
VEGFB Promotes Myoblasts Proliferation and Differentiation through VEGFR1-PI3K/Akt Signaling Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
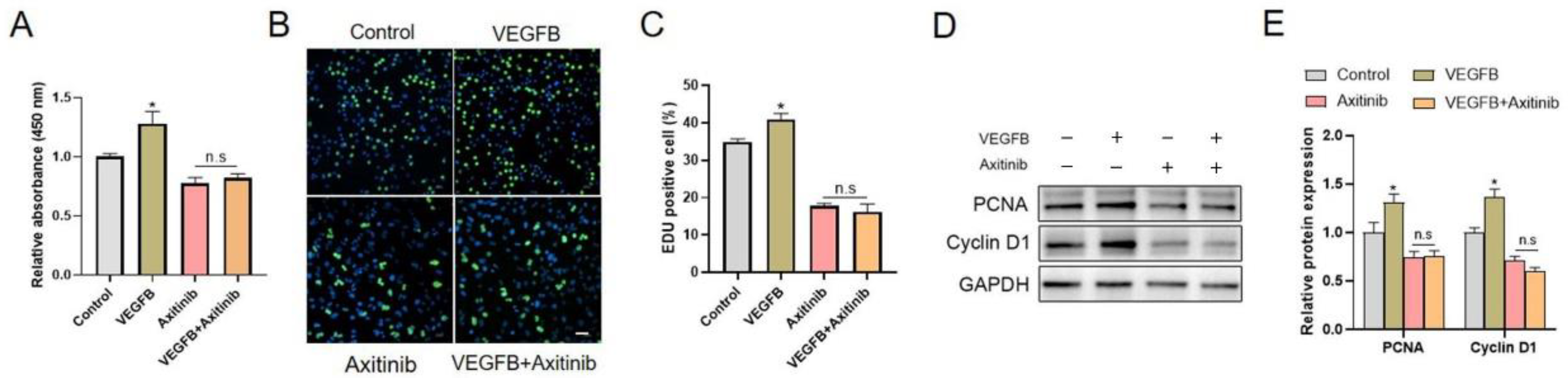
2.1. VEGFB Promoted the Proliferation of C2C12 Cells via VEGFRs Signaling
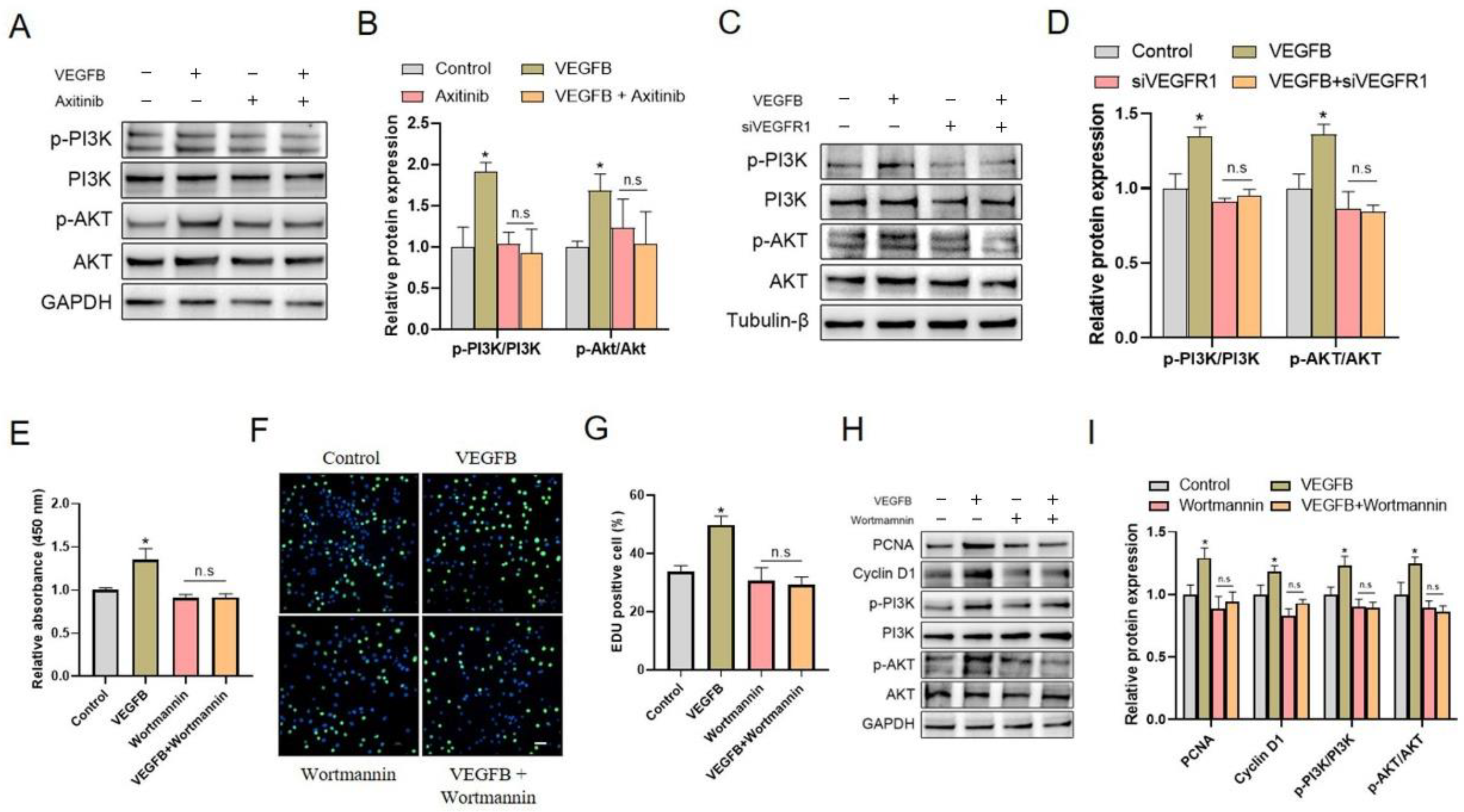
2.2. VEGFR1, but Not NRP1, Was Involved in VEGFB-Promoted Proliferation of C2C12 Cells
2.3. PI3K/Akt Signaling Pathway Was Involved in C2C12 Proliferation Induced by VEGFB
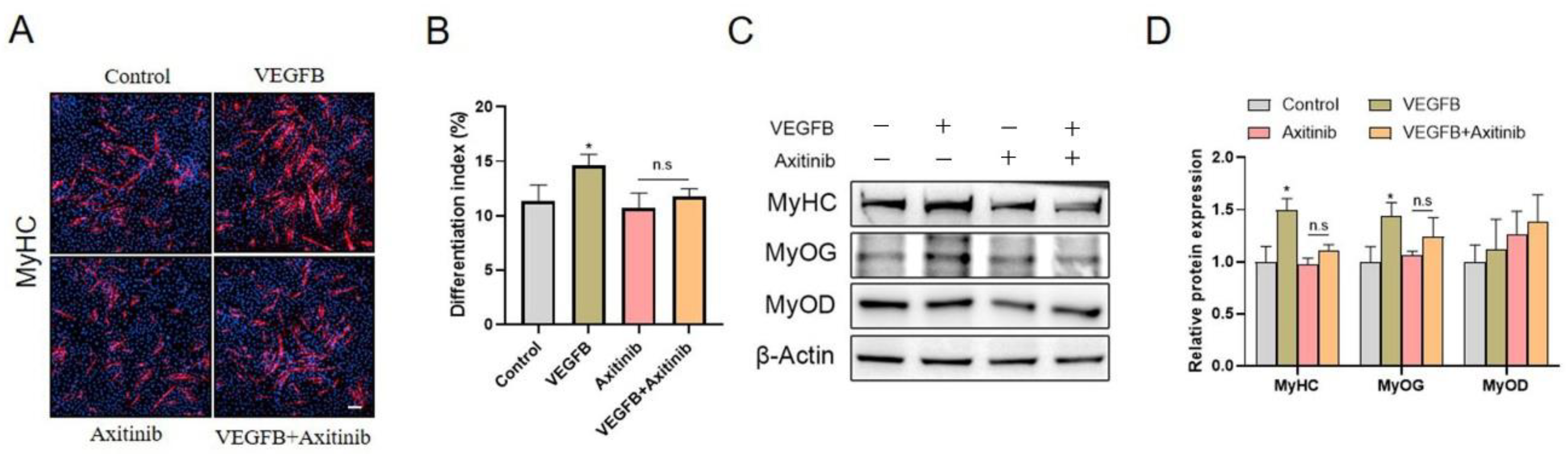
2.4. VEGFB Stimulated the Differentiation of C2C12 Cells through VEGFRs Signaling
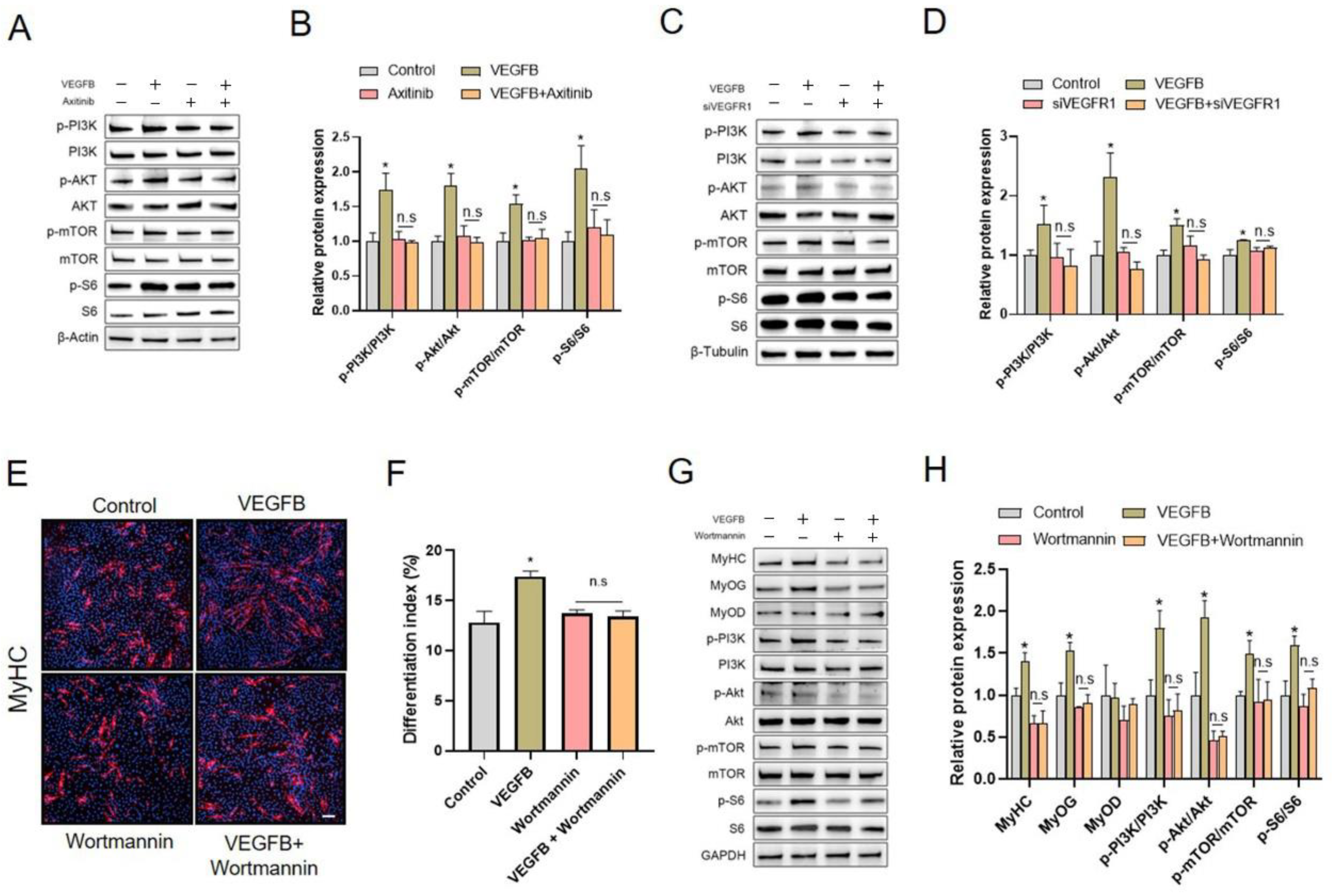
2.5. Knockdown of VEGFR1, Rather Than NRP1, Eliminated the Stimulation of C2C12 Differentiation Induced by VEGFB
2.6. PI3K/AKT/mTOR Signaling Pathway Was Involved in VEGFB-Stimulated C2C12 Differentiation
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Culture and Treatments
4.3. Cell Proliferation Assay
4.4. Transfection of C2C12 Cells with siRNA
4.5. Immunofluorescence Staining
4.6. Western Blotting Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chargé, S.B.P.; Rudnicki, M. Cellular and Molecular Regulation of Muscle Regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Dyar, K.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Schmidt, S.F.; Rohm, M.; Herzig, S.; Diaz, M.B. Cancer Cachexia: More Than Skeletal Muscle Wasting. Trends Cancer 2018, 4, 849–860. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building Muscle: Molecular Regulation of Myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Ewen, M.E. Where the cell cycle and histones meet: Figure 1. Genes Dev. 2000, 14, 2265–2270. [Google Scholar] [CrossRef] [Green Version]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2010, 107, 1127–1140. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Yang, Z.; Dong, D.; Xu, J.; Wang, J.; Li, H.; Huang, Y.; Lan, X.; Lei, C.; Ma, Y.; et al. miR-483 inhibits bovine myoblast cell proliferation and differentiation via IGF1/PI3K/AKT signal pathway. J. Cell Physiol. 2019, 234, 9839–9848. [Google Scholar] [CrossRef]
- Machida, S.; Booth, F.W. Insulin-like growth factor 1 and muscle growth: Implication for satellite cell proliferation. Proc. Nutr. Soc. 2004, 63, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite Cells and the Muscle Stem Cell Niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, M. Myogenic progenitor cells and skeletal myogenesis in vertebrates. Curr. Opin. Genet. Dev. 2006, 16, 525–532. [Google Scholar] [CrossRef]
- Devlin, R.B.; Emerson, C.P. Coordinate regulation of contractile protein synthesis during myoblast differentiation. Cell 1978, 13, 599–611. [Google Scholar] [CrossRef]
- White, R.B.; Biérinx, A.-S.; Gnocchi, V.F.; Zammit, P.S. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev. Biol. 2010, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Sato, K.; Iemitsu, M. The Role of Dehydroepiandrosterone (DHEA) in Skeletal Muscle. Vitam. Horm. 2018, 108, 205–221. [Google Scholar] [CrossRef]
- Tachtsis, B.; Camera, D.; Lacham-Kaplan, O. Potential Roles of n-3 PUFAs during Skeletal Muscle Growth and Regeneration. Nutrients 2018, 10, 309. [Google Scholar] [CrossRef] [Green Version]
- Huey, K.A. Potential Roles of Vascular Endothelial Growth Factor during Skeletal Muscle Hypertrophy. Exerc. Sport Sci. Rev. 2018, 46, 195–202. [Google Scholar] [CrossRef]
- Huusko, J.; Lottonen, L.; Merentie, M.; Gurzeler, E.; Anisimov, A.; Miyanohara, A.; Alitalo, K.; Tavi, P.; Ylä-Herttuala, S. AAV9-mediated VEGF-B Gene Transfer Improves Systolic Function in Progressive Left Ventricular Hypertrophy. Mol. Ther. 2012, 20, 2212–2221. [Google Scholar] [CrossRef] [Green Version]
- Zentilin, L.; Puligadda, U.; Lionetti, V.; Zacchigna, S.; Collesi, C.; Pattarini, L.; Ruozi, G.; Camporesi, S.; Sinagra, G.; Pepe, M.; et al. Cardiomyocyte VEGFR-1 activation by VEGF-B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB J. 2010, 24, 1467–1478. [Google Scholar] [CrossRef]
- Olofsson, B.; Pajusola, K.; Kaipainen, A.; von Euler, G.; Joukov, V.; Saksela, O.; Orpana, A.; Pettersson, R.F.; Alitalo, K.; Eriksson, U. Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc. Natl. Acad. Sci. USA 1996, 93, 2576–2581. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, I.; Shibuya, M.; Wennström, S. Differential activation of vascular genes by hypoxia in primary endothelial cells. Exp. Cell Res. 2004, 299, 476–485. [Google Scholar] [CrossRef]
- Li, X.; Tjwa, M.; Van Hove, I.; Enholm, B.; Neven, E.; Paavonen, K.; Jeltsch, M.; Juan, T.D.; Sievers, R.E.; Chorianopoulos, E.; et al. Reevaluation of the Role of VEGF-B Suggests a Restricted Role in the Revascularization of the Ischemic Myocardium. Arter. Thromb. Vasc. Biol. 2008, 28, 1614–1620. [Google Scholar] [CrossRef] [Green Version]
- Bry, M.; Kivelä, R.; Holopainen, T.; Anisimov, A.; Tammela, T.; Soronen, J.; Silvola, J.; Saraste, A.; Jeltsch, M.; Korpisalo, P.; et al. Vascular Endothelial Growth Factor-B Acts as a Coronary Growth Factor in Transgenic Rats without Inducing Angiogenesis, Vascular Leak, or Inflammation. Circulation 2010, 122, 1725–1733. [Google Scholar] [CrossRef]
- Koch, S.; Claesson-Welsh, L. Signal Transduction by Vascular Endothelial Growth Factor Receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef]
- Lal, N.; Puri, K.; Rodrigues, B. Vascular Endothelial Growth Factor B and Its Signaling. Front. Cardiovasc. Med. 2018, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Ambrosi, J.; Catalán, V.; Diez-Caballero, A.; Martínez-Cruz, L.A.; Gil, M.J.; García-Foncillas, J.; Cienfuegos, J.A.; Salvador, J.; Mato, J.M.; Frühbeck, G. Gene expression profile of omental adipose tissue in human obesity. FASEB J. 2003, 18, 215–217. [Google Scholar] [CrossRef]
- Sun, C.Y.; Lee, C.C.; Hsieh, M.F.; Chen, C.H.; Chou, K.M. Clinical association of circulating VEGF-B levels with hyperlipidemia and target organ damage in type 2 diabetic patients. J. Boil. Regul. Homeost. Agents 2014, 28, 225–236. [Google Scholar]
- Hagberg, C.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nat. Cell Biol. 2010, 464, 917–921. [Google Scholar] [CrossRef] [Green Version]
- Will, K.; Kalbe, C.; Kuzinski, J.; Lösel, D.; Viergutz, T.; Palin, M.-F.; Rehfeldt, C. Effects of leptin and adiponectin on proliferation and protein metabolism of porcine myoblasts. Histochem. Cell Biol. 2012, 138, 271–287. [Google Scholar] [CrossRef]
- Yanagiuchi, A.; Miyake, H.; Fujisawa, M. 206 Modulation of microenvironment by growth factors regulates in vivo growth of skeletal myoblasts. J. Urol. 2012, 187, e86. [Google Scholar] [CrossRef]
- Hsueh, T.-Y.; Baum, J.I.; Huang, Y. Effect of Eicosapentaenoic Acid and Docosahexaenoic Acid on Myogenesis and Mitochondrial Biosynthesis during Murine Skeletal Muscle Cell Differentiation. Front. Nutr. 2018, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Räsänen, M.; Sultan, I.; Paech, J.; Hemanthakumar, K.A.; Yu, W.; He, L.; Tang, J.; Sun, Y.; Hlushchuk, R.; Huan, X.; et al. VEGF-B Promotes Endocardium-Derived Coronary Vessel Development and Cardiac Regeneration. Circulation 2021, 143, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, Y.; Feng, Z.; Sheng, J.; Dong, A.; Zhang, M.; Cao, L. miR-345-5p regulates adipogenesis via targeting VEGF-B. Aging 2020, 12, 17114–17121. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, F.; Nagai, N.; Tang, Z.; Zhang, S.; Scotney, P.; Lennartsson, J.; Zhu, C.; Qu, Y.; Fang, C.; et al. VEGF-B inhibits apoptosis via VEGFR-1–mediated suppression of the expression of BH3-only protein genes in mice and rats. J. Clin. Investig. 2008, 118, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Jensen, L.D.; Nakamura, M.; Bräutigam, L.; Li, X.; Liu, Y.; Samani, N.J.; Cao, Y. VEGF-B-Neuropilin-1 signaling is spatiotemporally indispensable for vascular and neuronal development in zebrafish. Proc. Natl. Acad. Sci. USA 2015, 112, E5944–E5953. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Ai, W.; Zhang, F.; Zhu, X.; Shu, G.; Wang, L.; Gao, P.; Xi, Q.; Zhang, Y.; Jiang, Q.; et al. Enhanced Proliferation of Porcine Bone Marrow Mesenchymal Stem Cells Induced by Extracellular Calcium is Associated with the Activation of the Calcium-Sensing Receptor and ERK Signaling Pathway. Stem Cells Int. 2016, 2016, 6570671. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bai, M.; Ning, C.; Xie, B.; Zhang, J.; Liao, H.; Xiong, J.; Tao, X.; Yan, D.; Xi, X.; et al. Gankyrin facilitates follicle-stimulating hormone-driven ovarian cancer cell proliferation through the PI3K/AKT/HIF-1alpha/cyclin D1 pathway. Oncogene 2016, 35, 2506–2517. [Google Scholar] [CrossRef]
- Zhang, E.; Gao, B.; Yang, L.; Wu, X.; Wang, Z. Notoginsenoside Ft1 Promotes Fibroblast Proliferation via PI3K/Akt/mTOR Signaling Pathway and Benefits Wound Healing in Genetically Diabetic Mice. J. Pharmacol. Exp. Ther. 2015, 356, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Chen, X.; Zhong, G.; Chen, Q.; Hu, C. Mitofusin 2 Downregulation Triggers Pulmonary Artery Smooth Muscle Cell Proliferation and Apoptosis Imbalance in Rats with Hypoxic Pulmonary Hypertension via the PI3K/Akt and Mitochondrial Apoptosis Pathways. J. Cardiovasc. Pharmacol. 2016, 67, 164–174. [Google Scholar] [CrossRef]
- Glass, D.J. PI3 Kinase Regulation of Skeletal Muscle Hypertrophy and Atrophy. Natural Killer Cells 2010, 346, 267–278. [Google Scholar] [CrossRef]
- Matheny, R.W.; Adamo, M.L. Effects of PI3K catalytic subunit and Akt isoform deficiency on mTOR and p70S6K activation in myoblasts. Biochem. Biophys. Res. Commun. 2009, 390, 252–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tureckova, J.; Wilson, E.M.; Cappalonga, J.L.; Rotwein, P. Insulin-like growth factor-mediated muscle differentiation: Collaboration between phosphatidylinositol 3-kinase-Akt-signaling pathways and myogenin. J. Biol. Chem. 2001, 276, 39264–39270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi, T.; Matsui, T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011, 17, 1818–1824. [Google Scholar] [CrossRef] [Green Version]
- Kaliman, P.; Viñals, F.; Testar, X.; Palacín, M.; Zorzano, A. Phosphatidylinositol 3-Kinase Inhibitors Block Differentiation of Skeletal Muscle Cells. J. Biol. Chem. 1996, 271, 19146–19151. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-C.; Hung, S.-K.; Lin, H.-Y.; Chiou, W.-Y.; Lee, M.-S.; Liao, H.-F.; Huang, H.-B.; Ho, H.-C.; Hsu-Chueh, H. Targeting the PI3K/AKT/mTOR signaling pathway as an effectively radiosensitizing strategy for treating human oral squamous cell carcinoma in vitro and in vivo. Oncotarget 2017, 8, 68641–68653. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.-S.; Li, N.; Hu, X.-X.; Shi, G.-Q.; Liu, S.-R.; Liu, N. Expression of Myogenin and MCK genes regulated by PI3K/AKT pathway. Hereditas 2013, 35, 637–642. [Google Scholar] [CrossRef]
- Xu, Q.; Wu, Z. The Insulin-like Growth Factor-Phosphatidylinositol 3-Kinase-Akt Signaling Pathway Regulates Myogenin Expression in Normal Myogenic Cells but Not in Rhabdomyosarcoma-derived RD Cells. J. Biol. Chem. 2000, 275, 36750–36757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, C.; Palacios, D.; Mozzetta, C.; Forcales, S.V.; Morantte, I.; Ripani, M.; Jones, D.R.; Du, K.; Jhala, U.S.; Simone, C.; et al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol. Cell 2007, 28, 200–213. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, M.; Quan, L.; Lai, X.; Lang, L.; Li, F.; Yang, X.; Fu, Y.; Feng, S.; Yi, X.; Zhu, C.; et al. VEGFB Promotes Myoblasts Proliferation and Differentiation through VEGFR1-PI3K/Akt Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 13352. https://doi.org/10.3390/ijms222413352
Ling M, Quan L, Lai X, Lang L, Li F, Yang X, Fu Y, Feng S, Yi X, Zhu C, et al. VEGFB Promotes Myoblasts Proliferation and Differentiation through VEGFR1-PI3K/Akt Signaling Pathway. International Journal of Molecular Sciences. 2021; 22(24):13352. https://doi.org/10.3390/ijms222413352
Chicago/Turabian StyleLing, Mingfa, Lulu Quan, Xumin Lai, Limin Lang, Fan Li, Xiaohua Yang, Yiming Fu, Shengchun Feng, Xin Yi, Canjun Zhu, and et al. 2021. "VEGFB Promotes Myoblasts Proliferation and Differentiation through VEGFR1-PI3K/Akt Signaling Pathway" International Journal of Molecular Sciences 22, no. 24: 13352. https://doi.org/10.3390/ijms222413352