
Snapshots of the Fragmentation for C70@Single-Walled Carbon Nanotube: Tight-Binding Molecular Dynamics Simulations


Abstract
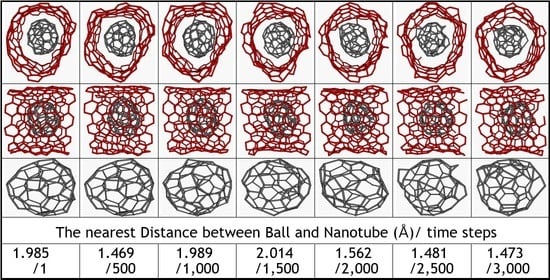
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Geometry of C70, SWCNT, And CNP
2.2. Distance Distribution Function of Adjacent Carbon Atoms in a CNP
3. Materials and Methods
3.1. Tight-Binding Molecular Dynamics Simulations
3.2. Hybrid Density Functional Calculations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kroto, H.W.; Allef, A.W.; Blum, S.P. C60: Buckminsterfullerene. Chem. Rev. 1991, 91, 1213–1235. [Google Scholar] [CrossRef]
- Iijima, S. Synthesis of Carbon Nanotubes. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Verberck, B.; Tarakina, N.V. Tubular fullerenes inside carbon nanotubes: Optimal molecular orientation versus tube radius. Eur. Phys. J. B 2011, 80, 355–362. [Google Scholar] [CrossRef]
- Shinohara, H. In pursuit of nanocarbons. Chem. Rec. 2012, 12, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Michel, K.H.; Verberck, B.; Nikolaev, A.V. Anisotropic Packing and One-Dimensional Fluctuations of C60 Molecules in Carbon Nanotubes. Phys. Rev. Lett. 2005, 95, 185506. [Google Scholar] [CrossRef]
- Rols, S.; Cambedouzou, J.; Chorro, M.; Schober, H.; Agafonov, V.; Launois, P.; Davydov, V.; Rakhmanina, A.V.; Kataura, H.; Sauvajol, J.-L. How confinement affects the dynamics of C60 in carbon nanopeapods. Phys. Rev. Lett. 2008, 101, 065507. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, C.; Osawa, E.; Lee, K.H. Dynamic Behavior of C60 Fullerene in Carbon Nanopeapods: Tight-Binding Molecular Dynamics Simulation. Bull. Korean Chem. Soc. 2019, 40, 724–728. [Google Scholar] [CrossRef]
- Lee, C.; Lee, J.Y.; Osawa, E.; Lee, K.H. A conversion dynamics of (C60)2 dimer fullerenes to a fused dimer cage in carbon nanopeapods: Tight-binding molecular dynamics simulation. Bull. Korean Chem. Soc. 2019, 40, 1054–1055. [Google Scholar] [CrossRef]
- Smith, B.W.; Luzzi, D.E. Formation mechanism of fullerene peapods and coaxial tubes: A path to large scale synthesis. Chem. Phys. Lett. 2000, 321, 169–174. [Google Scholar] [CrossRef]
- Bandow, S.; Takizawa, M.; Hirahara, K.; Yudasaka, M.; Iijima, S. Raman scattering study of double-wall carbon nanotubes derived from the chains of fullerenes in single-wall carbon nanotubes. Chem. Phys. Lett. 2001, 337, 48–54. [Google Scholar] [CrossRef]
- Li, J.; Shen, H. Effects of fullerene coalescence on the thermal conductivity of carbon nanopeapods. Mol. Phys. 2018, 116, 1297–1305. [Google Scholar] [CrossRef]
- Kalbac, M.; Kavan, L.; Juha, L.; Civis, S.; Zukalova, M.; Bittner, M.; Kubat, P.; Vorlicek, V.; Dunsch, L. Transformation of fullerene peapods to double-walled carbon nanotubes induced by UV radiation. Carbon 2005, 43, 1610–1616. [Google Scholar] [CrossRef]
- Xu, C.H.; Wang, C.Z.; Chan, C.T.; Ho, K.M. A transferable tight-binding potential for carbon. J. Phys. Condens. Matter 1992, 4, 6047–6054. [Google Scholar] [CrossRef]
- Wang, C.Z.; Ho, K.M.; Chan, C.T. Tight-binding molecular-dynamics study of amorphous carbon. Phys. Rev. Lett. 1993, 70, 611–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, G.; Gygi, F.; Golaz, J.C. Vibrational and electronic properties of neutral and negatively charged C20 Clusters. Phys. Rev. B 1998, 57, 1860–1867. [Google Scholar] [CrossRef]
- László, I. Molecular dynamics study of the C60 molecule. J. Mol. Struct. THEOCHEM 1999, 463, 181–184. [Google Scholar] [CrossRef]
- Xu, C.; Scuseria, G.E. Tight-binding molecular dynamics simulations of fullerene annealing and fragmentation. Phys. Rev. Lett. 1994, 31, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Lee, Y.H.; Lee, J.Y. Fragmentation of C60 and C70 clusters. Phys. Rev. B 1993, 48, 18230–18234. [Google Scholar] [CrossRef]
- Kim, E.; Oh, D.H.; Oh, C.W.; Lee, Y.H. Fragmentation of C60, C70 by tight-binding molecular-dynamics approach. Synth. Met. 1995, 70, 1495–1498. [Google Scholar] [CrossRef]
- Lee, C.; Lee, K.H. Decomposition Patterns of Three C20 Isomer Clusters: Tight-binding Molecular Dynamics Simulation. Bull. Korean Chem. Soc. 2016, 37, 911–916. [Google Scholar] [CrossRef]
- Musher, I. Comment on Some Theorems of Quantum Chemistry. Am. J. Phys. 1966, 34, 267–268. [Google Scholar] [CrossRef]
- Gear, C.W. Numerical Initial Value Problems in Ordinary Differential Equations; Prentice-Hall: Englewood Cliffs, NJ, USA, 1971. [Google Scholar]
- Kuznetsov, V. Stereochemistry of Simple Molecules inside Nanotubes and Fullerenes: Unusual Behavior of Usual Systems. Molecules 2020, 25, 2437. [Google Scholar] [CrossRef]
- Rols, S.; Cambedouzou, J.; Chorro, M.; Agafonov, V.; Rakhmanina, A.V.; Davydov, V.; Schober, H.; Sauvajol, J.-L. Probing the Dynamics of C60 Encaged Inside Single-Walled Carbon Nanotubes by Inelastic Neutron Scattering. Fuller. Nanotub. Carbon Nanostruct. 2008, 16, 463–470. [Google Scholar] [CrossRef]
- Glukhova, O.E.; Kolesnikova, A.S.; Kirillova, I.V. Investigation of the Effect of Bending on the Polymerization of Fullerenes Inside Carbon Nanotubes. Fuller. Nanotub. Carbon Nanostruct. 2012, 20, 391–394. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Ori, O. Skeletal Rearrangements of the C240 Fullerene: Efficient Topological Descriptors for Monitoring Stone–Wales Transformations. Mathematics 2020, 8, 968. [Google Scholar] [CrossRef]
- Beck, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. A 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephen, P.J.; Devlin, F.J.; Chablowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Herteing, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C. Gaussian 03; Revision B.04; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.Y.; Lee, C.; Osawa, E.; Choi, J.W.; Sur, J.C.; Lee, K.H. Snapshots of the Fragmentation for C70@Single-Walled Carbon Nanotube: Tight-Binding Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 3929. https://doi.org/10.3390/ijms22083929
Lee JY, Lee C, Osawa E, Choi JW, Sur JC, Lee KH. Snapshots of the Fragmentation for C70@Single-Walled Carbon Nanotube: Tight-Binding Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2021; 22(8):3929. https://doi.org/10.3390/ijms22083929
Chicago/Turabian StyleLee, Ji Young, Changhoon Lee, Eiji Osawa, Jong Woan Choi, Jung Chul Sur, and Kee Hag Lee. 2021. "Snapshots of the Fragmentation for C70@Single-Walled Carbon Nanotube: Tight-Binding Molecular Dynamics Simulations" International Journal of Molecular Sciences 22, no. 8: 3929. https://doi.org/10.3390/ijms22083929