
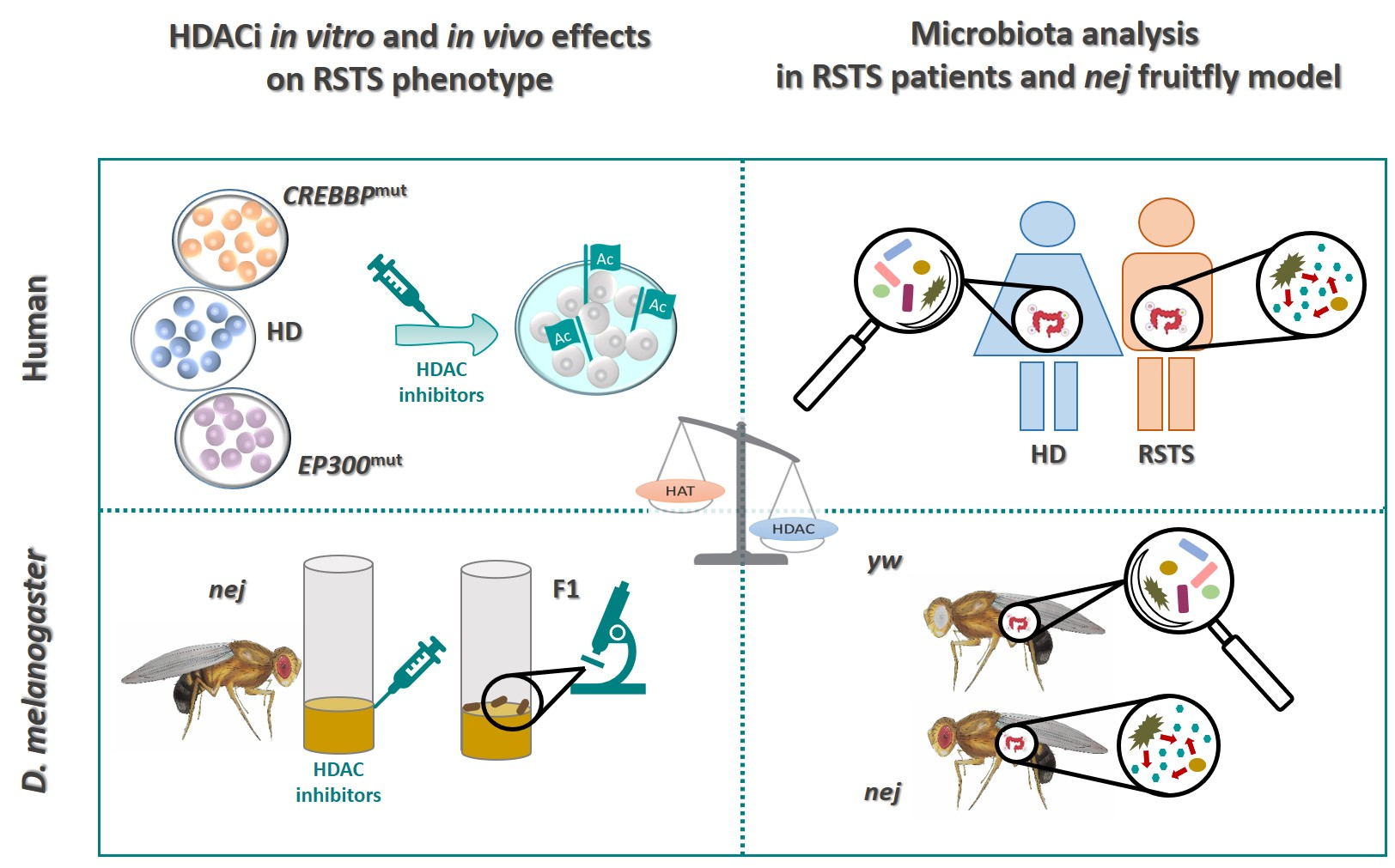
Insights into the Role of the Microbiota and of Short-Chain Fatty Acids in Rubinstein–Taybi Syndrome

 ,
,  , , , , , , ,
, , , , , , ,  , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. HDACi Exposure Counteracts Acetylation Imbalance in RSTS Lymphoblastoid Cell Lines (LCLs)
2.2. RSTS Patients Are Depleted in the Major Butyrate-Producer Faecalibacterium spp.
2.3. HDACi Exposure Leads to Partial Rescue of RSTS Phenotype in Drosophila CBP Mutants
2.4. The Fly Gut Microbiota of Heterozygous Drosophila CBP Mutants
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. AlphaLISA® Assay
4.3. Ki67 and TUNEL Assay
4.4. Subject Recruitment and Sampling for Gut Microbiota Profiling
4.5. Bacterial DNA Extraction and 16S rRNA Gene Sequencing of Human Gut Microbiota
4.6. Fecal Short-Chain Fatty Acid Quantification
4.7. Drosophila Melanogaster Stocks and HDACi Feeding
4.8. Fly Treatment and Embryo Immunostaining
4.9. Bacterial DNA Extraction and 16S rRNA Gene Sequencing of Drosophila Melanogaster Gut Microbiota
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atlasi, Y.; Stunnenberg, H.G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017, 18, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Xie, W. Epigenome in Early Mammalian Development: Inheritance, Reprogramming and Establishment. Trends Cell Biol. 2018, 28, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Uchida, S.; Shumyatsky, G.P. Epigenetic regulation of Fgf1 transcription by CRTC1 and memory enhancement. Brain Res. Bull. 2018, 141, 3–12. [Google Scholar] [CrossRef]
- Bjornsson, H.T. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015, 25, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, J.H.; Taybi, H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am. J. Dis. Child. 1963, 105, 588–608. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C.M. Rubinstein–Taybi syndrome. Eur. J. Hum. Genet. 2006, 14, 981–985. [Google Scholar] [CrossRef] [Green Version]
- Milani, D.; Manzoni, F.; Pezzani, L.; Ajmone, P.; Gervasini, C.; Menni, F.; Esposito, S. Rubinstein-Taybi syndrome: Clinical features, genetic basis, diagnosis, and management. Ital. J. Pediatr. 2015, 41, 4. [Google Scholar] [CrossRef] [Green Version]
- Fergelot, P.; Van Belzen, M.; Van Gils, J.; Afenjar, A.; Armour, C.M.; Arveiler, B.; Beets, L.; Burglen, L.; Busa, T.; Collet, M.; et al. Phenotype and genotype in 52 patients with Rubinstein–Taybi syndrome caused by EP300 mutations. Am. J. Med. Genet. Part A 2016, 170, 3069–3082. [Google Scholar] [CrossRef]
- Miller, R.W.; Rubinstein, J.H. Tumors in Rubinstein-Taybi syndrome. Am. J. Med. Genet. 1995, 56, 112–115. [Google Scholar] [CrossRef]
- Boot, M.V.; van Belzen, M.J.; Overbeek, L.I.; Hijmering, N.; Mendeville, M.; Waisfisz, Q.; Wesseling, P.; Hennekam, R.C.; de Jong, D. Benign and malignant tumors in Rubinstein-Taybi syndrome. Am. J. Med. Genet. Part A 2018, 176, 597–608. [Google Scholar] [CrossRef]
- Valor, L.M.; Viosca, J.; Lopez-Atalaya, J.P.; Barco, A. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr. Pharm. Des. 2013, 19, 5051–5064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, T.P.; Oh, S.P.; Fuchs, M.; Zhou, N.D.; Ch’ng, L.E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Oike, Y.; Hata, A.; Mamiya, T.; Kaname, T.; Noda, Y.; Suzuki, M.; Yasue, H.; Nabeshima, T.; Araki, K.; Yamamura, K. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: Implications for a dominant-negative mechanism. Hum. Mol. Genet. 1999, 8, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar]
- Dutto, I.; Scalera, C.; Prosperi, E. CREBBP and p300 lysine acetyl transferases in the DNA damage response. Cell. Mol. Life Sci. 2018, 75, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Narita, T.; Satpathy, S.; Srinivasan, B.; Hansen, B.K.; Schölz, C.; Hamilton, W.B.; Zucconi, B.E.; Wang, W.W.; Liu, W.R.; et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 2018, 174, 231–244.e12. [Google Scholar] [CrossRef] [Green Version]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Atalaya, J.P.; Gervasini, C.; Mottadelli, F.; Spena, S.; Piccione, M.; Scarano, G.; Selicorni, A.; Barco, A.; Larizza, L. Histone acetylation deficits in lymphoblastoid cell lines from patients with Rubinstein-Taybi syndrome. J. Med. Genet. 2012, 49, 66–74. [Google Scholar] [CrossRef]
- Alarcón, J.M.; Malleret, G.; Touzani, K.; Vronskaya, S.; Ishii, S.; Kandel, E.R.; Barco, A. Chromatin Acetylation, Memory, and LTP Are Impaired in CBP+/− Mice. Neuron 2004, 42, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Simon, G.M.; Cheng, J.; Gordon, J.I. Quantitative assessment of the impact of the gut microbiota on lysine epsilon-acetylation of host proteins using gnotobiotic mice. Proc. Natl. Acad. Sci. USA 2012, 109, 11133–11138. [Google Scholar] [CrossRef] [Green Version]
- Stilling, R.M.; van de Wouw, M.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem. Int. 2016, 99, 110–132. [Google Scholar] [CrossRef] [PubMed]
- Astbury, S.M.; Corfe, B.M. Uptake and metabolism of the short-chain fatty acid butyrate, a critical review of the literature. Curr. Drug Metab. 2012, 13, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Italian Society of Human Nutrition Nutrients and Energy Reference Intake Levels for Italian Population. Available online: https://sinu.it/tabelle-larn-2014/ (accessed on 18 February 2021).
- Akimaru, H.; Chen, Y.; Dai, P.; Hou, D.-X.; Nonaka, M.; Smolik, S.M.; Armstrong, S.; Goodman, R.H.; Ishii, S. Drosophila CBP is a co-activator of cubitus interruptus in hedgehog signalling. Nature 1997, 386, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Akimaru, H.; Hou, D.-X.; Ishii, S. Drosophila CBP is required for dorsal–dependent twist gene expression. Nat. Genet. 1997, 17, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Cobos, S.N.; Bennett, S.A.; Torrente, M.P. The impact of histone post-translational modifications in neurodegenerative diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- José-Enériz, E.S.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC inhibitors in acute myeloid leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef] [Green Version]
- Spartalis, E.; Athanasiadis, D.I.; Chrysikos, D.; Spartalis, M.; Boutzios, G.; Schizas, D.; Garmpis, N.; Damaskos, C.; Paschou, S.A.; Ioannidis, A.; et al. Histone deacetylase inhibitors and anaplastic thyroid carcinoma. Anticancer Res. 2019, 39, 1119–1127. [Google Scholar] [CrossRef]
- Tarnowski, M.; Tkacz, M.; Kopytko, P.; Bujak, J.; Piotrowska, K.; Pawlik, A. Trichostatin A Inhibits Rhabdomyosarcoma Proliferation and Induces Differentiation through MyomiR Reactivation. Folia Biol. 2019, 65, 43–52. [Google Scholar]
- Lipska, K.; Gumieniczek, A.; Filip, A.A. Anticonvulsant valproic acid and other short-chain fatty acids as novel anticancer therapeutics: Possibilities and challenges. Acta Pharm. 2020, 70, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.G.; Huang, Y.R.; Yu, T.; Jiang, H.W.; Xu, Y.; Zhao, X.Y. Chidamide, a histone deacetylase inhibitor, induces growth arrest and apoptosis in multiple myeloma cells in a caspase-dependent manner. Oncol. Lett. 2019, 18, 411–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannitti, T.; Palmieri, B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D 2011, 11, 227–249. [Google Scholar] [CrossRef]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent Progress in Histone Deacetylase Inhibitors as Anticancer Agents. Curr. Med. Chem. 2018, 27, 2449–2493. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R. Inhibition of Histone Deacetylase Activity by Butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Borghi, E.; Vignoli, A. Rett syndrome and other neurodevelopmental disorders share common changes in gut microbial community: A descriptive review. Int. J. Mol. Sci. 2019, 20, 4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindefeldt, M.; Eng, A.; Darban, H.; Bjerkner, A.; Zetterström, C.K.; Allander, T.; Andersson, B.; Borenstein, E.; Dahlin, M.; Prast-Nielsen, S. The ketogenic diet influences taxonomic and functional composition of the gut microbiota in children with severe epilepsy. Npj Biofilms Microbiomes 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, J.S.; Pilarowski, G.O.; Carosso, G.A.; Zhang, L.; Huso, D.L.; Goff, L.A.; Vernon, H.J.; Hansen, K.D.; Bjornsson, H.T. A ketogenic diet rescues hippocampal memory defects in a mouse model of Kabuki syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijtz, R.D.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach Knudsen, K.E.; Lærke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Theil, P.K.; Purup, S.; Hald, S.; Schioldan, A.G.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Naruse, I.; Maekawa, T.; Masuya, H.; Shiroishi, T.; Ishii, S. Abnormal skeletal patterning in embryos lacking a single Cbp allele: A partial similarity with Rubinstein-Taybi syndrome. Proc. Natl. Acad. Sci. USA 1997, 94, 10215–10220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzkin, L.M.; Johnson, S.; Paight, C.; Markow, T.A. Preadult Parental Diet Affects Offspring Development and Metabolism in Drosophila melanogaster. PLoS ONE 2013, 8, e59530. [Google Scholar] [CrossRef]
- Capo, F.; Wilson, A.; Di Cara, F. The intestine of Drosophila melanogaster: An emerging versatile model system to study intestinal epithelial homeostasis and host-microbial interactions in humans. Microorganisms 2019, 7, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fast, D.; Duggal, A.; Foley, E. Monoassociation with Lactobacillus plantarum Disrupts Intestinal Homeostasis in Adult Drosophila melanogaster. mBio 2018, 9, e01114–e01118. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.C.; Kim, S.-H.; You, H.; Kim, B.; Kim, A.C.; Lee, K.-A.; Yoon, J.-H.; Ryu, J.-H.; Lee, W.-J. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 2011, 334, 670–674. [Google Scholar] [CrossRef] [Green Version]
- Sharon, G.; Segal, D.; Ringo, J.M.; Hefetz, A.; Zilber-Rosenberg, I.; Rosenberg, E. Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2010, 107, 20051–20056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, A.; Kamaraj, M.; Basu, M.; Mukherjee, D.; Kapoor, S.; Ranjan, S.; Swamy, M.M.; Kaypee, S.; Scaria, V.; Kundu, T.K.; et al. Chemical and genetic rescue of an ep300 knockdown model for Rubinstein Taybi Syndrome in zebrafish. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1203–1215. [Google Scholar] [CrossRef]
- Spena, S.; Milani, D.; Rusconi, D.; Negri, G.; Colapietro, P.; Elcioglu, N.; Bedeschi, F.; Pilotta, A.; Spaccini, L.; Ficcadenti, A.; et al. Insights into genotype-phenotype correlations from CREBBP point mutation screening in a cohort of 46 Rubinstein-Taybi syndrome patients. Clin. Genet. 2015, 88, 431–440. [Google Scholar] [CrossRef]
- Negri, G.; Milani, D.; Colapietro, P.; Forzano, F.; Della Monica, M.; Rusconi, D.; Consonni, L.; Caffi, L.G.; Finelli, P.; Scarano, G.; et al. Clinical and molecular characterization of Rubinstein-Taybi syndrome patients carrying distinct novel mutations of the EP300 gene. Clin. Genet. 2015, 87, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Negri, G.; Magini, P.; Milani, D.; Colapietro, P.; Rusconi, D.; Scarano, E.; Bonati, M.T.; Priolo, M.; Crippa, M.; Mazzanti, L.; et al. From Whole Gene Deletion to Point Mutations of EP300-Positive Rubinstein-Taybi Patients: New Insights into the Mutational Spectrum and Peculiar Clinical Hallmarks. Hum. Mutat. 2016, 37, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Schölz, C.; Weinert, B.T.; Wagner, S.A.; Beli, P.; Miyake, Y.; Qi, J.; Jensen, L.J.; Streicher, W.; McCarthy, A.R.; Westwood, N.J.; et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 2015, 33, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.C.; Chen, Y.J.; Lian, Y.C.; Chang, B.E.; Huang, C.C.; Huang, W.L.; Pan, Y.H.; Jeng, J.H. Butyrate stimulates histone H3 acetylation, 8-isoprostane production, RANKL expression, and regulated osteoprotegerin expression/secretion in MG-63 osteoblastic cells. Int. J. Mol. Sci. 2018, 19, 4071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freese, K.; Seitz, T.; Dietrich, P.; Lee, S.M.L.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Histone deacetylase expressions in hepatocellular carcinoma and functional effects of histone deacetylase inhibitors on liver cancer cells in vitro. Cancers 2019, 11, 1587. [Google Scholar] [CrossRef] [Green Version]
- Tarasenko, N.; Chekroun-Setti, H.; Nudelman, A.; Rephaeli, A. Comparison of the anticancer properties of a novel valproic acid prodrug to leading histone deacetylase inhibitors. J. Cell. Biochem. 2018, 119, 3417–3428. [Google Scholar] [CrossRef]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. PubMed NCBI EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [Green Version]
- Chriett, S.; Dąbek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verduci, E.; Moretti, F.; Bassanini, G.; Banderali, G.; Rovelli, V.; Casiraghi, M.C.; Morace, G.; Borgo, F.; Borghi, E. Phenylketonuric diet negatively impacts on butyrate production. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinf. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pẽa, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassanini, G.; Ceccarani, C.; Borgo, F.; Severgnini, M.; Rovelli, V.; Morace, G.; Verduci, E.; Borghi, E. Phenylketonuria Diet Promotes Shifts in Firmicutes Populations. Front. Cell. Infect. Microbiol. 2019, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M. Drosophila: A Laboratory Handbook; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Fede, E.; Ottaviano, E.; Grazioli, P.; Ceccarani, C.; Galeone, A.; Parodi, C.; Colombo, E.A.; Bassanini, G.; Fazio, G.; Severgnini, M.; et al. Insights into the Role of the Microbiota and of Short-Chain Fatty Acids in Rubinstein–Taybi Syndrome. Int. J. Mol. Sci. 2021, 22, 3621. https://doi.org/10.3390/ijms22073621
Di Fede E, Ottaviano E, Grazioli P, Ceccarani C, Galeone A, Parodi C, Colombo EA, Bassanini G, Fazio G, Severgnini M, et al. Insights into the Role of the Microbiota and of Short-Chain Fatty Acids in Rubinstein–Taybi Syndrome. International Journal of Molecular Sciences. 2021; 22(7):3621. https://doi.org/10.3390/ijms22073621
Chicago/Turabian StyleDi Fede, Elisabetta, Emerenziana Ottaviano, Paolo Grazioli, Camilla Ceccarani, Antonio Galeone, Chiara Parodi, Elisa Adele Colombo, Giulia Bassanini, Grazia Fazio, Marco Severgnini, and et al. 2021. "Insights into the Role of the Microbiota and of Short-Chain Fatty Acids in Rubinstein–Taybi Syndrome" International Journal of Molecular Sciences 22, no. 7: 3621. https://doi.org/10.3390/ijms22073621