
Impaired Retromer Function in Niemann-Pick Type C Disease Is Dependent on Intracellular Cholesterol Accumulation

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Result
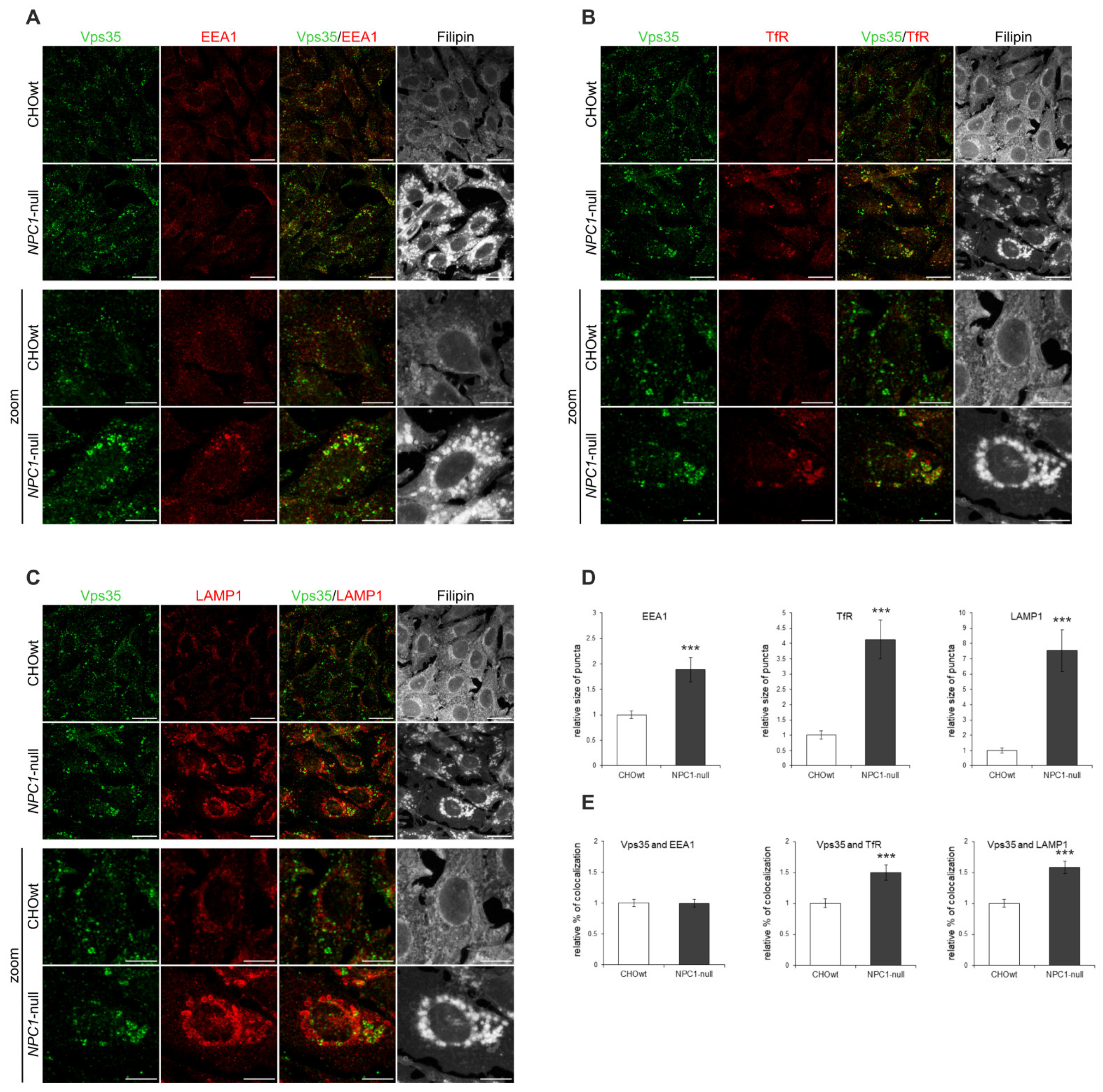
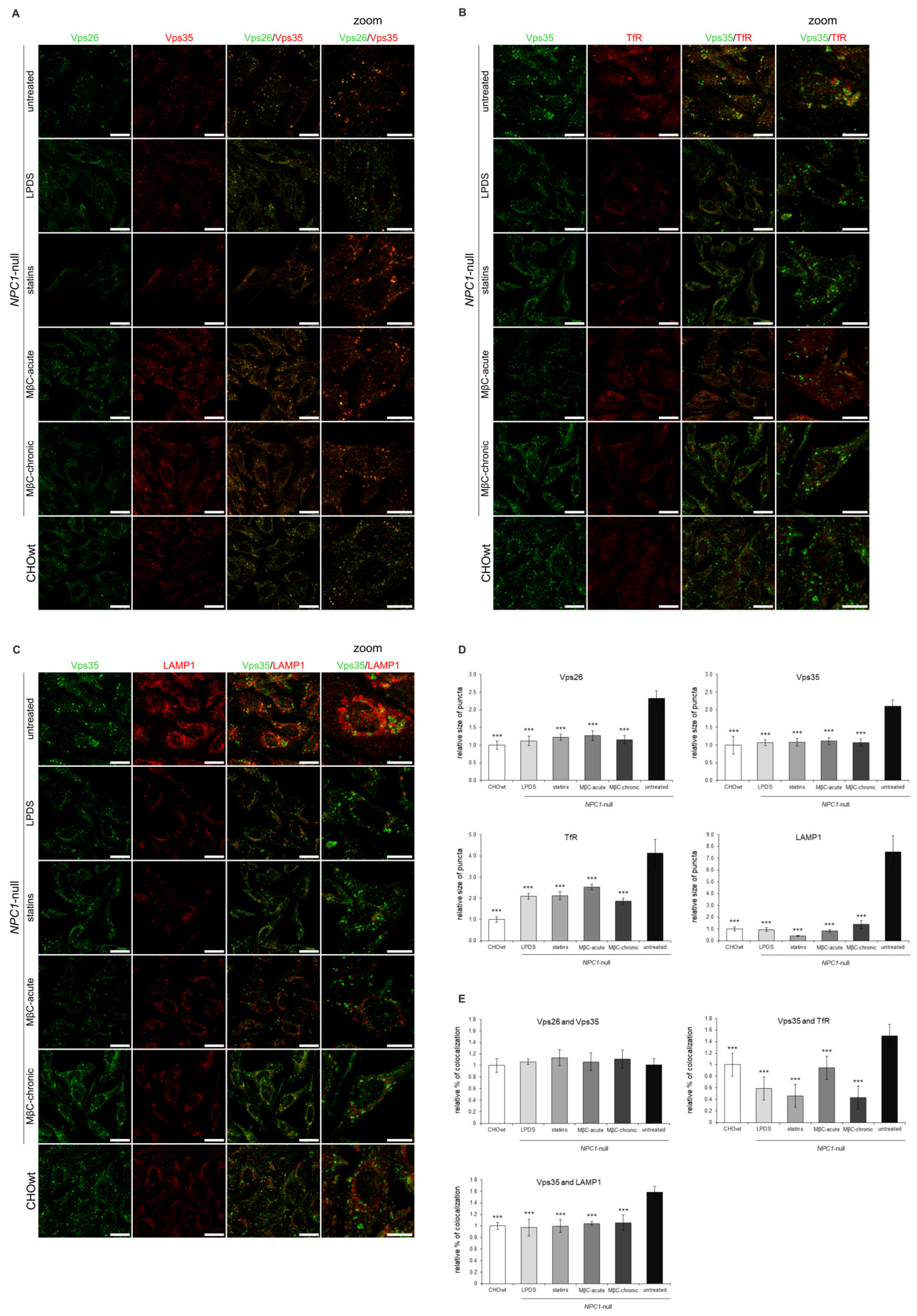
2.1. Retromer Trafficking and Distribution Is Impaired in CHO NPC1-Null Cells
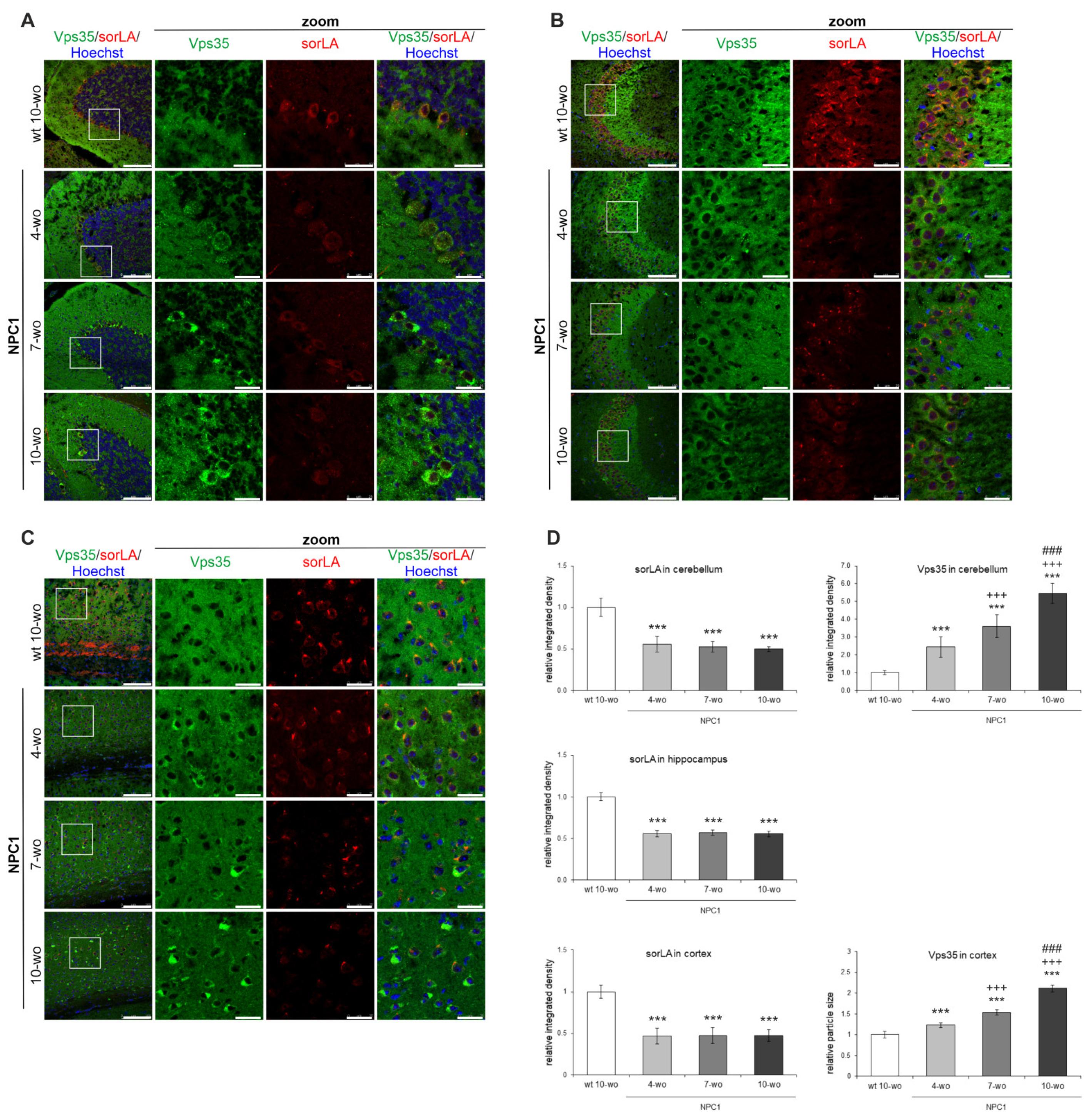
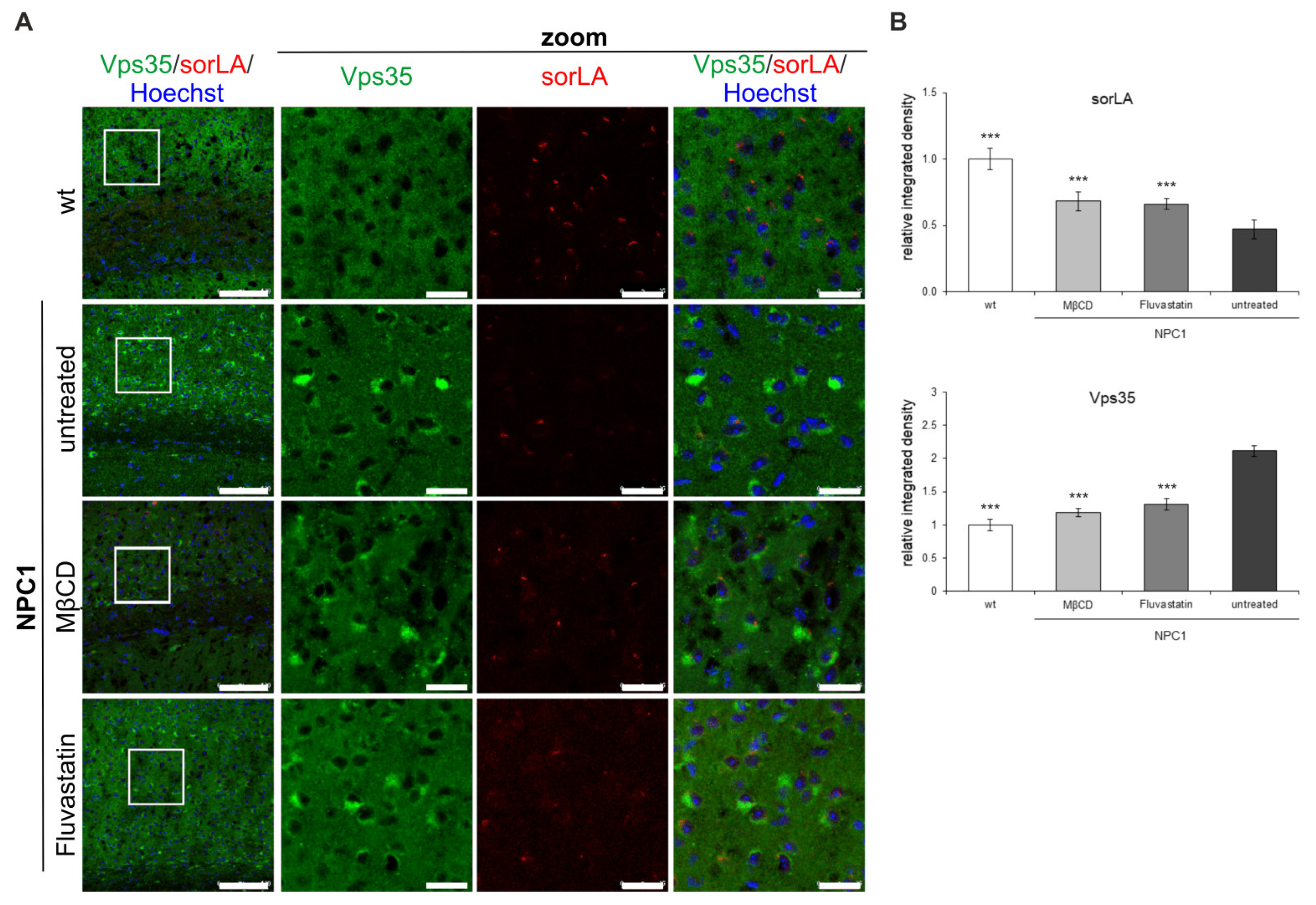
2.2. Retromer Distribution Is Altered in NPC1-Mouse Brains
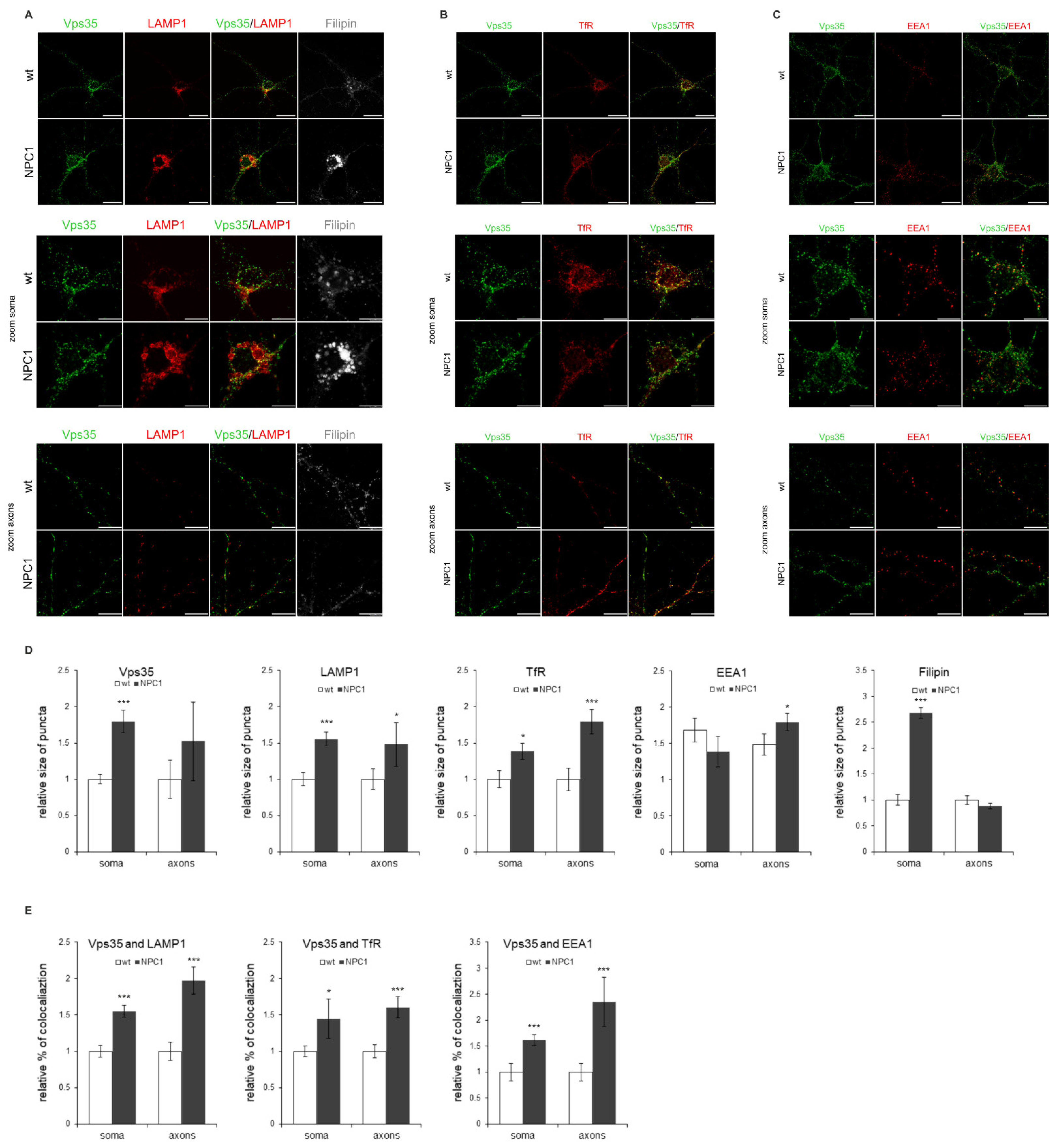
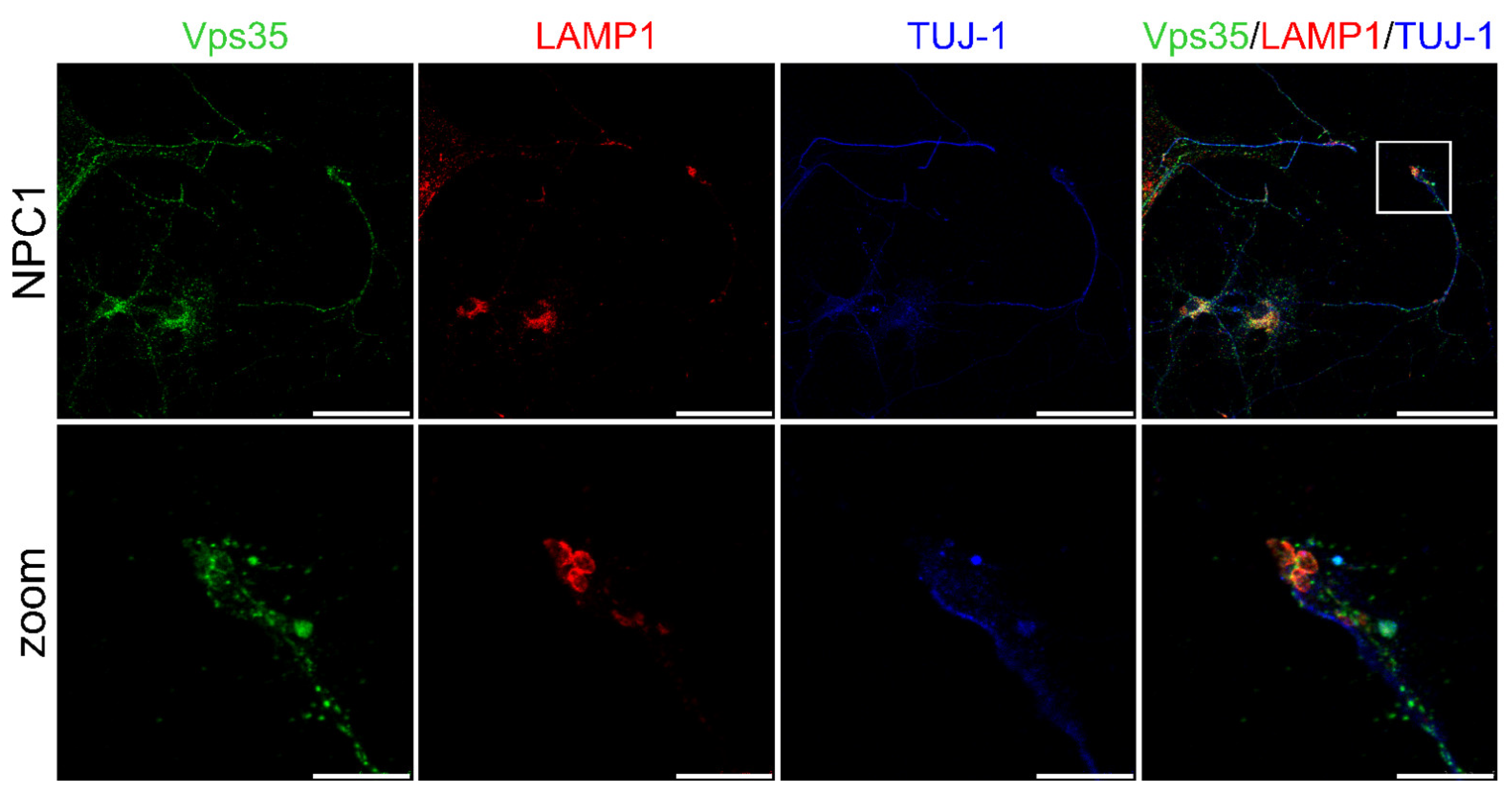
2.3. NPC1-Mouse Primary Neurons Show Retromer Transport Defect
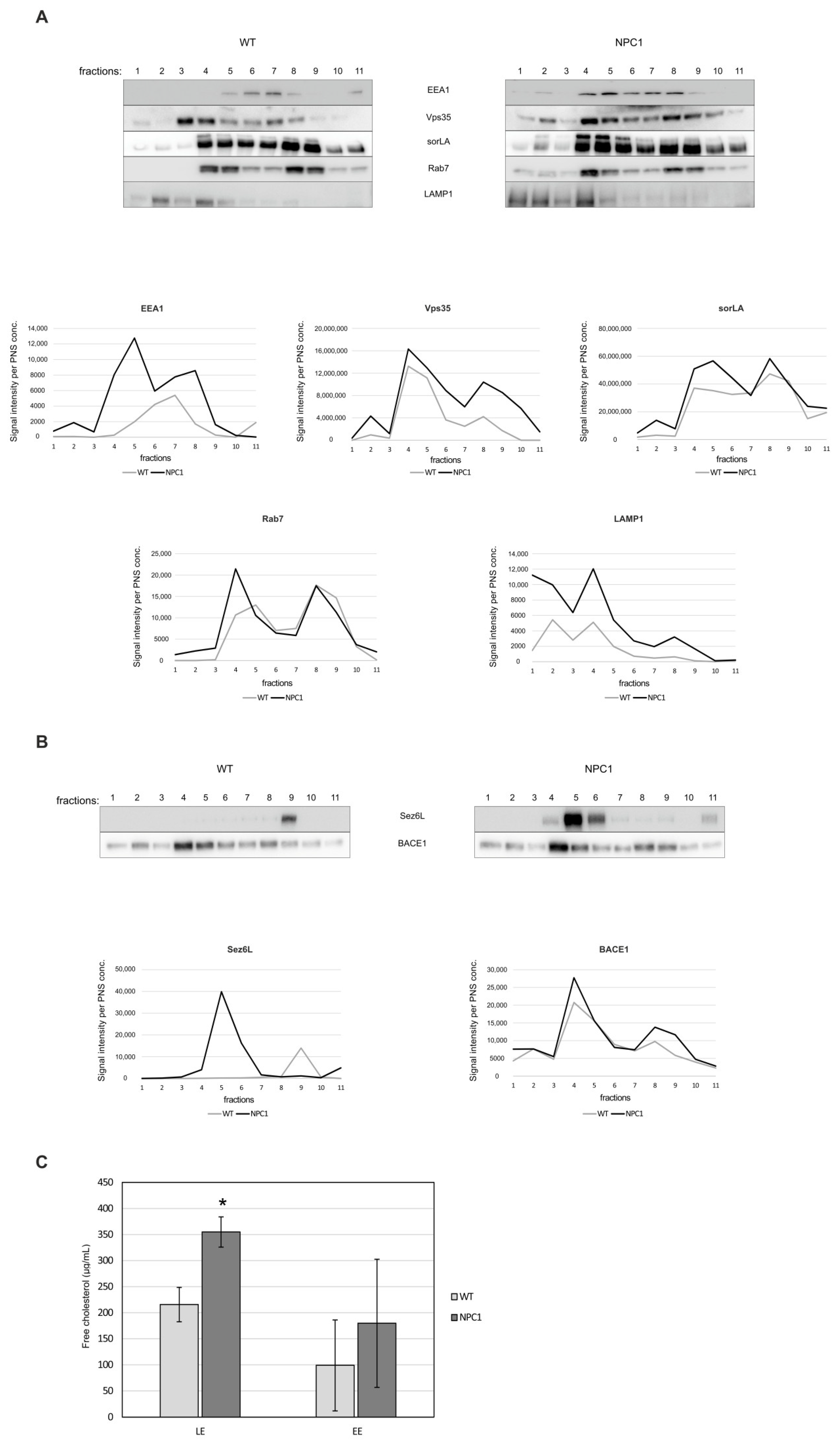
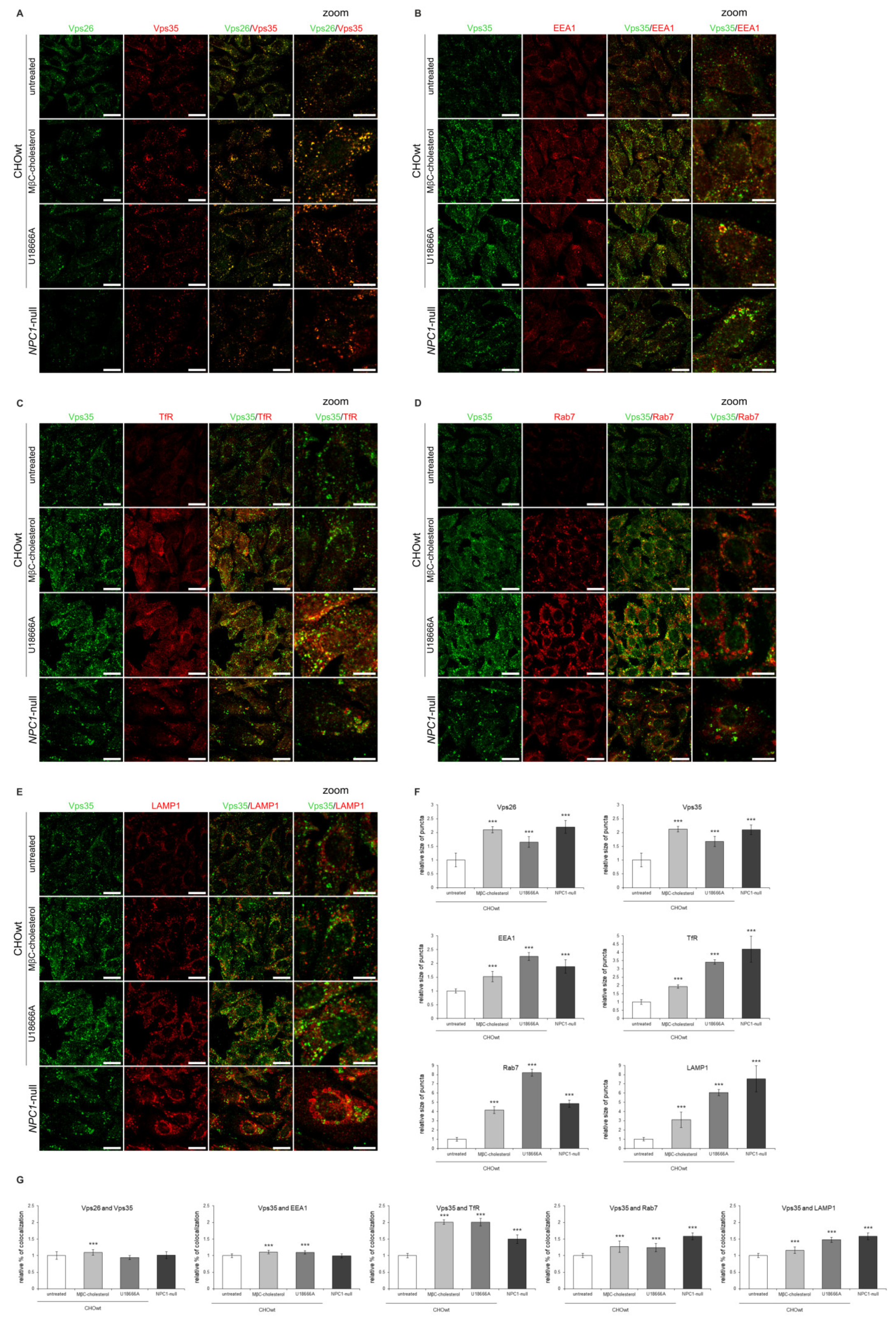
2.4. Retromer Impairment Is Dependent on Cholesterol Levels
3. Discussion
4. Material and Methods
4.1. Cell Culture and Primary Neurons
4.2. NPC1 Mouse Model
4.3. Antibodies
4.4. Cell Lysate Preparation
4.5. Western Blot
4.6. Immunocytochemistry and Confocal Microscopy
4.7. Immunohistochemistry of Mouse Brain Cryosections
4.8. Cholesterol Depletion/Loading
4.9. Endosome Fractionation
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Cataldo, A.M.; Barnett, J.L.; Pieroni, C.; Nixon, R.A. Increased Neuronal Endocytosis and Protease Delivery to Early Endosomes in Sporadic Alzheimer’s Disease: Neuropathologic Evidence for a Mechanism of Increased β-Amyloidogenesis. J. Neurosci. 1997, 17, 6142–6151. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Petanceska, S.; Terio, N.B.; Peterhoff, C.M.; Durham, R.; Mercken, M.; Mehta, P.D.; Buxbaum, J.; Haroutunian, V.; Nixon, R.A. Aβ localization in abnormal endosomes: Association with earliest Aβ elevations in AD and Down syndrome. Neurobiol. Aging 2004, 25, 1263–1272. [Google Scholar] [CrossRef]
- Nixon, R.A.; Cataldo, A.M.; Mathews, P.M. The endosomal-lysosomal system of neurons in Alzheimer’s disease pathogenesis: A review. Neurochem. Res. 2000, 25, 1161–1172. [Google Scholar] [CrossRef]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaman, M.N.; McCaffery, J.M.; Emr, S.D. A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J. Cell Biol. 1998, 142, 665–681. [Google Scholar] [CrossRef] [Green Version]
- Arighi, C.N.; Hartnell, L.M.; Aguilar, R.C.; Haft, C.R.; Bonifacino, J.S. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 2004, 165, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaman, M.N.J.J. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J. Cell Biol. 2004, 165, 111–122. [Google Scholar] [CrossRef]
- Temkin, P.; Lauffer, B.; Jäger, S.; Cimermancic, P.; Krogan, N.J.; von Zastrow, M. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat. Cell Biol. 2011, 13, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burd, C.; Cullen, P.J. Retromer: A Master Conductor of Endosome Sorting. Cold Spring Harb. Perspect. Biol. 2014, 6, a016774. [Google Scholar] [CrossRef]
- Steinberg, F.; Gallon, M.; Winfield, M.; Thomas, E.C.; Bell, A.J.; Heesom, K.J.; Tavaré, J.M.; Cullen, P.J. A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat. Cell Biol. 2013, 15, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, S.A.; Petsko, G.A. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat. Rev. Neurosci. 2015, 16, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Muzio, L.; Sirtori, R.; Gornati, D.; Eleuteri, S.; Fossaghi, A.; Brancaccio, D.; Manzoni, L.; Ottoboni, L.; Feo, L.D.; Quattrini, A.; et al. Retromer stabilization results in neuroprotection in a model of Amyotrophic Lateral Sclerosis. Nat. Commun. 2020, 11, 3848. [Google Scholar] [CrossRef]
- Qureshi, Y.H.; Baez, P.; Reitz, C. Endosomal Trafficking in Alzheimer’ s Disease, Parkinson’ s Disease, and Neuronal Ceroid Lipofuscinosis. Mol Cell Biol. 2020, 40, e00262-20. [Google Scholar] [CrossRef]
- Follett, J.; Norwood, S.J.; Hamilton, N.A.; Mohan, M.; Kovtun, O.; Tay, S.; Zhe, Y.; Wood, S.A.; Mellick, G.D.; Silburn, P.A.; et al. The Vps35 D620N Mutation Linked to Parkinson’s Disease Disrupts the Cargo Sorting Function of Retromer. Traffic 2014, 15, 230–244. [Google Scholar] [CrossRef]
- Vardarajan, B.N.; Bruesegem, S.Y.; Harbour, M.E.; Inzelberg, R.; Friedland, R.; St George-Hyslop, P.; Seaman, M.N.J.; Farrer, L.A. Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol. Aging 2012, 33, 2231.e15–2231.e30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Rogaeva, E.; Meng, Y.; Lee, J.H.; Gu, Y.; Kawarai, T.; Zou, F.; Katayama, T.; Baldwin, C.T.; Cheng, R.; Hasegawa, H.; et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007, 39, 168–177. [Google Scholar] [CrossRef]
- Kölsch, H.; Jessen, F.; Wiltfang, J.; Lewczuk, P.; Dichgans, M.; Teipel, S.J.; Kornhuber, J.; Frölich, L.; Heuser, I.; Peters, O.; et al. Association of SORL1 gene variants with Alzheimer’s disease. Brain Res. 2009, 1264, 1–6. [Google Scholar] [CrossRef]
- Pottier, C.; Hannequin, D.; Coutant, S.; Wallon, D.; Rousseau, S.; Legallic, S. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol. Psychiatry 2012, 17, 875–879. [Google Scholar] [CrossRef]
- Small, S.A.; Kent, K.; Pierce, A.; Leung, C.; Kang, M.S.; Okada, H.; Honig, L.; Vonsattel, J.P.; Kim, T.W. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann. Neurol. 2005, 58, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.M.; Reiche, J.; Schmidt, V.; Gotthardt, M.; Spoelgen, R.; Behlke, J.; von Arnim, C.A.F.; Breiderhoff, T.; Jansen, P.; Wu, X.; et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2005, 102, 13461–13466. [Google Scholar] [CrossRef] [Green Version]
- Dodson, S.E.; Gearing, M.; Lippa, C.F.; Montine, T.J.; Levey, A.I.; Lah, J.J. LR11/SorLA expression is reduced in sporadic Alzheimer disease but not in familial Alzheimer disease. J. Neuropathol. Exp. Neurol. 2006, 65, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. National Institute on Aging, National Institutes of Health. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer’s Disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, C.H.; Kumar, S.; Pinotsi, D.; Tunnacliffe, A.; St George-Hyslop, P.; Mandelkow, E.; Mandelkow, E.-M.; Kaminski, C.F.; Kaminski Schierle, G.S. Extracellular Monomeric Tau Protein Is Sufficient to Initiate the Spread of Tau Protein Pathology. J. Biol. Chem. 2014, 289, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Carosi, J.M.; Denton, D.; Kumar, S.; Sargeant, T.J. Retromer dysfunction at the nexus of tauopathies. Cell Death Differ. 2021, 28, 884–899. [Google Scholar] [CrossRef]
- Mecozzi, V.J.; Berman, D.E.; Simoes, S.; Vetanovetz, C.; Awal, M.R.; Patel, V.M.; Schneider, R.T.; Petsko, G.A.; Ringe, D.; Small, S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014, 10, 443–449. [Google Scholar] [CrossRef]
- Young, J.E.; Fong, L.K.; Frankowski, H.; Petsko, G.A.; Small, S.A.; Goldstein, L.S.B. Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer’s Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem Cell Rep. 2018, 10, 1046–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.G.; Chiu, J.; Ramanjulu, M.; Blass, B.E.; Praticò, D. A pharmacological chaperone improves memory by reducing Aβ and tau neuropathology in a mouse model with plaques and tangles. Mol. Neurodegener. 2020, 15, 1. [Google Scholar] [CrossRef]
- Nixon, R.A. Niemann-Pick Type C disease and Alzheimer’s disease: The APP-endosome connection fattens up. Am. J. Pathol. 2004, 164, 757–761. [Google Scholar] [CrossRef]
- Malnar, M.; Hecimovic, S.; Mattsson, N.; Zetterberg, H. Bidirectional links between Alzheimer’s disease and Niemann-Pick type C disease. Neurobiol. Dis. 2014, 72, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Wraith, J.E.; Baumgartner, M.R.; Bembi, B.; Covanis, A.; Levade, T.; Mengel, E.; Pineda, M.; Sedel, F.; Topçu, M.; Vanier, M.T.; et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol. Genet. Metab. 2009, 98, 152–165. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick disease type C Review. Orphanet J. Rare Dis. 2013, 8, 1–18. [Google Scholar]
- Colombo, A.; Dinkel, L.; Müller, S.A.; Sebastian Monasor, L.; Schifferer, M.; Cantuti-Castelvetri, L.; König, J.; Vidatic, L.; Bremova-Ertl, T.; Lieberman, A.P.; et al. Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia. Nat. Commun. 2021, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem. 2019, 294, 1706–1709. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Evans, E.; Platt, F.M. Lipids on trial: The search for the offending metabolite in Niemann-Pick type C disease. Traffic 2010, 11, 419–428. [Google Scholar] [CrossRef]
- Liu, B. Therapeutic potential of cyclodextrins in the treatment of Niemann–Pick type C disease. Clin. Lipidol. 2012, 7, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Davidson, C.D.; Fishman, Y.I.; Puskás, I.; Szemán, J.; Sohajda, T.; Mccauliff, L.A.; Sikora, J.; Storch, J.; Vanier, M.T.; Szente, L.; et al. Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. Ann. Clin. Transl. Neurol. 2016, 3, 366–380. [Google Scholar] [CrossRef]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A.; et al. Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: A non-randomised, open-label, phase 1–2 trial. Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef] [Green Version]
- Santos-Lozano, A.; García, D.V.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Gadea, G.N.; Lucia, A. Niemann-Pick disease treatment: A systematic review of clinical trials. Ann. Transl. Med. 2015, 3, 360. [Google Scholar] [CrossRef] [PubMed]
- Hastings, C.; Vieira, C.; Liu, B.; Bascon, C.; Gao, C.; Wang, R.Y.; Casey, A.; Hrynkow, S. Expanded access with intravenous hydroxypropyl-β-cyclodextrin to treat children and young adults with Niemann-Pick disease type C1: A case report analysis. Orphanet J. Rare Dis. 2019, 14, 228. [Google Scholar] [CrossRef]
- German, D.C.; Quintero, E.M.; Liang, C.L.; Ng, B.; Punia, S.; Xie, C.; Dietschy, J.M. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J. Comp. Neurol. 2001, 433, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, D.C.; Milenkovic, L.; Beier, S.M.; Manuel, H.; Buchanan, J.; Scott, M.P. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005, 1, 81–95. [Google Scholar] [CrossRef]
- Malnar, M.; Kosicek, M.; Lisica, A.; Posavec, M.; Krolo, A.; Njavro, J.; Omerbasic, D.; Tahirovic, S.; Hecimovic, S. Cholesterol-depletion corrects APP and BACE1 misstrafficking in NPC1-deficient cells. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 1270–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malnar, M.; Kosicek, M.; Mitterreiter, S.; Omerbasic, D.; Lichtenthaler, S.F.; Goate, A.; Hecimovic, S. Niemann-Pick type C cells show cholesterol dependent decrease of APP expression at the cell surface and its increased processing through the β-secretase pathway. Biochim. Biophys. Acta-Mol. Basis Dis. 2010, 1802, 682–691. [Google Scholar] [CrossRef] [Green Version]
- Causevic, M.; Dominko, K.; Malnar, M.; Vidatic, L.; Cermak, S.; Pigoni, M.; Kuhn, P.H.; Colombo, A.; Havas, D.; Flunkert, S.; et al. BACE1-cleavage of Sez6 and Sez6L is elevated in Niemann-Pick type C disease mouse brains. PLoS ONE 2018, 13, e0200344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, S.A. Retromer sorting: A pathogenic pathway in late-onset Alzheimer disease. Arch. Neurol. 2008, 65, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalla, A.; Vetanovetz, C.P.; Morel, E.; Chamoun, Z.; Di Paolo, G.; Small, S.A. The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol. Dis. 2012, 47, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Pentchev, P.G.; Gal, A.E.; Booth, A.D.; Omodeo-Sale, F.; Fours, J.; Neumeyer, B.A.; Quirk, J.M.; Dawson, G.; Brady, R.O. A lysosomal storage disorder in mice characterized by a dual deficiency of sphingomyelinase and glucocerebrosidase. Biochim. Biophys. Acta-Lipids Lipid Metab. 1980, 619, 669–679. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Koroniak, K.; Hogl, S.; Colombo, A.; Zeitschel, U.; Willem, M.; Volbracht, C.; Schepers, U.; Imhof, A.; Hoffmeister, A.; et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012, 31, 3157–3168. [Google Scholar] [CrossRef]
- Pigoni, M.; Wanngren, J.; Kuhn, P.H.; Munro, K.M.; Gunnersen, J.M.; Takeshima, H.; Feederle, R.; Voytyuk, I.; De Strooper, B.; Levasseur, M.D.; et al. Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol. Neurodegener. 2016, 11, 67. [Google Scholar] [CrossRef]
- Small, S.A.; Petsko, G.A. Endosomal recycling reconciles the Alzheimer’s disease paradox. Sci. Transl. Med. 2020, 12, eabb1717. [Google Scholar] [CrossRef] [PubMed]
- Marquer, C.; Tian, H.; Yi, J.; Bastien, J.; Dall’Armi, C.; Yang-Klingler, Y.J.; Zhou, B.; Chan, R.B.; Di Paolo, G. Arf6 controls retromer traffic and intracellular cholesterol distribution via a phosphoinositide-based mechanism. Nat. Commun. 2016, 7, 11919. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, J.K.; Pietrini, S.D.; D’Souza-Schorey, C. ARF6-mediated endosome recycling reverses lipid accumulation defects in Niemann-Pick type C disease. PLoS ONE 2009, 4, e5193. [Google Scholar] [CrossRef] [Green Version]
- Kutchukian, C.; Vivas, O.; Casas, M.; Jones, J.G.; Tiscione, S.A.; Simó, S.; Ory, D.S.; Dixon, R.E.; Dickson, E.J. NPC1 regulates the distribution of phosphatidylinositol 4-kinases at Golgi and lysosomal membranes. EMBO J. 2021, 40, e105990. [Google Scholar] [CrossRef]
- Lucin, K.M.; O’Brien, C.E.; Bieri, G.; Czirr, E.; Mosher, K.I.; Abbey, R.J.; Mastroeni, D.F.; Rogers, J.; Spencer, B.; Masliah, E.; et al. Microglial Beclin 1 Regulates Retromer Trafficking and Phagocytosis and Is Impaired in Alzheimer’s Disease. Neuron 2013, 79, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudoin, G.M.J.; Lee, S.-H.; Singh, D.; Yuan, Y.; Ng, Y.-G.; Reichardt, L.F.; Arikkath, J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Platt, N.; Speak, A.O.; Colaco, A.; Gray, J.; Smith, D.A.; Williams, I.M.; Wallom, K.L.; Platt, F.M. Immune dysfunction in Niemann-Pick disease type C. J. Neurochem. 2016, 136, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.B.; Wastney, M.; Patel, S.; Suresh, S.; Cooney, A.M.; Dwyer, N.K.; Roff, C.F.; Ohno, K.; Morris, J.A.; Carstea, E.D.; et al. The Niemann-Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J. Biol. Chem. 1999, 274, 9627–9635. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.L.; Grudzien, A.; Manhart, I.O.; Kelly, B.L.; Oakley, H.; Vassar, R. Statins cause intracellular accumulation of amyloid precursor protein, β-secretase-cleaved fragments, and amyloid β-peptide via an isoprenoid-dependent mechanism. J. Biol. Chem. 2005, 280, 18755–18770. [Google Scholar] [CrossRef] [Green Version]
- Christian, A.E.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997, 38, 2264–2272. [Google Scholar] [CrossRef]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta-Biomembr. 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, N.; Olsson, M.; Gustavsson, M.K.; Kosicek, M.; Malnar, M.; Månsson, J.E.; Blomqvist, M.; Gobom, J.; Andreasson, U.; Brinkmalm, G.; et al. Amyloid-β metabolism in Niemann-Pick C disease models and patients. Metab. Brain Dis. 2012, 27, 573–585. [Google Scholar] [CrossRef]
- De Araújo, M.E.G.; Lamberti, G.; Huber, L.A. Isolation of early and late endosomes by density gradient centrifugation. Cold Spring Harb. Protoc. 2015, 2015, 1013–1016. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dominko, K.; Rastija, A.; Sobocanec, S.; Vidatic, L.; Meglaj, S.; Lovincic Babic, A.; Hutter-Paier, B.; Colombo, A.-V.; Lichtenthaler, S.F.; Tahirovic, S.; et al. Impaired Retromer Function in Niemann-Pick Type C Disease Is Dependent on Intracellular Cholesterol Accumulation. Int. J. Mol. Sci. 2021, 22, 13256. https://doi.org/10.3390/ijms222413256
Dominko K, Rastija A, Sobocanec S, Vidatic L, Meglaj S, Lovincic Babic A, Hutter-Paier B, Colombo A-V, Lichtenthaler SF, Tahirovic S, et al. Impaired Retromer Function in Niemann-Pick Type C Disease Is Dependent on Intracellular Cholesterol Accumulation. International Journal of Molecular Sciences. 2021; 22(24):13256. https://doi.org/10.3390/ijms222413256
Chicago/Turabian StyleDominko, Kristina, Ana Rastija, Sandra Sobocanec, Lea Vidatic, Sarah Meglaj, Andrea Lovincic Babic, Birgit Hutter-Paier, Alessio-Vittorio Colombo, Stefan F. Lichtenthaler, Sabina Tahirovic, and et al. 2021. "Impaired Retromer Function in Niemann-Pick Type C Disease Is Dependent on Intracellular Cholesterol Accumulation" International Journal of Molecular Sciences 22, no. 24: 13256. https://doi.org/10.3390/ijms222413256