
Investigation of Receptor Heteromers Using NanoBRET Ligand Binding

 , , and
, , and
Abstract
:1. Introduction
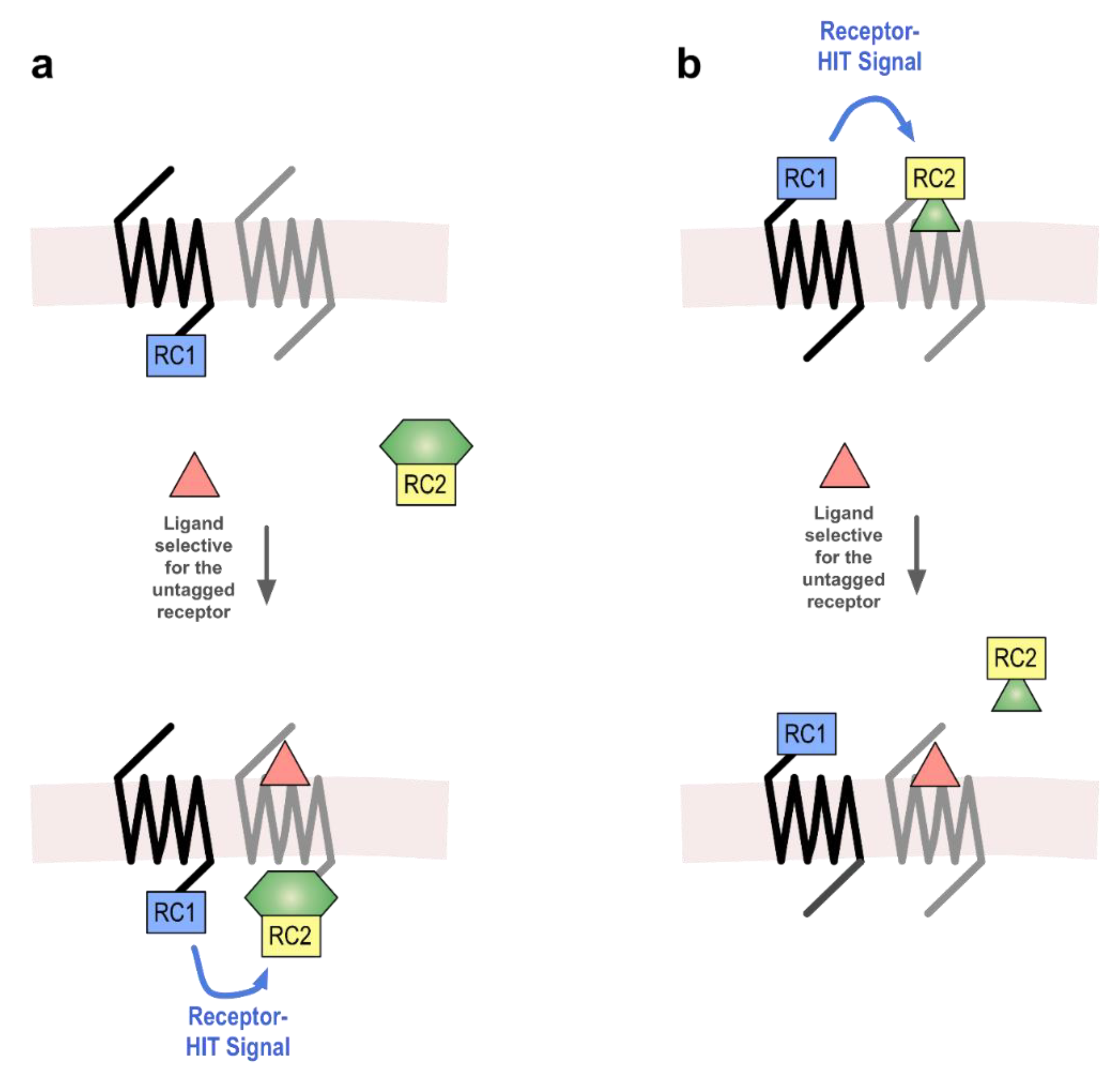
2. Results
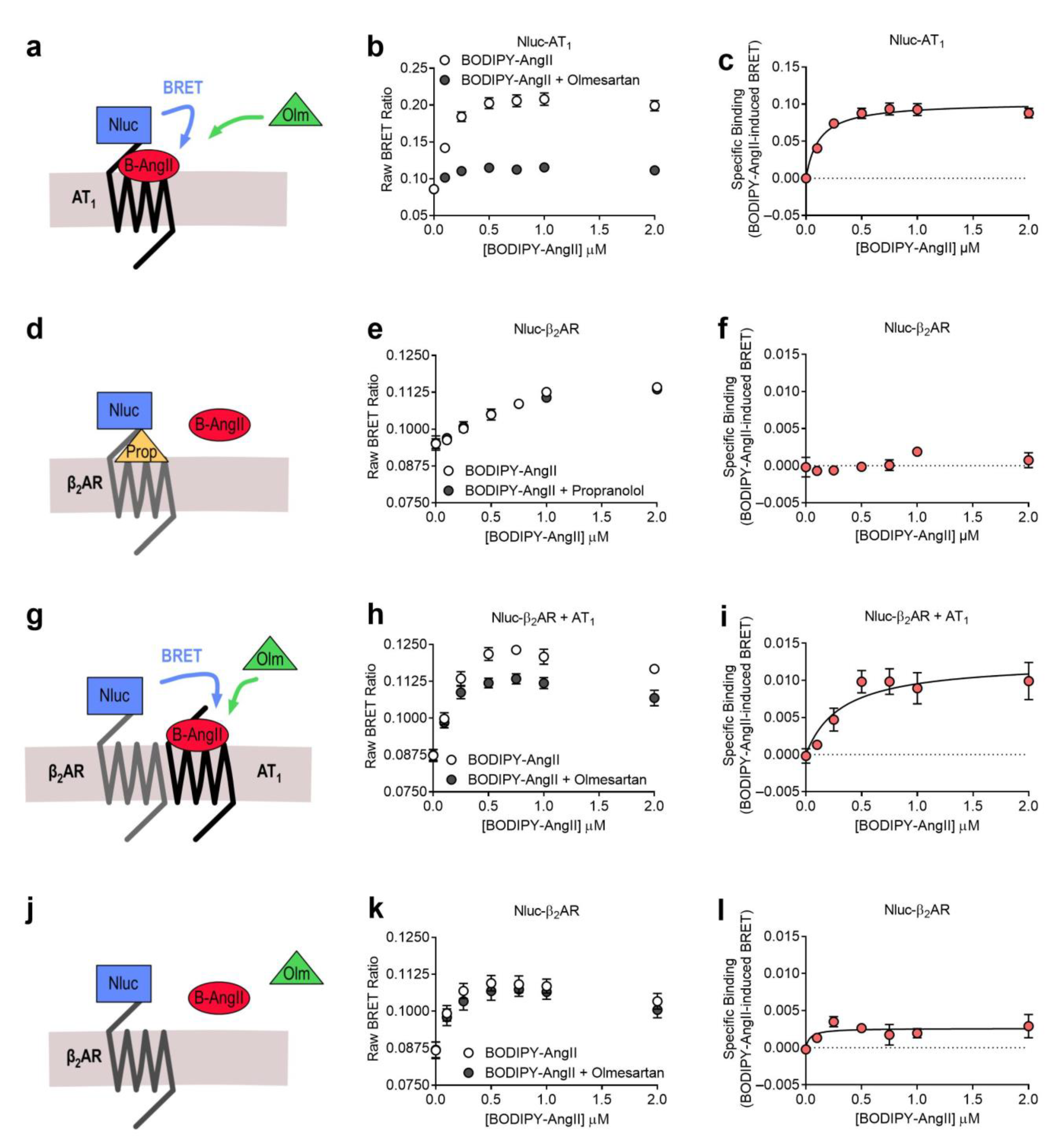
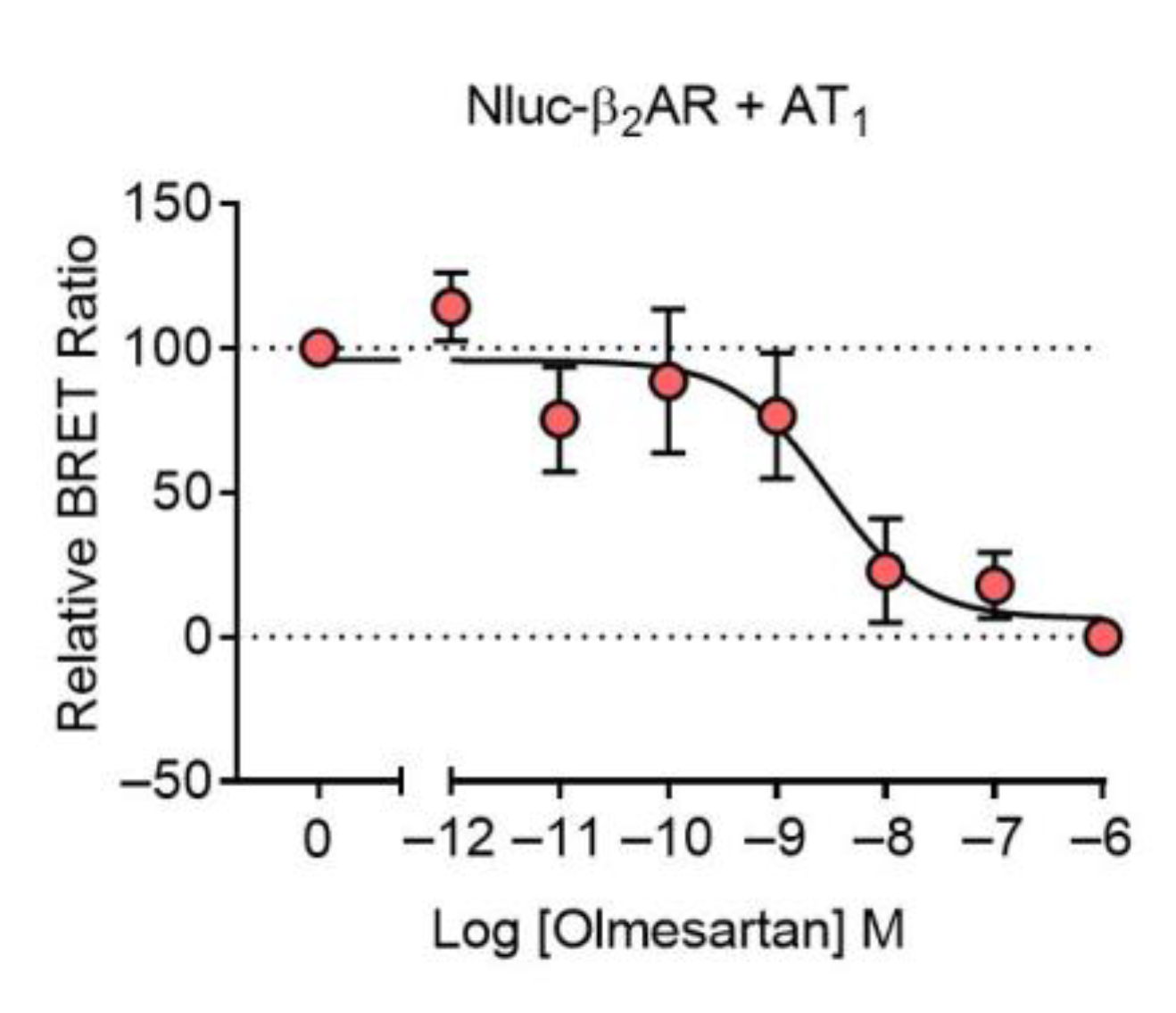
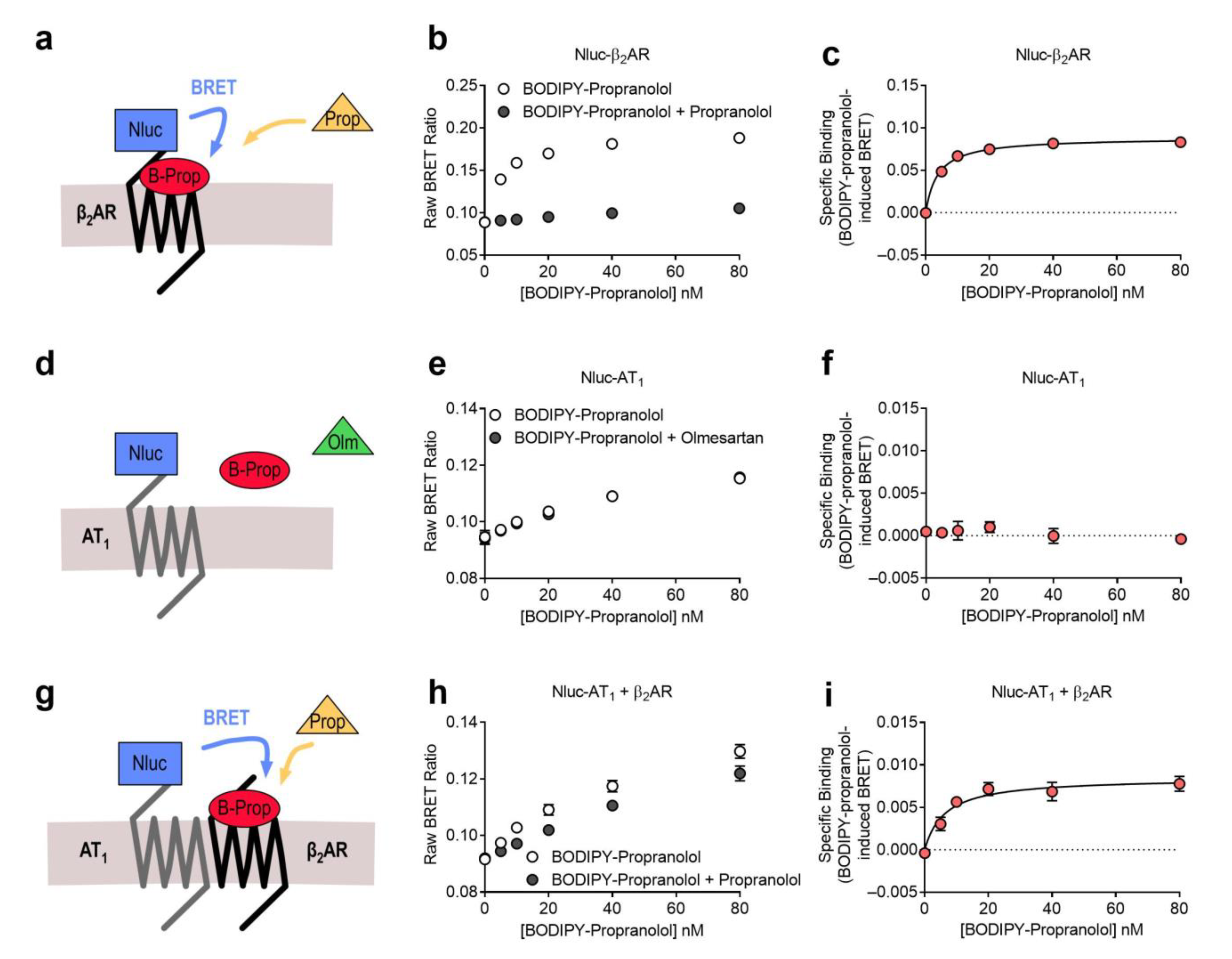
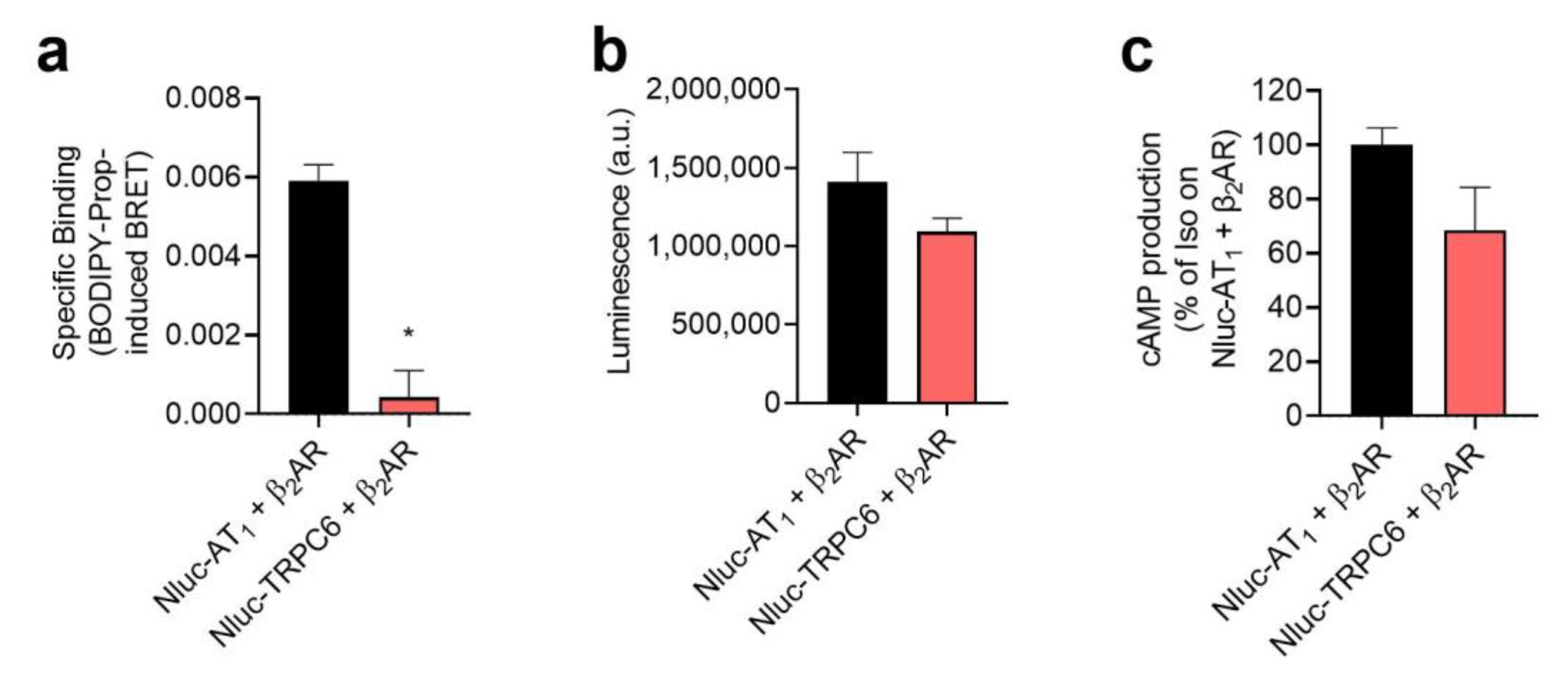
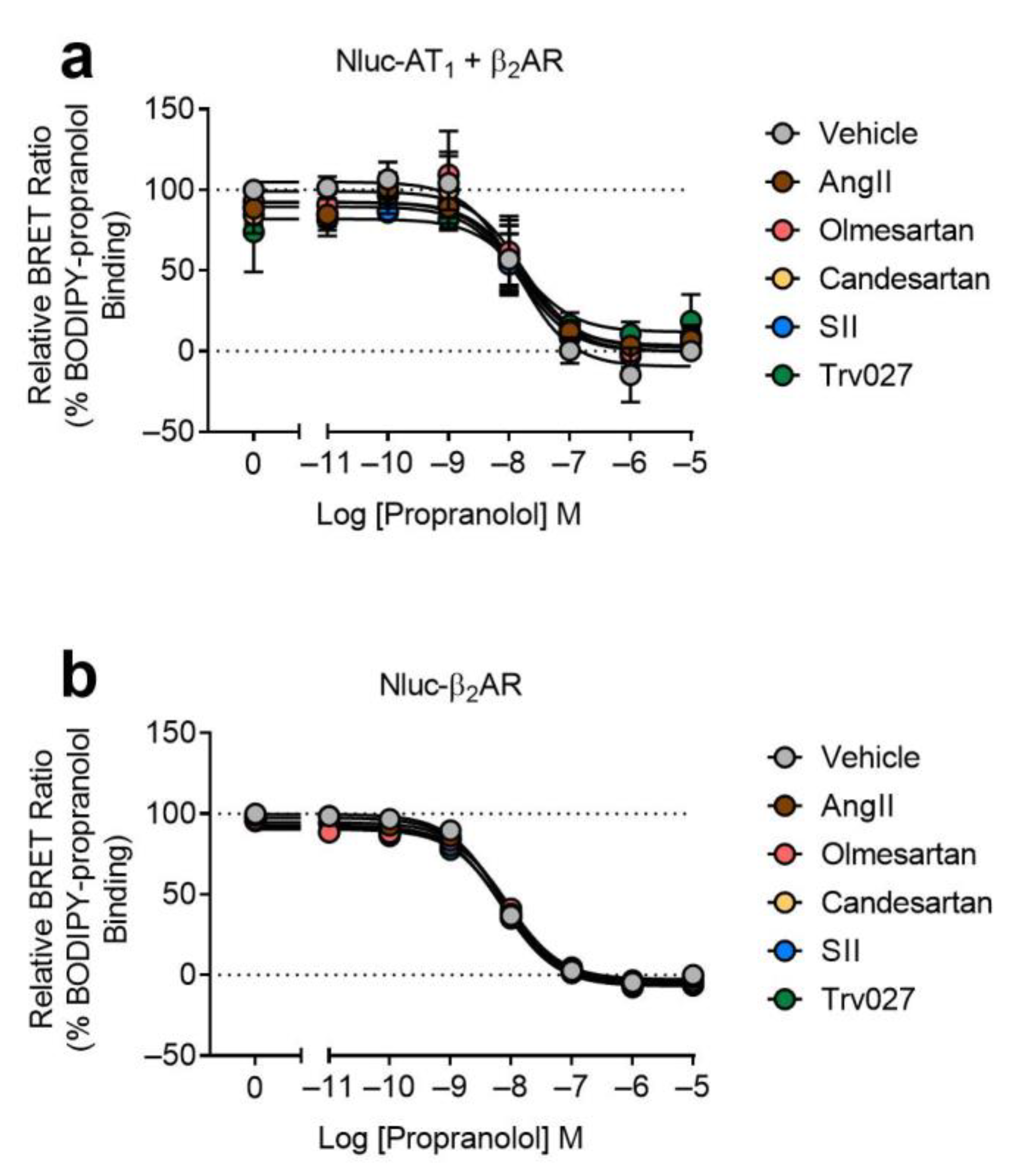
2.1. AT1-β2AR Heteromer
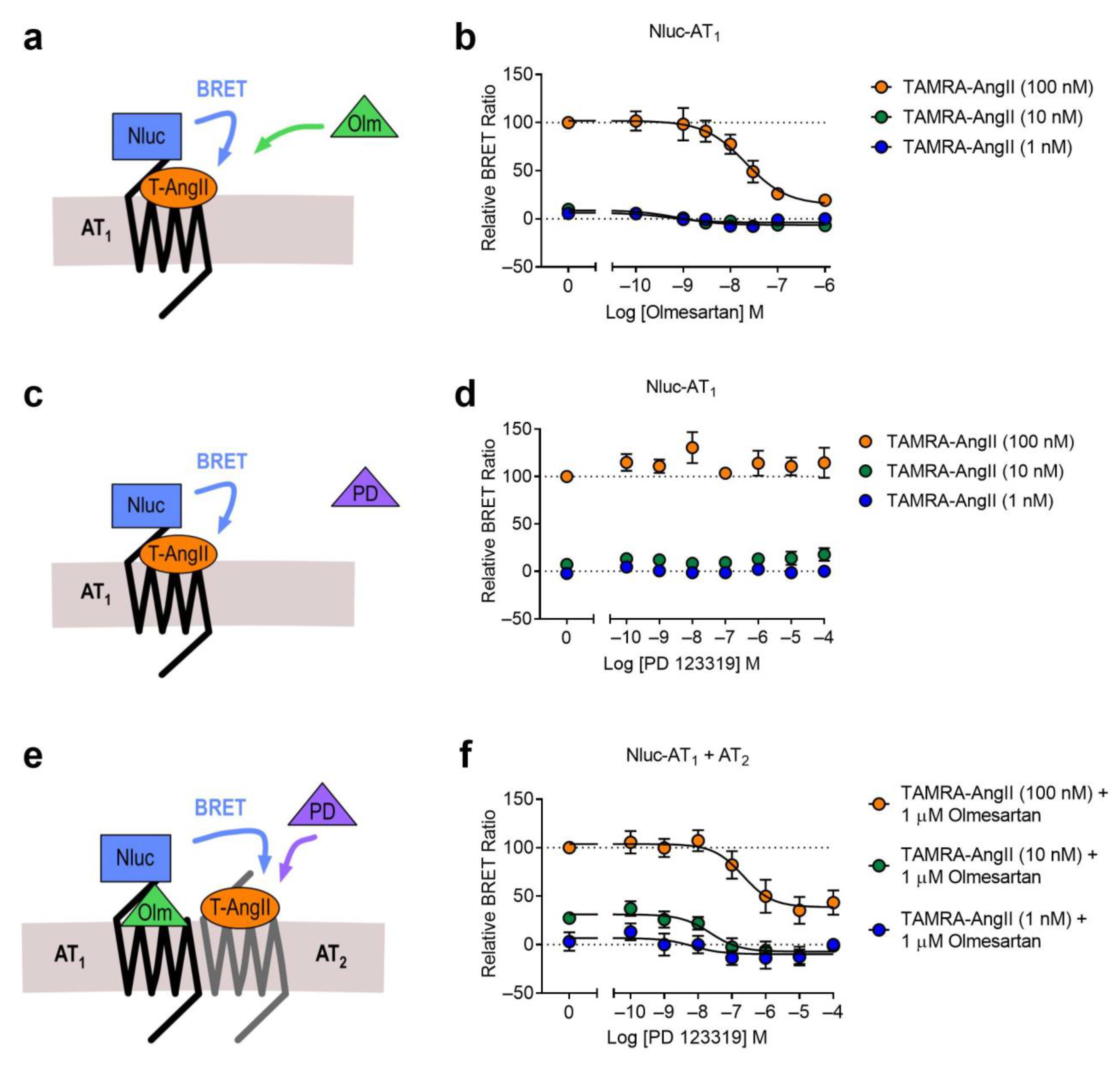
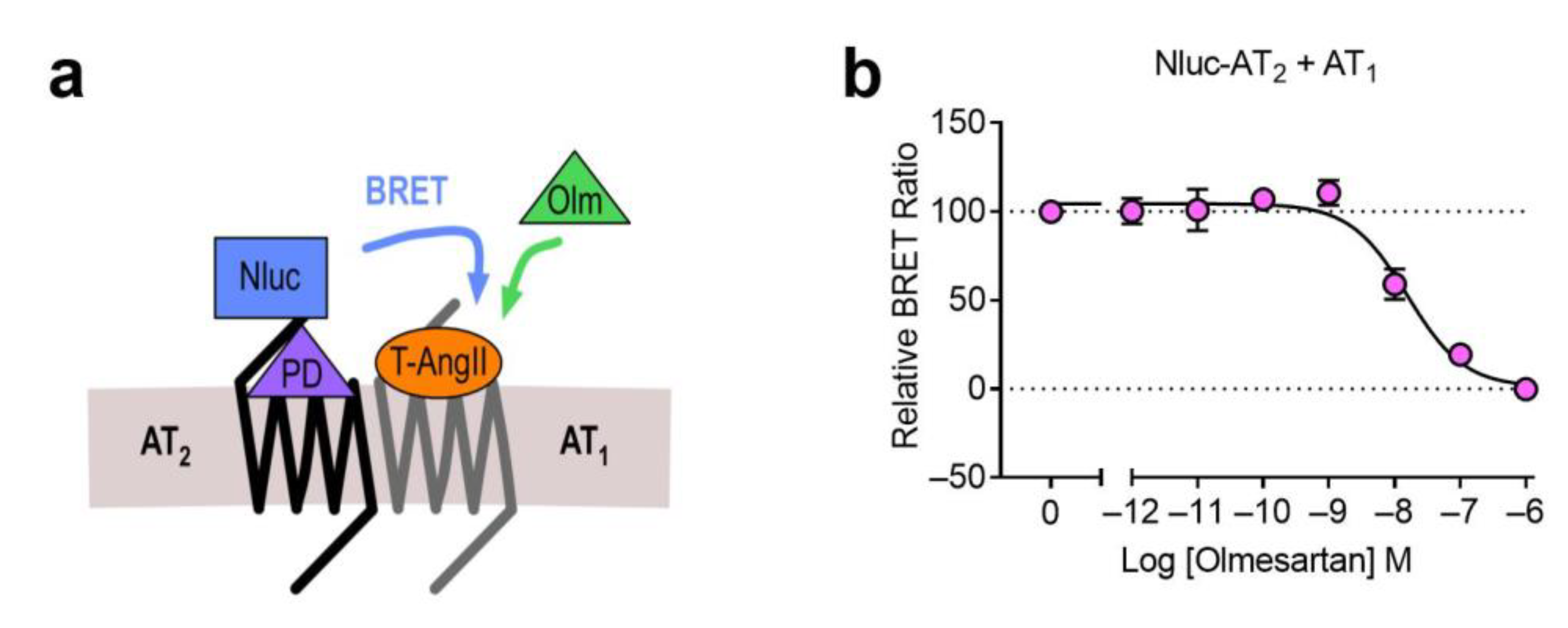
2.2. AT1-AT2 Heteromer
3. Discussion
4. Materials and Methods
4.1. cDNA Constructs
4.2. Ligands, and Generation of BODIPY-AngII
4.3. Cell Culture and Transfection
4.4. NanoBRET Saturation Ligand Binding Assays
4.5. NanoBRET Competition Ligand Binding Assays
4.6. Inositol Monophosphate (IP1) Accumulation Assays
4.7. cAMP Accumulation Assays
4.8. Data Presentation and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferre, S.; Baler, R.; Bouvier, M.; Caron, M.G.; Devi, L.A.; Durroux, T.; Fuxe, K.; George, S.R.; Javitch, J.A.; Lohse, M.J.; et al. Building a new conceptual framework for receptor heteromers. Nat. Chem. Biol. 2009, 5, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- See, H.B.; Seeber, R.M.; Kocan, M.; Eidne, K.A.; Pfleger, K.D.G. Application of G protein-coupled receptor-heteromer identification technology to monitor β-arrestin recruitment to G protein-coupled receptor heteromers. Assay Drug Dev. Technol. 2011, 9, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, E.K.M.; Pfleger, K.D.G. Receptor-Heteromer Investigation Technology and its application using BRET. Front. Endocrinol. 2012, 3, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porrello, E.R.; Pfleger, K.D.; Seeber, R.M.; Qian, H.; Oro, C.; Abogadie, F.; Delbridge, L.M.; Thomas, W.G. Heteromerization of angiotensin receptors changes trafficking and arrestin recruitment profiles. Cell Signal. 2011, 23, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Schelshorn, D.; Joly, F.; Mutel, S.; Hampe, C.; Breton, B.; Mutel, V.; Lutjens, R. Lateral allosterism in the glucagon receptor family: Glucagon-like peptide 1 induces G-protein-coupled receptor heteromer formation. Mol. Pharmacol. 2012, 81, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, S.; See, H.B.; Seeber, R.M.; Armstrong, S.P.; White, C.W.; Ventura, S.; Ayoub, M.A.; Pfleger, K.D. Identification and profiling of novel alpha1A-adrenoceptor-CXC chemokine receptor 2 heteromer. J. Biol. Chem. 2012, 287, 12952–12965. [Google Scholar] [CrossRef] [Green Version]
- Watts, A.O.; van Lipzig, M.M.; Jaeger, W.C.; Seeber, R.M.; van Zwam, M.; Vinet, J.; van der Lee, M.M.; Siderius, M.; Zaman, G.J.; Boddeke, H.W.; et al. Identification and profiling of CXCR3-CXCR4 chemokine receptor heteromer complexes. Br. J. Pharmacol. 2013, 168, 1662–1674. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, M.A.; Zhang, Y.; Kelly, R.S.; See, H.B.; Johnstone, E.K.; McCall, E.A.; Williams, J.H.; Kelly, D.J.; Pfleger, K.D. Functional interaction between angiotensin II receptor type 1 and chemokine (C-C motif) receptor 2 with implications for chronic kidney disease. PLoS ONE 2015, 10, e0119803. [Google Scholar] [CrossRef] [Green Version]
- Toth, A.D.; Gyombolai, P.; Szalai, B.; Varnai, P.; Turu, G.; Hunyady, L. Angiotensin type 1A receptor regulates beta-arrestin binding of the beta2-adrenergic receptor via heterodimerization. Mol. Cell Endocrinol. 2017, 442, 113–124. [Google Scholar] [CrossRef] [Green Version]
- White, C.W.; Vanyai, H.K.; See, H.B.; Johnstone, E.K.M.; Pfleger, K.D.G. Using nanoBRET and CRISPR/Cas9 to monitor proximity to a genome-edited protein in real-time. Sci. Rep. 2017, 7, 3187. [Google Scholar] [CrossRef] [Green Version]
- Fillion, D.; Devost, D.; Sleno, R.; Inoue, A.; Hébert, T.E. Asymmetric Recruitment of β-Arrestin1/2 by the Angiotensin II Type I and Prostaglandin F2α Receptor Dimer. Front. Endocrinol. 2019, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, M.A.; See, H.B.; Seeber, R.M.; Armstrong, S.P.; Pfleger, K.D. Profiling epidermal growth factor receptor and heregulin receptor 3 heteromerization using receptor tyrosine kinase heteromer investigation technology. PLoS ONE 2013, 8, e64672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susec, M.; Sencanski, M.; Glisic, S.; Veljkovic, N.; Pedersen, C.; Drinovec, L.; Stojan, J.; Nøhr, J.; Vrecl, M. Functional characterization of β2-adrenergic and insulin receptor heteromers. Neuropharmacology 2019, 152, 78–89. [Google Scholar] [CrossRef]
- O’Brien, S.L.; Johnstone, E.K.M.; Devost, D.; Conroy, J.; Reichelt, M.E.; Purdue, B.W.; Ayoub, M.A.; Kawai, T.; Inoue, A.; Eguchi, S.; et al. BRET-based assay to monitor EGFR transactivation by the AT1R reveals Gq/11 protein-independent activation and AT1R-EGFR complexes. Biochem. Pharmacol. 2018, 158, 232–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpatrick, L.E.; Alcobia, D.C.; White, C.W.; Peach, C.J.; Glenn, J.R.; Zimmerman, K.; Kondrashov, A.; Pfleger, K.D.G.; Ohana, R.F.; Robers, M.B.; et al. Complex Formation between VEGFR2 and the beta2-Adrenoceptor. Cell Chem. Biol. 2019, 26, 830–841 e839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, R.J.; Tikellis, C.; Rosado, C.J.; Tsorotes, D.; Dimitropoulos, A.; Smith, M.; Huet, O.; Seeber, R.M.; Abhayawardana, R.; Johnstone, E.K.; et al. Transactivation of RAGE mediates angiotensin-induced inflammation and atherogenesis. J. Clin. Investig. 2019, 129, 406–421. [Google Scholar] [CrossRef]
- Jaeger, W.C.; Armstrong, S.P.; Hill, S.J.; Pfleger, K.D.G. Biophysical Detection of Diversity and Bias in GPCR Function. Front. Endocrinol. 2014, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Stoddart, L.A.; Johnstone, E.K.M.; Wheal, A.J.; Goulding, J.; Robers, M.B.; Machleidt, T.; Wood, K.V.; Hill, S.J.; Pfleger, K.D.G. Application of BRET to monitor ligand binding to GPCRs. Nat. Methods 2015, 12, 661–663. [Google Scholar] [CrossRef]
- Ali, A.; Johnstone, E.K.M.; Baby, B.; See, H.B.; Song, A.; Rosengren, K.J.; Pfleger, K.D.G.; Ayoub, M.A.; Vijayan, R. Insights into the Interaction of LVV-Hemorphin-7 with Angiotensin II Type 1 Receptor. Int. J. Mol. Sci. 2020, 22, 209. [Google Scholar] [CrossRef]
- Soave, M.; Stoddart, L.A.; Brown, A.; Woolard, J.; Hill, S.J. Use of a new proximity assay (NanoBRET) to investigate the ligand-binding characteristics of three fluorescent ligands to the human beta1-adrenoceptor expressed in HEK-293 cells. Pharmacol. Res. Perspect. 2016, 4, e00250. [Google Scholar] [CrossRef]
- White, C.W.; Johnstone, E.K.M.; See, H.B.; Pfleger, K.D.G. NanoBRET ligand binding at a GPCR under endogenous promotion facilitated by CRISPR/Cas9 genome editing. Cell Signal. 2019, 54, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Dale, N.C.; Johnstone, E.K.M.; White, C.W.; Pfleger, K.D.G. NanoBRET: The Bright Future of Proximity-Based Assays. Front. Bioeng. Biotechnol. 2019, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; White, C.W.; Nguyen, K.; Hill, S.J.; Pfleger, K.D.G. Fluorescence- and bioluminescence-based approaches to study GPCR ligand binding. Br. J. Pharmacol. 2016, 173, 3028–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barki-Harrington, L.; Luttrell, L.M.; Rockman, H.A. Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: A functional role for receptor-receptor interaction in vivo. Circulation 2003, 108, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammad, M.M.; Dupre, D.J. Chaperones contribute to G protein coupled receptor oligomerization, but do not participate in assembly of the G protein with the receptor signaling complex. J. Mol. Signal. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AbdAlla, S.; Lother, H.; Abdel-tawab, A.M.; Quitterer, U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J. Biol. Chem. 2001, 276, 39721–39726. [Google Scholar] [CrossRef] [Green Version]
- Rivas-Santisteban, R.; Rodriguez-Perez, A.I.; Muñoz, A.; Reyes-Resina, I.; Labandeira-García, J.L.; Navarro, G.; Franco, R. Angiotensin AT1 and AT2 receptor heteromer expression in the hemilesioned rat model of Parkinson’s disease that increases with levodopa-induced dyskinesia. J. Neuroinflamm. 2020, 17, 243. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Knowle, D.; Gavini, N.; Pulakat, L. Identification of the region of AT2 receptor needed for inhibition of the AT1 receptor-mediated inositol 1,4,5-triphosphate generation. FEBS Lett. 2002, 532, 379–386. [Google Scholar] [CrossRef]
- Axelband, F.; Assuncao-Miranda, I.; de Paula, I.R.; Ferrao, F.M.; Dias, J.; Miranda, A.; Miranda, F.; Lara, L.S.; Vieyra, A. Ang-(3-4) suppresses inhibition of renal plasma membrane calcium pump by Ang II. Regul. Pept. 2009, 155, 81–90. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Dupre, D.J.; Feng, Y.; Robitaille, M.; Lazartigues, E.; Feng, Y.H.; Hebert, T.E.; Wu, G. Rab1 GTPase and dimerization in the cell surface expression of angiotensin II type 2 receptor. J. Pharmacol. Exp. Ther. 2009, 330, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Chen, C.; Ren, H.; Han, Y.; He, D.; Zhou, L.; Hopfer, U.; Jose, P.A.; Zeng, C. Angiotensin II AT(2) receptor decreases AT(1) receptor expression and function via nitric oxide/cGMP/Sp1 in renal proximal tubule cells from Wistar-Kyoto rats. J. Hypertens 2012, 30, 1176–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrao, F.M.; Lara, L.S.; Axelband, F.; Dias, J.; Carmona, A.K.; Reis, R.I.; Costa-Neto, C.M.; Vieyra, A.; Lowe, J. Exposure of luminal membranes of LLC-PK1 cells to ANG II induces dimerization of AT1/AT2 receptors to activate SERCA and to promote Ca2+ mobilization. Am. J. Physiol. Renal. Physiol. 2012, 302, F875–F883. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.; Ferrao, F.M.; Axelband, F.; Carmona, A.K.; Lara, L.S.; Vieyra, A. ANG-(3-4) inhibits renal Na+-ATPase in hypertensive rats through a mechanism that involves dissociation of ANG II receptors, heterodimers, and PKA. Am. J. Physiol. Renal. Physiol. 2014, 306, F855–F863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inuzuka, T.; Fujioka, Y.; Tsuda, M.; Fujioka, M.; Satoh, A.O.; Horiuchi, K.; Nishide, S.; Nanbo, A.; Tanaka, S.; Ohba, Y. Attenuation of ligand-induced activation of angiotensin II type 1 receptor signaling by the type 2 receptor via protein kinase C. Sci. Rep. 2016, 6, 21613. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, F.M.; Cardoso, L.H.D.; Drummond, H.A.; Li, X.C.; Zhuo, J.L.; Gomes, D.S.; Lara, L.S.; Vieyra, A.; Lowe, J. Luminal ANG II is internalized as a complex with AT1R/AT2R heterodimers to target endoplasmic reticulum in LLC-PK1 cells. Am. J. Physiol. Renal. Physiol. 2017, 313, F440–F449. [Google Scholar] [CrossRef] [Green Version]
- Wangler, N.J.; Santos, K.L.; Schadock, I.; Hagen, F.K.; Escher, E.; Bader, M.; Speth, R.C.; Karamyan, V.T. Identification of membrane-bound variant of metalloendopeptidase neurolysin (EC 3.4.24.16) as the non-angiotensin type 1 (non-AT1), non-AT2 angiotensin binding site. J. Biol. Chem. 2012, 287, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Birdsall, N.J. Class A GPCR heterodimers: Evidence from binding studies. Trends Pharmacol. Sci. 2010, 31, 499–508. [Google Scholar] [CrossRef]
- Rashid, A.J.; So, C.H.; Kong, M.M.; Furtak, T.; El-Ghundi, M.; Cheng, R.; O’Dowd, B.F.; George, S.R. D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci. USA 2007, 104, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentini, C.; Busi, C.; Gorruso, E.; Gotti, C.; Spano, P.; Missale, C. Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol. Pharmacol. 2008, 74, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Marcellino, D.; Ferre, S.; Casado, V.; Cortes, A.; Le Foll, B.; Mazzola, C.; Drago, F.; Saur, O.; Stark, H.; Soriano, A.; et al. Identification of dopamine D1-D3 receptor heteromers. Indications for a role of synergistic D1-D3 receptor interactions in the striatum. J. Biol. Chem. 2008, 283, 26016–26025. [Google Scholar] [CrossRef] [Green Version]
- Ferre, S.; von Euler, G.; Johansson, B.; Fredholm, B.B.; Fuxe, K. Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc. Natl. Acad. Sci. USA 1991, 88, 7238–7241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.; Strange, P.G. Dopamine D2 receptor dimer formation: Evidence from ligand binding. J. Biol. Chem. 2001, 276, 22621–22629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Asmar, L.; Springael, J.Y.; Ballet, S.; Andrieu, E.U.; Vassart, G.; Parmentier, M. Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol. Pharmacol. 2005, 67, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.P.; Nikolaev, V.O.; Lorenz, K.; Ferrandon, S.; Zhuang, Z.; Lohse, M.J. Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat. Chem. Biol. 2008, 4, 126–131. [Google Scholar] [CrossRef]
- Hounsou, C.; Margathe, J.F.; Oueslati, N.; Belhocine, A.; Dupuis, E.; Thomas, C.; Mann, A.; Ilien, B.; Rognan, D.; Trinquet, E.; et al. Time-resolved FRET binding assay to investigate hetero-oligomer binding properties: Proof of concept with dopamine D1/D3 heterodimer. ACS Chem. Biol. 2015, 10, 466–474. [Google Scholar] [CrossRef]
- Oueslati, N.; Hounsou, C.; Belhocine, A.; Rodriguez, T.; Dupuis, E.; Zwier, J.M.; Trinquet, E.; Pin, J.-P.; Durroux, T. Time-Resolved FRET Strategy to Screen GPCR Ligand Library. In G Protein-Coupled Receptor Screening Assays: Methods and Protocols; Prazeres, D.M.F., Martins, S.A.M., Eds.; Springer New York: New York, NY, USA, 2015; pp. 23–36. [Google Scholar]
- Alcobia, D.C.; Ziegler, A.I.; Kondrashov, A.; Comeo, E.; Mistry, S.; Kellam, B.; Chang, A.; Woolard, J.; Hill, S.J.; Sloan, E.K. Visualizing Ligand Binding to a GPCR In Vivo Using NanoBRET. In iScience; Elsevier: Amsterdam, The Netherlands, 2018; Volume 6, pp. 280–288. [Google Scholar]
- Trifilieff, P.; Rives, M.-L.; Urizar, E.; Piskorowski, R.A.; Vishwasrao, H.D.; Castrillon, J.; Schmauss, C.; Slttman, M.; Gullberg, M.; Javitch, J.A. Detection of antigen interactions ex vivo by proximity ligation assay: Endogenous dopamine D2-adenosine A2A receptor complexes in the striatum. BioTechniques 2011, 51, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Borroto-Escuela, D.O.; Craenenbroeck, K.V.; Romero-Fernandez, W.; Guidolin, D.; Woods, A.S.; Rivera, A.; Haegeman, G.; Agnati, L.F.; Tarakanov, A.O.; Fuxe, K. Dopamine D2 and D4 receptor heteromerization and its allosteric receptor–receptor interactions. Biochem. Biophys. Res. Commun. 2011, 404, 928–934. [Google Scholar] [CrossRef]
- Parhamifar, L.; Sime, W.; Yudina, Y.; Vilhardt, F.; Mörgelin, M.; Sjölander, A. Ligand-Induced Tyrosine Phosphorylation of Cysteinyl Leukotriene Receptor 1 Triggers Internalization and Signaling in Intestinal Epithelial Cells. PLoS ONE 2010, 5, e14439. [Google Scholar] [CrossRef] [Green Version]
- Dacres, H.; Michie, M.; Wang, J.; Pfleger, K.D.; Trowell, S.C. Effect of enhanced Renilla luciferase and fluorescent protein variants on the Forster distance of Bioluminescence resonance energy transfer (BRET). Biochem. Biophys. Res. Commun. 2012, 425, 625–629. [Google Scholar] [CrossRef] [Green Version]
- Weihs, F.; Wang, J.; Pfleger, K.D.G.; Dacres, H. Experimental determination of the bioluminescence resonance energy transfer (BRET) Förster distances of NanoBRET and red-shifted BRET pairs. Anal. Chim. Acta X 2020, 6, 100059. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR monomers and oligomers: It takes all kinds. Trends Neurosci. 2008, 31, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayoub, M.A.; Pfleger, K.D. Recent advances in bioluminescence resonance energy transfer technologies to study GPCR heteromerization. Curr. Opin. Pharmacol. 2010, 10, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, D.; Carr, I.C.; Pediani, J.; Lopez-Gimenez, J.F.; Thurlow, R.; Fidock, M.; Milligan, G. High-affinity interactions between human alpha1A-adrenoceptor C-terminal splice variants produce homo- and heterodimers but do not generate the alpha1L-adrenoceptor. Mol. Pharmacol. 2004, 66, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goin, J.C.; Nathanson, N.M. Quantitative analysis of muscarinic acetylcholine receptor homo- and heterodimerization in live cells: Regulation of receptor down-regulation by heterodimerization. J. Biol. Chem. 2006, 281, 5416–5425. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, M.A.; Levoye, A.; Delagrange, P.; Jockers, R. Preferential formation of MT1/MT2 melatonin receptor heterodimers with distinct ligand interaction properties compared with MT2 homodimers. Mol. Pharmacol. 2004, 66, 312–321. [Google Scholar] [CrossRef]
- Small, K.M.; Schwarb, M.R.; Glinka, C.; Theiss, C.T.; Brown, K.M.; Seman, C.A.; Liggett, S.B. Alpha2A- and alpha2C-adrenergic receptors form homo- and heterodimers: The heterodimeric state impairs agonist-promoted GRK phosphorylation and beta-arrestin recruitment. Biochemistry 2006, 45, 4760–4767. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle | AngII | Olmesartan | Candesartan | SII | Trv027 | |
|---|---|---|---|---|---|---|
| Nluc-AT1 + β2AR | 7.96 ± 0.18 | 7.94 ± 0.31 | 7.91 ± 0.26 | 7.92 ± 0.32 | 7.98 ± 0.31 | 7.88 ± 0.35 |
| Nluc-β2AR | 8.17 ± 0.02 | 8.14 ± 0.02 | 8.06 ± 0.02 | 8.10 ± 0.04 | 8.16 ± 0.11 | 8.12 ± 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnstone, E.K.M.; See, H.B.; Abhayawardana, R.S.; Song, A.; Rosengren, K.J.; Hill, S.J.; Pfleger, K.D.G. Investigation of Receptor Heteromers Using NanoBRET Ligand Binding. Int. J. Mol. Sci. 2021, 22, 1082. https://doi.org/10.3390/ijms22031082
Johnstone EKM, See HB, Abhayawardana RS, Song A, Rosengren KJ, Hill SJ, Pfleger KDG. Investigation of Receptor Heteromers Using NanoBRET Ligand Binding. International Journal of Molecular Sciences. 2021; 22(3):1082. https://doi.org/10.3390/ijms22031082
Chicago/Turabian StyleJohnstone, Elizabeth K. M., Heng B. See, Rekhati S. Abhayawardana, Angela Song, K. Johan Rosengren, Stephen J. Hill, and Kevin D. G. Pfleger. 2021. "Investigation of Receptor Heteromers Using NanoBRET Ligand Binding" International Journal of Molecular Sciences 22, no. 3: 1082. https://doi.org/10.3390/ijms22031082