green Version]
- Quiroz, S.; Yustis, J.C.; Chávez-Hernández, E.C.; Martínez, T.; García-Ponce, B. Beyond the genetic pathways, flowering regulation complexity in Arabidopsis thaliana. Int. J. Mol. Sci. 2021, 22, 5716. [Google Scholar] [CrossRef]
- Newton, L.A.; Runkle, E.S. High-temperature inhibition of flowering of Phalaenopsis and Doritaenopsis Orchids. HortScience 2009, 44, 1271–1276. [Google Scholar] [CrossRef]
- Kemi, U.; Leinonen, P.H.; Savolainen, O.; Kuittinen, H. Inflorescence shoot elongation, but not flower primordia formation, is photoperiodically regulated in Arabidopsis lyrata. Ann. Bot. 2019, 124, 91–102. [Google Scholar] [CrossRef]
- Abba, M.; Idris, S. Mechanisms of photoperiod in regulation of Rice flowering. Int. J. Tech. Res. Appl. 2014, 2, 56–58. [Google Scholar]
- Zeng, X.R.; Liu, H.L.; Du, H.Y.; Wang, S.J.; Yang, W.M.; Chi, Y.J.; Wang, J.; Huang, F.; Yu, D.Y. Soybean MADS-box gene GmAGL1 promotes flowering via the photoperiod pathway. BMC Genom. 2018, 19, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazakis, C.M.; Coneva, V.; Colasanti, J. ZCN8 encodes a potential orthologue of Arabidopsis FT florigen that integrates both endogenous and photoperiod flowering signals in maize. J. Exp. Bot. 2011, 62, 4833–4842. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.; Lienqueo, I.; Llanes, A.; Rojas, P.; Perez, J.; Correa, F.; Sagredo, B.; Masciarelli, O.; Luna, V.; Almada, R. Comparative transcriptomic analysis reveals novel roles of transcription factors and hormones during the flowering induction and floral bud differentiation in sweet cherry trees (Prunus avium L. cv. Bing). PLoS ONE 2020, 15, e0230110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.; Pan, C.; Wang, M.X.; Cui, L.; Han, B.Y. Progress on the mechanism of hormones regulating plant flower formation. Hereditas 2020, 42, 739–751. [Google Scholar] [PubMed]
- Fukazawa, J.; Ohashi, Y.; Takahashi, R.; Nakai, K.; Takahashi, Y. DELLA degradation by gibberellin promotes flowering via GAF1-TPR-dependent repression of floral repressors in Arabidopsis. Plant Cell 2021, 33, 2258–2272. [Google Scholar] [CrossRef]
- Zhang, H.L.; Wang, W.; Huang, J.F.; Wang, Y.T.; Hu, L.; Yuan, Y.; Lyu, M.L.; Wu, B.H. Role of gibberellin and its three GID1 receptors in Jasminum sambac stem elongation and flowering. Planta 2021, 255, 17. [Google Scholar] [CrossRef]
- Zhang, S.; Gottschalk, C.; van Nocker, S. Genetic mechanisms in the repression of flowering by gibberellins in apple (Malus x domestica Borkh.). BMC Genom. 2019, 20, 747. [Google Scholar] [CrossRef]
- He, J.; Xin, P.Y.; Ma, X.T.; Chu, J.F.; Wang, G.D. Gibberellin metabolism in flowering plants: An update and perspectives. Front. Plant Sci. 2020, 11, 532. [Google Scholar] [CrossRef]
- Song, Y.; Dou, L.D.; Zhang, H.J. Molecular and genetic mechanisms of control of floral induction in higher plants. Plant Physiol. J. 2012, 50, 56. [Google Scholar]
- Porri, A.; Torti, S.; Romera-Branchat, M.; Coupland, G. Spatially distinct regulatory roles for gibberellins in the promotion of flowering of Arabidopsis under long photoperiods. Development 2012, 139, 2198–2209. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.K.; Zhan, W.; Zhang, H.; Song, S.L.; Qu, X.B.; Wang, J.Y.; Tigabu, M.; Li, X.; Zhang, Q.H.; Zhao, M.H.; et al. Genetic improvement in Juglans mandshurica and its uses in China: Current status and future prospects. Phyton-Int. J. Exp. Bot. 2022, 91, 489–505. [Google Scholar] [CrossRef]
- Li, X.; Cai, K.W.; Pei, X.N.; Li, Y.; Hu, Y.B.; Meng, F.J.; Song, X.S.; Tigabu, M.; Ding, C.J.; Zhao, X.Y. Genome-wide identification of NAC transcription factor family in Juglans mandshurica and their expression analysis during the fruit development and ripening. Int. J. Mol. Sci. 2021, 22, 12414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, C.; Lu, X.; Sun, X.; Liu, C.; Zhou, Q.; Deng, J. Flower development of heterodichogamous Juglans mandshurica (Juglandaceae). Front. Plant Sci. 2021, 12, 541163. [Google Scholar] [CrossRef] [PubMed]
- Luan, F.; Wang, Z.; Yang, Y.; Ji, Y.; Lv, H.Z.; Han, K.Q.; Liu, D.H.; Shang, X.F.; He, X.R.; Zeng, N. Juglans mandshurica Maxim.: A review of its traditional usages, phytochemical constituents, and pharmacological properties. Front. Pharmacol. 2021, 11, 569800. [Google Scholar] [CrossRef]
- Zhang, R.; Yang, C.; Jiang, Y.; Li, L. A PIF7-CONSTANS-centered molecular regulatory network underlying shade-accelerated flowering. Mol. Plant 2019, 12, 1587–1597. [Google Scholar] [CrossRef]
- Kinmonth-Schultz, H.; Lewandowska-Sabat, A.; Imaizumi, T.; Ward, J.K.; Rognli, O.A.; Fjellheim, S. Flowering times of wild Arabidopsis accessions from across norway correlate with expression levels of FT, CO, and FLC genes. Front. Plant Sci. 2021, 12, 747740. [Google Scholar] [CrossRef]
- Liu, C.Y.; Zhang, Q.Q.; Zhu, H.; Cai, C.M.; Li, S. Characterization of mungbean CONSTANS-LIKE genes and functional analysis of CONSTANS-LIKE 2 in the regulation of flowering time in Arabidopsis. Front. Plant Sci. 2021, 12, 608603. [Google Scholar] [CrossRef]
- Pan, G.; Li, Z.; Yin, M.; Huang, S.Q.; Tao, J.; Chen, A.G.; Li, J.J.; Tang, H.J.; Chang, L.; Deng, Y.; et al. Genome-wide identification, expression, and sequence analysis of CONSTANS-like gene family in cannabis reveals a potential role in plant flowering time regulation. BMC Plant Biol. 2021, 21, 142. [Google Scholar] [CrossRef]
- Xi, C.Y.; Jin, Z.R.; Qiong, H.; Liang, W.W.; Jia, L. Conservation and divergence of the CONSTANS-Like (COL) genes related to flowering and circadian rhythm in Brassica napus. Front. Plant Sci. 2021, 12, 760379. [Google Scholar]
- Li, J.; Gao, K.; Yang, X.Y.; Khan, W.U.; Guo, B.; Guo, T.; An, X.M. Identification and characterization of the CONSTANS-like gene family and its expression profiling under light treatment in Populus. Int. J. Biol. Macromol. 2020, 161, 999–1010. [Google Scholar] [CrossRef]
- Xiao, G.; Li, B.J.; Chen, H.J.; Chen, W.; Wang, Z.Y.; Gui, R.Y.; Guo, X.Q. Overexpression of PvCO1, a bamboo CONSTANS-LIKE gene, delays flowering by reducing expression of the FT gene in transgenic Arabidopsis. BMC Plant Biol. 2018, 18, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Hettiarachchi, G.H.C.M.; Deng, X.W.; Holm, M.; Notes, A. Arabidopsis CONSTANS-LIKE3 is a positive regulator of red light signaling and root growth. Plant Cell 2006, 18, 70–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.H.; Ju, Y.; Seo, P.J.; Lee, J.H.; Park, C.M. The SOC1-SPL module integrates photoperiod and gibberellic acid signals to control flowering time in Arabidopsis. Plant J. 2012, 69, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Hong, J.; Chen, X.F.; Zhang, C.Q.; Chen, M.J.; Luo, Z.J.; Chang, S.W.; Bai, S.X.; Liang, W.Q.; Liu, Q.Q.; et al. Gibberellins orchestrate panicle architecture mediated by DELLA-KNOX signalling in rice. Plant Biotechnol. J. 2021, 19, 2304–2318. [Google Scholar] [CrossRef]
- Hauvermale, A.L.; Ariizumi, T.; Steber, C.M. Gibberellin signaling: A theme and variations on DELLA repression. Plant Physiol. 2012, 160, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Qin, L.; Lee, S.; Fu, X.; Richards, D.E.; Cao, D.; Luo, D.; Harberd, N.P.; Peng, J. Gibberellin regulates Arabidopsis floral development via suppression of DELLA protein function. Development 2004, 131, 1055–1064. [Google Scholar] [CrossRef] [Green Version]
- Tyler, L.; Thomas, S.G.; Hu, J.; Dill, A.; Alonso, J.M.; Ecker, J.R.; Sun, T.P. Della proteins and gibberellin-regulated seed germination and floral development in Arabidopsis. Plant Physiol. 2004, 135, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.P.; Pan, J.J.; Li, Y.; Lou, D.J.; Hu, Y.R.; Yu, D.Q. The DELLA-CONSTANS Transcription factor cascade integrates Gibberellic acid and photoperiod signaling to regulate flowering. Plant Physiol. 2016, 172, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.B.; Hou, J.L.; Cui, J.; He, C.; Li, W.B.; Chen, X.Y.; Li, M.; Wang, W.Q. The effects of endogenous hormones on the flowering and fruiting of glycyrrhiza uralensis. Plants 2019, 8, 519. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Ma, N.; Jia, Y.C.; Zhang, Y.; Feng, M.; Jiang, C.Z.; Ma, C.; Gao, J.P. An ethylene-induced regulatory module delays flower senescence by regulating cytokinin content. Plant Physiol. 2016, 173, 853–862. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Chang, X.X.; Kasuga, T.; Bui, M.; Reid, M.S.; Jiang, C.Z. A basic helix-loop-helix transcription factor, PhFBH4, regulates flower senescence by modulating ethylene biosynthesis pathway in petunia. Hortic. Res. 2015, 2, 15059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciniak, K.; Przedniczek, K. Gibberellin signaling repressor LlDELLA1 controls the flower and pod development of Yellow Lupine (Lupinus luteus L.). Int. J. Mol. Sci. 2020, 21, 1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenreira, T.; Lange, M.J.P.; Lange, T.; Bres, C.; Labadie, M.; Monfort, A.; Hernould, M.; Rothan, C.; Denoyes, B. A Specific Gibberellin 20-Oxidase dictates the flowering-runnering decision in diploid Strawberry. Plant Cell 2017, 29, 2168–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, H.L.; Huang, X.M.; Zhu, Y.N.; Xie, B.X.; Liu, H.C.; Song, S.W.; Hao, Y.W.; Chen, R.Y. Identification of DELLA genes and key stage for GA sensitivity in bolting and flowering of flowering Chinese Cabbage. Int. J. Mol. Sci. 2021, 22, 12092. [Google Scholar] [CrossRef] [PubMed]
- Martignago, D.; Siemiatkowska, B.; Lombardi, A.; Conti, L. Abscisic acid and flowering regulation: Many targets, different places. Int. J. Mol. Sci. 2020, 21, 9700. [Google Scholar] [CrossRef]
- D’Aloia, M.; Bonhomme, D.; Bouché, F.; Tamseddak, K.; Ormenese, S.; Torti, S.; Coupland, G.; Périlleux, C. Cytokinin promotes flowering of Arabidopsis via transcriptional activation of the FT paralogue TSF. Plant J. 2011, 65, 972–979. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.L.; Sui, X.L.; Lucas, W.J.; Li, Y.X.; Feng, S.; Ma, S.; Fan, J.W.; Gao, L.H.; Zhang, Z.X. Down-regulation of the sucrose transporter CsSUT1 causes male sterility by altering carbohydrate supply. Plant Physiol. 2019, 180, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.S.; Li, Z.W.; Wu, S.W.; Wan, X.Y. The essential roles of sugar metabolism for pollen development and male fertility in plants. Crop J. 2021, 9, 1223–1236. [Google Scholar] [CrossRef]
- Gu, J.H.; Zeng, Z.; Wang, Y.R.; Lyu, Y.M. Transcriptome analysis of carbohydrate metabolism genes and molecular regulation of sucrose transport gene LoSUT on the flowering process of developing oriental Hybrid Lily ‘Sorbonne’ Bulb. Int. J. Mol. Sci. 2020, 21, 3092. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, L.L.; Yan, J.P.; Ye, J.B.; Cheng, S.Y.; Xu, F.; Wang, G.Y.; Zhang, W.W.; Liao, Y.L.; Liu, X.M. Functional characterization of the EMBRYONIC FLOWER 2 gene involved in flowering in Ginkgo biloba. Front. Plant Sci. 2021, 12, 681166. [Google Scholar] [CrossRef]
- Zhu, W.W.; Yang, L.; Wu, D.; Meng, Q.C.; Deng, X.; Huang, G.Q.; Zhang, J.; Chen, X.F.; Ferrándiz, C.; Liang, W.Q.; et al. Rice SEPALLATA Genes OsMADS5 and OsMADS34 cooperate to limit inflorescence branching by repressing the TERMINAL FLOWER1-like gene RCN4. New Phytol. 2021, 233, 1682–1700. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.T.; Shi, Q.Q.; Qi, Y.C.; Wang, S.J.; Lei, Y.; Hu, H.C.; Liu, Y.L.; Yang, T.X.; Wei, A.Z. ZbAGL11, a class D MADS-box transcription factor of Zanthoxylum bungeanum, is involved in sporophytic apomixis. Hortic. Res. 2021, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Asquini, E.; Miolli, G.V.; Weigl, K.; Hanke, M.; Flachowsky, H.; Si-Ammour, A. The MADS-Box gene MdDAM1 controls growth cessation and bud dormancy in Apple. Front. Plant Sci. 2020, 11, 1003. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.M.; Deng, H.; Ma, W.X.; Zhou, Q.; Liu, Z.P. Genome-wide identification of the MADS-box transcription factor family in autotetraploid cultivated alfalfa (Medicago sativa L.) and expression analysis under abiotic stress. BMC Genom. 2021, 22, 603. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.W.; He, M.H.; Zeng, Z.P.; Chen, Y.Z.; Hanna, A.D.; Zhu, H.B. Genome-wide identification and expression analysis of the MADS-Box gene family in sweet Potato [Ipomoea batatas (L.) Lam]. Front. Genet. 2021, 12, 750137. [Google Scholar] [CrossRef]
- Ren, L.L.; Sun, H.W.; Dai, S.Y.; Feng, S.; Qiao, K.; Wang, J.G.; Gong, S.F.; Zhou, A.M. Identification and characterization of MIKCc-Type MADS-Box genes in the flower organs of Adonis amurensis. Int. J. Mol. Sci. 2021, 22, 9362. [Google Scholar] [CrossRef]
- Lin, Z.Y.; Cao, D.D.; Damaris, R.N.; Yang, P.F. Genome-wide identification of MADS-box gene family in sacred lotus (Nelumbo nucifera) identifies a SEPALLATA homolog gene involved in floral development. BMC Plant Biol. 2020, 20, 497. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Tang, D.Q.; Lin, X.C.; Ding, M.Q.; Tong, Z.K. Genome-wide identification of MADS-box family genes in moso bamboo (Phyllostachys edulis) and a functional analysis of PeMADS5 in flowering. BMC Plant Biol. 2018, 18, 176. [Google Scholar] [CrossRef]
- Xu, K.D.; Liu, K.; Wu, J.X.; Wang, W.; Zhu, Y.L.; Li, C.J.; Zhao, M.Z.; Wang, Y.W.; Li, C.W.; Zhao, L.J. A MADS-box gene associated with protocorm-like body formation in Rosa canina alters floral organ development in Arabidopsis. Can. J. Plant Sci. 2018, 98, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Song, G.Q.; Han, X.; Ryner, J.T.; Thompson, A.; Wang, K. Utilizing MIKC-type MADS-box protein SOC1 for yield potential enhancement in maize. Plant Cell Rep. 2021, 40, 1679–1693. [Google Scholar] [CrossRef]
- Moon, J.; Suh, S.S.; Lee, H.; Choi, K.R.; Hong, C.B.; Paek, N.C.; Kim, S.G.; Lee, I. The SOC1 MADS-box gene integrates vernalization and gibberellin signals for flowering in Arabidopsis. Plant J. 2003, 35, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Li, L.; Ma, T.F.; Pei, J.L.; Zhao, Z.Y.; Lu, M.Z.; Wu, A.M.; Lin, X.C. The SOC1-like gene BoMADS50 is associated with the flowering of Bambusa oldhamii. Hortic. Res. 2021, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Jaudal, M.; Zhang, L.L.; Che, C.; Li, G.F.; Tang, Y.H.; Wen, J.Q.; Mysore, K.S.; Putterill, J. A SOC1-like gene MtSOC1a promotes flowering and primary stem elongation in Medicago. J. Exp. Bot. 2018, 69, 4867–4880. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, Z.C.; Dong, W.; Wang, Z.J.; Zhang, L.S. Expansion and functional divergence of the SHORT VEGETATIVE PHASE (SVP) genes in Eudicots. Genome Biol. Evol. 2018, 10, 3026–3037. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, U.; Höhmann, S.; Nettesheim, K.; Wisman, E.; Saedler, H.; Huijser, P. Molecular cloning of SVP: A negative regulator of the floral transition in Arabidopsis. Plant J. 2000, 21, 351–360. [Google Scholar] [CrossRef]
- Li, C.C.; Gu, H.Y.; Jiang, W.; Zou, C.H.; Wei, D.Y.; Wang, Z.M.; Tang, Q.L. Protein interactions of SOC1 with SVP are regulated by a few crucial amino acids in flowering pathways of Brassica juncea. Acta Physiol. Plant. 2019, 41, 43. [Google Scholar] [CrossRef]
- Jaudal, M.; Monash, J.; Zhang, L.; Wen, J.; Mysore, K.S.; Macknight, R.; Putterill, J. Overexpression of Medicago SVP genes causes floral defects and delayed flowering in Arabidopsis but only affects floral development in Medicago. J. Exp. Bot. 2014, 65, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.M.; Zhang, J.Z.; Hu, C.G. Characterization and Expression Analysis of PtAGL24, a SHORT VEGETATIVE PHASE/AGAMOUS-LIKE 24 (SVP/AGL24)-Type MADS-Box Gene from Trifoliate Orange (Poncirus trifoliata L. Raf.). Front. Plant Sci. 2016, 7, 823. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.E.; Singh, M.B.; Bhalla, P.L. Novel members of the AGAMOUS LIKE 6 subfamily of MIKCC-type MADS-box genes in soybean. BMC Plant Biol. 2013, 13, 105. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.K.; Wu, X.; Lee, J.S.; Ahn, J.H. AGAMOUS-LIKE 6 is a floral promoter that negatively regulates the FLC/MAF clade genes and positively regulates FT in Arabidopsis. Plant J. 2011, 65, 62–76. [Google Scholar] [CrossRef]
- Lu, H.; Klocko, A.L.; Brunner, A.M.; Ma, C.; Magnuson, A.C.; Howe, G.T.; An, X.; Strauss, S.H. RNA interference suppression of AGAMOUS and SEEDSTICK alters floral organ identity and impairs floral organ determinacy, ovule differentiation, and seed-hair development in Populus. New Phytol. 2019, 222, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.D.; Jiang, W.; Dong, Y.; Ma, X.; Zhou, W.W.; Wang, Z.M.; Fan, Y.H.; Wei, D.Y.; Tang, Q.L. AGL19 directly interacts with floral signal integrator AGL24 in flowering time control of Brassica juncea. Acta Physiol. Plant. 2020, 42, 175. [Google Scholar] [CrossRef]
- Gregis, V. AGL24, SHORT VEGETATIVE PHASE, and APETALA1 redundantly control AGAMOUS during early stages of flower development in Arabidopsis. Plant Cell 2006, 18, 1373–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, P.; García-Ponce, B.; Fonseca-Salazar, G.; Alvarez-Buylla, E.R.; Yu, H. AGAMOUS-LIKE 17, a novel flowering promoter, acts in a FT-independent photoperiod pathway. Plant J. 2008, 55, 253–265. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Smyth, G.K.; Wei, S. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zeng, M.X.; Ju, H.J.; Yang, D.; Min, W.J.; Shan, D.; Lei, K.; Ge, G.; Yun, L.C.; Ping, W.L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Han, R.; Cai, K.; Guo, R.; Pei, X.; Zhao, X. Characterization of Phytohormones and Transcriptomic Profiling of the Female and Male Inflorescence Development in Manchurian Walnut (Juglans mandshurica Maxim.). Int. J. Mol. Sci. 2022, 23, 5433. https://doi.org/10.3390/ijms23105433
Li X, Han R, Cai K, Guo R, Pei X, Zhao X. Characterization of Phytohormones and Transcriptomic Profiling of the Female and Male Inflorescence Development in Manchurian Walnut (Juglans mandshurica Maxim.). International Journal of Molecular Sciences. 2022; 23(10):5433. https://doi.org/10.3390/ijms23105433
Chicago/Turabian StyleLi, Xiang, Rui Han, Kewei Cai, Ruixue Guo, Xiaona Pei, and Xiyang Zhao. 2022. "Characterization of Phytohormones and Transcriptomic Profiling of the Female and Male Inflorescence Development in Manchurian Walnut (Juglans mandshurica Maxim.)" International Journal of Molecular Sciences 23, no. 10: 5433. https://doi.org/10.3390/ijms23105433