
BRCA Mutations in Prostate Cancer: Assessment, Implications and Treatment Considerations

 ,
,  , and
, and 
Abstract
:1. Introduction
2. DNA Repair Pathways
2.1. Base Excision Repair (BER)
2.2. Nucleotide Excision Repair (NER)
2.3. Mismatch Repair
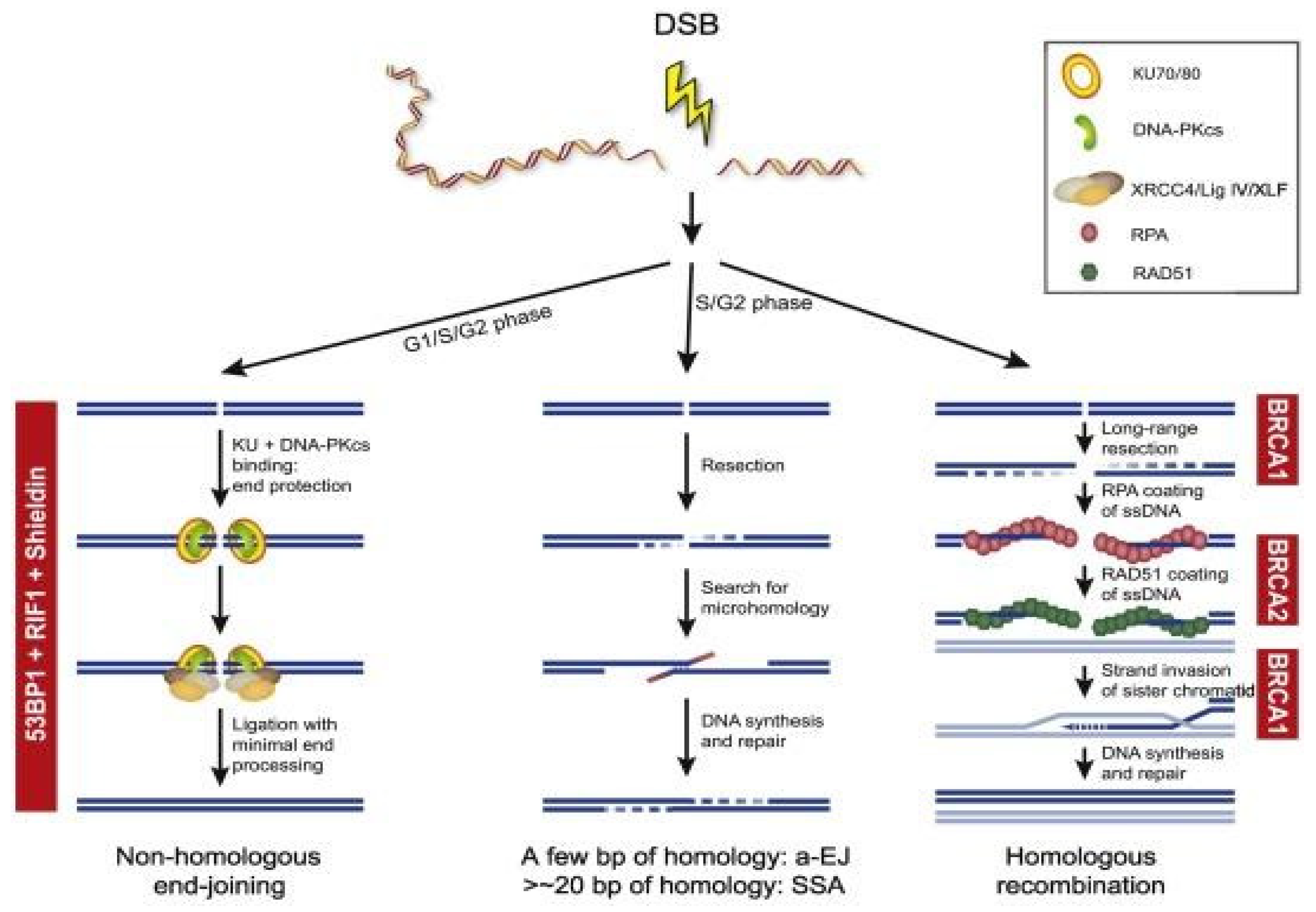
2.4. Homologous Recombination and Non-Homologous End Joining
2.5. Synthetic Lethality
3. Genomic Analysis of Prostate Cancer
4. DDR Mutations in Prostate Cancer
4.1. Germline Mutations in DDR Genes
4.2. Somatic Mutations in DDR Genes
4.3. BRCA Genes-Functional Similarities and Differences between BRCA1 and BRCA2
4.4. Inherited Mutations in DDR Genes and Prostate Cancer Risk
5. Implications for the Treatment
5.1. Clinical Development of PARP Inhibitors in Prostate Cancer
5.2. PARP Inhibitors and Synthetic Lethality
5.2.1. TOPARP-A and TOPARP-B Studies
5.2.2. PROFOUND Study
5.2.3. TRITON2 and GALAHAD Studies
5.3. PARP Inhibitors and AR Signalling Inhibitors
5.4. PARP Inhibitors and Immune Checkpoint Inhibitors
6. Safety and Toxicity Profile of PARP Inhibitors
7. Resistance Mechanisms to PARP Inhibitors
7.1. Reversion Mutation
7.2. Increase of Drug Efflux
7.3. Decreased PARP Trapping through Disruption of PARP1 and PARG Proteins
7.4. Stabilization of Stalled Forks
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Koochekpour, S. Genetic and epigenetic changes in human prostate cancer. Iran. Red. Crescent. Med. J. 2011, 13, 80–98. [Google Scholar] [PubMed]
- Castro, E.; Eeles, R. The role of BRCA1 and BRCA2 in prostate cancer. Asian. J. Androl. 2012, 14, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; Boysen, G.; Barbieri, C.E.; Bryant, H.E.; Castro, E.; Nelson, P.S.; Olmos, D.; Pritchard, C.C.; Rubin, M.A.; de Bono, J.S. DNA Repair in Prostate Cancer: Biology and Clinical Implications. Eur. Urol. 2017, 71, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussios, S.; Rassy, E.; Shah, S.; Ioannidou, E.; Sheriff, M.; Pavlidis, N. Aberrations of DNA repair pathways in prostate cancer: A cornerstone of precision oncology. Expert. Opin. Ther. Targets 2021, 25, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; van Gent, D.C.; Incrocci, L.; van Weerden, W.M.; Nonnekens, J. Role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate. Cancer Prostatic. Dis. 2020, 23, 24–37. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.; Moschetta, M.; Pappas-Gogos, G.; Sheriff, M.; Boussios, S. Genetic Aberrations of DNA Repair Pathways in Prostate Cancer: Translation to the Clinic. Int. J. Mol. Sci. 2021, 22, 9783. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nombela, P.; Lozano, R.; Aytes, A.; Mateo, J.; Olmos, D.; Castro, E. BRCA2 and Other DDR Genes in Prostate Cancer. Cancers 2019, 11, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Knudsen, K.E. DNA Damage Response in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2019, 9, a030486. [Google Scholar] [CrossRef]
- Dianov, G.L.; Hübscher, U. Mammalian Base Excision Repair: The Forgotten Archangel. Nucleic. Acids. Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef]
- Abbotts, R.; Wilson, D.M., III. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair. 2007, 6, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Kuasne, H.; Rodrigues, I.S.; Losi-Guembarovski, R.; Reis, M.B.; Fuganti, P.E.; Gregório, E.P.; Libos Junior, F.; Matsuda, H.M.; Rodrigues, M.A.; Kishima, M.O.; et al. Base excision repair genes XRCC1 and APEX1 and the risk for prostate cancer. Mol. Biol. Rep. 2011, 38, 1585–1591. [Google Scholar] [CrossRef]
- Rybicki, B.A.; Conti, D.V.; Moreira, A.; Cicek, M.; Casey, G.; Witte, J.S. DNA Repair Gene XRCC1 and XPD Polymorphisms and Risk of Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2004, 3, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Raja, S.; Van Houten, B. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic. Acids. Res. 2020, 48, 11227–11243. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Sugasawa, K.; Yanagisawa, J.; Sonoyama, T.; Ui, M.; Enomoto, T.; Takio, K.; Tanaka, K.; van der Spek, P.J.; Bootsma, D.; et al. Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. EMBO J. 1994, 13, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Nishi, R.; Okuda, Y.; Watanabe, E.; Mori, T.; Iwai, S.; Masutani, C.; Sugasawa, K.; Hanaoka, F. Centrin 2 Stimulates Nucleotide Excision Repair by Interacting with Xeroderma Pigmentosum Group C Protein. Mol. Cell. Biol. 2005, 25, 5664–5674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fousteri, M.; Vermeulen, W.; van Zeeland, A.A.; Mullenders, L.H. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol. Cell 2006, 23, 471–482. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell. Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Mandal, R.K.; Gangwar, R.; Kapoor, R.; Mittal, R.D. Polymorphisms in base-excision & nucleotide-excision repair genes & prostate cancer risk in north Indian population. Indian J. Med. Res. 2012, 135, 64–71. [Google Scholar] [PubMed]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair. 2016, 38, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell. Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Lipkin, S.M.; Wang, V.; Jacoby, R.; Banerjee-Basu, S.; Baxevanis, A.D.; Lynch, H.T.; Elliott, R.M.; Collins, F.S. MLH3: A DNA mismatch repair gene associated with mammalian microsatellite instability. Nat. Genet. 2000, 24, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.; Wu, X.; Her, C. MutS Homologues hMSH4 and hMSH5: Genetic Variations, Functions, and Implications in Human Diseases. Curr. Genom. 2013, 14, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral-Silva, G.K.; Martins, M.D.; Pontes, H.A.; Fregnani, E.R.; Lopes, M.A.; Fonseca, F.P.; Vargas, P.A. Mismatch repair system proteins in oral benign and malignant lesions. J. Oral. Pathol. Med. 2017, 46, 241–245. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Fishel, R. Mismatch Repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.A.; Lord, C.J.; Ashworth, A. Therapeutic targeting of the DNA mismatch repair pathway. Clin. Cancer Res. 2010, 16, 5107–5113. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. Rev. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA Mismatch Repair: Functions and Mechanisms. Chem. Rev. 2006, 106, 302–323. [Google Scholar] [CrossRef]
- Graham, L.S.; Montgomery, B.; Cheng, H.H.; Yu, E.Y.; Nelson, P.S.; Pritchard, C.; Erickson, S.; Alva, A.; Schweizer, M.T. Mismatch repair deficiency in metastatic prostate cancer: Response to PD-1 blockade and standard therapies. PLoS ONE 2020, 15, e0233260. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, V.; Paunu, K.; Ahlskog, J.K.; Varnai, R.; Sipeky, C.; Sundvall, M. PARP Inhibitors in Prostate Cancer—The Preclinical Rationale and Current Clinical Development. Genes 2019, 10, 565. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.; Pascal, L.E.; Zhou, Y.; Qiu, X.; Wei, L.; Ai, J.; Nelson, J.B.; Zhong, M.; Xue, B.; Wang, S.; et al. ELL2 regulates DNA non-homologous end joining (NHEJ) repair in prostate cancer cells. Cancer Lett. 2018, 415, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Koch, C.A.; Agyei, R.; Galicia, S.; Metalnikov, P.; O’Donnell, P.; Starostine, A.; Weinfeld, M.; Durocher, D. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 2004, 23, 3874–3885. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lu, H.; Tippin, B.; Goodman, M.F.; Shimazaki, N.; Koiwai, O.; Hsieh, C.L.; Schwarz, K.; Lieber, M.R. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell 2004, 16, 701–713. [Google Scholar] [CrossRef]
- Hsu, H.L.; Yannone, S.M.; Chen, D.J. Defining interactions between DNA-PK and ligase IV/XRCC4. DNA Repair. 2002, 1, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Verdun, R.E.; Karlseder, J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 2006, 127, 709–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural basis of homologous recombination. Cell. Mol. Life Sci. 2020, 77, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Krajewska, M.; Fehrmann, R.S.; de Vries, E.G.; van Vugt, M.A. Regulators of homologous recombination repair as novel targets for cancer treatment. Front. Genet. 2015, 6, 96. [Google Scholar] [CrossRef] [Green Version]
- Bednarz-Knoll, N.; Eltze, E.; Semjonow, A.; Brandt, B. BRCAness in prostate cancer. Oncotarget 2019, 10, 2421–2422. [Google Scholar] [CrossRef]
- Ladan, M.M.; van Gent, D.C.; Jager, A. Homologous Recombination Deficiency Testing for BRCA-Like Tumors: The Road to Clinical Validation. Cancers 2021, 13, 1004. [Google Scholar] [CrossRef]
- Castro, E.; Goh, C.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Dadaev, T.; Govindasami, K.; Guy, M.; Ellis, S.; Frost, D.; et al. Effect of BRCA Mutations on Metastatic Relapse and Cause-specific Survival After Radical Treatment for Localised Prostate Cancer. Eur. Urol. 2015, 68, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef]
- Sigorski, D.; Iżycka-Świeszewska, E.; Bodnar, L. Poly(ADP-Ribose) Polymerase Inhibitors in Prostate Cancer: Molecular Mechanisms, and Preclinical and Clinical Data. Target. Oncol. 2020, 15, 709–722. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Abson, C.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Ryan, J.E.; Sheriff, M.; Rassy, E.; Pavlidis, N. Veliparib in ovarian cancer: A new synthetically lethal therapeutic approach. Investig. New Drugs 2020, 38, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Boussios, S.; Moschetta, M.; Karihtala, P.; Samartzis, E.P.; Sheriff, M.; Pappas-Gogos, G.; Ozturk, M.A.; Uccello, M.; Karathanasi, A.; Tringos, M.; et al. Development of new poly(ADP-ribose) polymerase (PARP) inhibitors in ovarian cancer: Quo Vadis? Ann. Transl. Med. 2020, 8, 1706. [Google Scholar] [CrossRef] [PubMed]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef]
- Pilarski, R. The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 79–86. [Google Scholar] [CrossRef]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Castro, E.; Romero-Laorden, N.; Del Pozo, A.; Lozano, R.; Medina, A.; Puente, J.; Piulats, J.M.; Lorente, D.; Saez, M.I.; Morales-Barrera, R.; et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Messina, C.; Cattrini, C.; Soldato, D.; Vallome, G.; Caffo, O.; Castro, E.; Olmos, D.; Boccardo, F.; Zanardi, E. BRCA Mutations in Prostate Cancer: Prognostic and Predictive Implications. J. Oncol. 2020, 2020, 4986365. [Google Scholar] [CrossRef]
- Cheng, H.H.; Sokolova, A.O.; Schaeffer, E.M.; Small, E.J.; Higano, C.S. Germline and Somatic Mutations in Prostate Cancer for the Clinician. J. Natl. Compr. Cancer Netw. 2019, 17, 515–521. [Google Scholar] [CrossRef] [Green Version]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network Clinical Guidelines in Oncology (NCCN Guidelines): Prostate Cancer (Version 1.2022). Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1459 (accessed on 18 October 2021).
- National Comprehensive Cancer Network Clinical Guidelines in Oncology (NCCN Guidelines): Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic (Version 1.2022). Available online: https://www.nccn.org/guidelines/guidelines-detail?category=2&id=1503 (accessed on 18 October 2021).
- Giri, V.N.; Knudsen, K.E.; Kelly, W.K.; Cheng, H.H.; Cooney, K.A.; Cookson, M.S.; Dahut, W.; Weissman, S.; Soule, H.R.; Petrylak, D.P.; et al. Implementation of Germline Testing for Prostate Cancer: Philadelphia Prostate Cancer Consensus Conference 2019. J. Clin. Oncol. 2020, 38, 2798–2811. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Castro, E.; Fizazi, K.; Heidenreich, A.; Ost, P.; Procopio, G.; Tombal, B.; Gillessen, S.; ESMO Guidelines Committee. Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1119–1134. [Google Scholar] [CrossRef]
- Mottet, N.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer-2020 Update. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2021, 79, 243–262. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence. Olaparib for Previously Treated BRCA-Mutation Positive Hormone-Relapsed Metastatic Prostate Cancer. February 2021. Available online: https://www.nice.org.uk/guidance/gid-ta10584/documents/129 (accessed on 13 October 2021).
- Barbieri, C.E.; Rubin, M.A. Genomic rearrangements in prostate cancer. Curr. Opin. Urol. 2015, 25, 71–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef] [Green Version]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Chiu, Y.T.; Liu, J.; Tang, K.; Wong, Y.C.; Khanna, K.K.; Ling, M.T. Inactivation of ATM/ATR DNA damage checkpoint promotes androgen induced chromosomal instability in prostate epithelial cells. PLoS ONE 2012, 7, e51108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, M.T.; Antonarakis, E.S.; Bismar, T.A.; Guedes, L.B.; Cheng, H.H.; Tretiakova, M.S.; Vakar-Lopez, F.; Klemfuss, N.; Konnick, E.Q.; Mostaghel, E.A.; et al. Genomic Characterization of Prostatic Ductal Adenocarcinoma Identifies a High Prevalence of DNA Repair Gene Mutations. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef]
- Isaacsson Velho, P.; Silberstein, J.L.; Markowski, M.C.; Luo, J.; Lotan, T.L.; Isaacs, W.B.; Antonarakis, E.S. Intraductal/ductal histology and lymphovascular invasion are associated with germline DNA-repair gene mutations in prostate cancer. Prostate 2018, 78, 401–407. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Gurova, K.V.; Roklin, O.W.; Krivokrysenko, V.I.; Chumakov, P.M.; Cohen, M.B.; Feinstein, E.; Gudkov, A.V. Expression of prostate specific antigen (PSA) is negatively regulated by p53. Oncogene 2002, 21, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Li, L.C.; Carroll, P.R.; Dahiya, R. Epigenetic changes in prostate cancer: Implication for diagnosis and treatment. J. Natl. Cancer Inst. 2005, 97, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Collins, N.; Poot, R.A.; Kukimoto, I.; García-Jiménez, C.; Dellaire, G.; Varga-Weisz, P.D. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat. Genet. 2002, 32, 627–632. [Google Scholar] [CrossRef]
- Deuring, R.; Fanti, L.; Armstrong, J.A.; Sarte, M.; Papoulas, O.; Prestel, M.; Daubresse, G.; Verardo, M.; Moseley, S.L.; Berloco, M.; et al. The ISWI chromatin-remodeling protein is required for gene expression and the maintenance of higher order chromatin structure in vivo. Mol. Cell 2000, 5, 355–365. [Google Scholar] [CrossRef]
- Havas, K.; Whitehouse, I.; Owen-Hughes, T. ATP-dependent chromatin remodeling activities. Cell. Mol. Life Sci. 2001, 58, 673–682. [Google Scholar] [CrossRef]
- Morova, T.; McNeill, D.R.; Lallous, N.; Gönen, M.; Dalal, K.; Wilson, D.M., III; Gürsoy, A.; Keskin, Ö.; Lack, N.A. Androgen receptor-binding sites are highly mutated in prostate cancer. Nat. Commun. 2020, 11, 832. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Welcsh, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Tulinius, H.; Egilsson, V.; Olafsdóttir, G.H.; Sigvaldason, H. Risk of prostate, ovarian, and endometrial cancer among relatives of women with breast cancer. BMJ 1992, 305, 855–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arason, A.; Barkardóttir, R.B.; Egilsson, V. Linkage analysis of chromosome 17q markers and breast-ovarian cancer in Icelandic families, and possible relationship to prostatic cancer. Am. J. Hum. Genet. 1993, 52, 711–717. [Google Scholar]
- Anderson, D.E.; Badzioch, M.D. Familial effects of prostate and other cancers on lifetime breast cancer risk. Breast Cancer Res. Treat. 1993, 28, 107–113. [Google Scholar] [CrossRef]
- Sellers, T.A.; Potter, J.D.; Rich, S.S.; Drinkard, C.R.; Bostick, R.M.; Kushi, L.H.; Zheng, W.; Folsom, A.R. Familial clustering of breast and prostate cancers and risk of postmenopausal breast cancer. J. Natl. Cancer Inst. 1994, 86, 1860–1865. [Google Scholar] [CrossRef]
- Easton, D.F.; Steele, L.; Fields, P.; Ormiston, W.; Averill, D.; Daly, P.A.; McManus, R.; Neuhausen, S.L.; Ford, D.; Wooster, R.; et al. Cancer risks in two large breast cancer families linked to BRCA2 on chromosome 13q12-13. Am. J. Hum. Genet. 1997, 61, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsdottir, K.; Ashworth, A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006, 25, 5864–5874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.J.; Irvine, R.A.; Buchanan, G.; Koh, S.S.; Park, J.M.; Tilley, W.D.; Stallcup, M.R.; Press, M.F.; Coetzee, G.A. Breast cancer susceptibility gene 1 (BRCAI) is a coactivator of the androgen receptor. Cancer Res. 2000, 60, 5946–5949. [Google Scholar] [PubMed]
- Urbanucci, A.; Waltering, K.K.; Suikki, H.E.; Helenius, M.A.; Visakorpi, T. Androgen regulation of the androgen receptor coregulators. BMC Cancer 2008, 8, 219. [Google Scholar] [CrossRef] [Green Version]
- Schayek, H.; Haugk, K.; Sun, S.; True, L.D.; Plymate, S.R.; Werner, H. Tumor suppressor BRCA1 is expressed in prostate cancer and controls insulin-like growth factor I receptor (IGF-IR) gene transcription in an androgen receptor-dependent manner. Clin. Cancer Res. 2009, 15, 1558–1565. [Google Scholar] [CrossRef] [Green Version]
- Bednarz, N.; Eltze, E.; Semjonow, A.; Rink, M.; Andreas, A.; Mulder, L.; Hannemann, J.; Fisch, M.; Pantel, K.; Weier, H.U.; et al. BRCA1 loss preexisting in small subpopulations of prostate cancer is associated with advanced disease and metastatic spread to lymph nodes and peripheral blood. Clin. Cancer Res. 2010, 16, 3340–3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cussiol, J.R.; Dibitetto, D.; Sims, J.R.; Twayana, S.; Weiss, R.S.; Freire, R.; Marini, F.; Pellicioli, A.; Smolka, M.B. TOPBP1 Dpb11 plays a conserved role in homologous recombination DNA repair through the coordinated recruitment of 53BP1 Rad9. J. Cell Biol. 2017, 216, 623–639. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.L.; Busch, E.L.; Friebel, T.M.; Cronin, A.; Leslie, G.; McGuffog, L.; Adlard, J.; Agata, S.; Agnarsson, B.A.; Ahmed, M.; et al. Association of Genomic Domains in BRCA1 and BRCA2 with Prostate Cancer Risk and Aggressiveness. Cancer Res. 2020, 80, 624–638. [Google Scholar] [CrossRef] [Green Version]
- Giri, V.N.; Knudsen, K.E.; Kelly, W.K.; Abida, W.; Andriole, G.L.; Bangma, C.H.; Bekelman, J.E.; Benson, M.C.; Blanco, A.; Burnett, A.; et al. Role of Genetic Testing for Inherited Prostate Cancer Risk: Philadelphia Prostate Cancer Consensus Conference 2017. J. Clin. Oncol. 2018, 36, 414–424. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.C.; McCarthy, A.; Thomsen, M.K.; Ashworth, A.; Swain, A. Brca2 and Trp53 deficiency cooperate in the progression of mouse prostate tumourigenesis. PLoS Genet. 2010, 6, e1000995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, L.; Arbini, A.A.; Yao, J.L.; di Sant’Agnese, P.A.; Marra, E.; Greco, M. Loss of BRCA2 promotes prostate cancer cell invasion through up-regulation of matrix metalloproteinase-9. Cancer Sci. 2008, 99, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.O.; Cheng, H.H. Genetic Testing in Prostate Cancer. Curr. Oncol. Rep. 2020, 22, 5. [Google Scholar] [CrossRef]
- Leongamornlert, D.; Mahmud, N.; Tymrakiewicz, M.; Saunders, E.; Dadaev, T.; Castro, E.; Goh, C.; Govindasami, K.; Guy, M.; O’Brien, L.; et al. Germline BRCA1 mutations increase prostate cancer risk. Br. J. Cancer 2012, 106, 1697–1701. [Google Scholar] [CrossRef] [Green Version]
- Page, E.C.; Bancroft, E.K.; Brook, M.N.; Assel, M.; Hassan Al Battat, M.; Thomas, S.; Taylor, N.; Chamberlain, A.; Pope, J.; Raghallaigh, H.N.; et al. Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 2019, 76, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Bancroft, E.K.; Page, E.C.; Castro, E.; Lilja, H.; Vickers, A.; Sjoberg, D.; Assel, M.; Foster, C.S.; Mitchell, G.; Drew, K.; et al. Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: Results from the initial screening round of the IMPACT study. Eur. Urol. 2014, 66, 489–499. [Google Scholar] [CrossRef]
- Saxby, H.; Mikropoulos, C.; Boussios, S. An Update on the Prognostic and Predictive Serum Biomarkers in Metastatic Prostate Cancer. Diagnostics 2020, 10, 549. [Google Scholar] [CrossRef]
- Ioannidou, E.; Moschetta, M.; Shah, S.; Parker, J.S.; Ozturk, M.A.; Pappas-Gogos, G.; Sheriff, M.; Rassy, E.; Boussios, S. Angiogenesis and Anti-Angiogenic Treatment in Prostate Cancer: Mechanisms of Action and Molecular Targets. Int. J. Mol. Sci. 2021, 22, 9926. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R&D 2020, 20, 55–73. [Google Scholar]
- Rubin, M.A.; Maher, C.A.; Chinnaiyan, A.M. Common gene rearrangements in prostate cancer. J. Clin. Oncol. 2011, 29, 3659–3668. [Google Scholar] [CrossRef] [Green Version]
- Rubin, M.A. ETS rearrangements in prostate cancer. Asian J. Androl. 2012, 14, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Loeb, L.A. Human Cancers Express a Mutator Phenotype: Hypothesis, Origin, and Consequences. Cancer Res. 2016, 76, 2057–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Sandhu, S.K.; Kelly, W.K.; Scher, H.I.; Efstathiou, E.; Lara, P.; Yu, E.Y.; George, D.J.; Chi, K.N.; Summa, J.; et al. Pre-specified interim analysis of GALAHAD: A phase II study of niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD). Ann. Oncol. 2019, 30, v884–v885. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Salami, S.S.; Spratt, D.E.; Kaffenberger, S.D.; Jacobs, M.F.; Morgan, T.M. Bringing Prostate Cancer Germline Genetics into Clinical Practice. J. Urol. 2019, 202, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Campbell, D.; Patnaik, A.; Sautois, B.; Shapiro, J.; Vogelzang, N.J.; Bryce, A.H.; McDermott, R.; Ricci, F.; Rowe, J.; et al. Preliminary results from the TRITON2 study of rucaparib in patients (pts) with DNA damage repair (DDR)-deficient metastatic castration-resistant prostate cancer (mCRPC): Updated analyses. Ann. Oncol. 2019, 30, v325–v355. [Google Scholar] [CrossRef]
- Ramakrishnan Geethakumari, P.; Schiewer, M.J.; Knudsen, K.E.; Kelly, W.K. PARP Inhibitors in Prostate Cancer. Curr. Treat. Options Oncol. 2017, 18, 37. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Cooke, D.; Neille, C.; Sadauskaite, A.; Moschetta, M.; Zakynthinakis-Kyriakou, N.; Pavlidis, N. PARP Inhibitors in Ovarian Cancer: The Route to “Ithaca”. Diagnostics 2019, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.; Nelson, P.; Vasioukhin, V. Recent advances in prostate cancer research: Large-scale genomic analyses reveal novel driver mutations and DNA repair defects. F1000Research 2018, 7, 1173. [Google Scholar] [CrossRef]
- Stopsack, K.H. Efficacy of PARP Inhibition in Metastatic Castration-resistant Prostate Cancer is Very Different with Non-BRCA DNA Repair Alterations: Reconstructing Prespecified Endpoints for Cohort B from the Phase 3 PROfound Trial of Olaparib. Eur. Urol. 2021, 79, 442–445. [Google Scholar] [CrossRef]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced "BRCAness" and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479. [Google Scholar] [CrossRef] [Green Version]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Fay, A.P.; Antonarakis, E.S. Blocking the PD-1/PD-L1 axis in advanced prostate cancer: Are we moving in the right direction? Ann. Transl. Med. 2019, 7, S7. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. ImmunoTherapy Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Yu, E.Y.; Hoon Park, S.; Huang, Y.H.; Bennamoun, M.; Xu, L.; Kim, J.; Antonarakis, E.S. Phase III study of pembrolizumab (pembro) plus olaparib versus enzalutamide (enza) or abiraterone acetate (abi) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) who progressed on chemotherapy: KEYLYNK-010. J. Clin. Oncol. 2020, 38, TPS256. [Google Scholar] [CrossRef]
- Nizialek, E.; Antonarakis, E.S. PARP Inhibitors in Metastatic Prostate Cancer: Evidence to Date. Cancer Manag. Res. 2020, 12, 8105–8114. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Guo, Z.; Hu, Z. The Role of PARP Inhibitors in the Treatment of Prostate Cancer: Recent Advances in Clinical Trials. Biomolecules 2021, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Posadas, E.; Bhowmick, N.; Kim, H.; Daskivich, T.; Gupta, A.; Sandler, H.; Kamrava, M.; Zumsteg, Z.; Freedland, S.; et al. Integrating PARP Inhibitors Into Advanced Prostate Cancer Therapeutics. Oncology 2021, 35, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.M.; et al. A Phase I-II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise Management of Ovarian Cancer: On the Cutting Edge. J. Pers. Med. 2020, 10, 41. [Google Scholar] [CrossRef]
- Nitecki, R.; Gockley, A.A.; Floyd, J.L.; Coleman, R.L.; Melamed, A.; Rauh-Hain, J.A. The incidence of myelodysplastic syndrome in patients receiving poly-ADP ribose polymerase inhibitors for treatment of solid tumors: A meta-analysis. J. Clin. Oncol. 2020, 38, 3641. [Google Scholar] [CrossRef]
- Kikuchi, R.; Lao, Y.; Bow, D.A.; Chiou, W.J.; Andracki, M.E.; Carr, R.A.; Voorman, R.L.; De Morais, S.M. Prediction of clinical drug-drug interactions of veliparib (ABT-888) with human renal transporters (OAT1, OAT3, OCT2, MATE1, and MATE2K). J. Pharm. Sci. 2013, 102, 4426–4432. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis Filho, J.S.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013, 229, 422–429. [Google Scholar] [CrossRef]
- Ter Brugge, P.; Kristel, P.; van der Burg, E.; Boon, U.; de Maaker, M.; Lips, E.; Mulder, L.; de Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Weigelt, B.; Comino-Méndez, I.; de Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [Green Version]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [Green Version]
- Jaspers, J.E.; Sol, W.; Kersbergen, A.; Schlicker, A.; Guyader, C.; Xu, G.; Wessels, L.; Borst, P.; Jonkers, J.; Rottenberg, S. BRCA2-deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Res. 2015, 75, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rottenberg, S.; Nygren, A.O.; Pajic, M.; van Leeuwen, F.W.; van der Heijden, I.; van de Wetering, K.; Liu, X.; de Visser, K.E.; Gilhuijs, K.G.; van Tellingen, O.; et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12117–12122. [Google Scholar] [CrossRef] [Green Version]
- Hanzlikova, H.; Gittens, W.; Krejcikova, K.; Zeng, Z.; Caldecott, K.W. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017, 45, 2546–2557. [Google Scholar] [CrossRef]
- Amé, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Höger, T.; Ménissier-de Murcia, J.; de Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Llobet, S.G.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2019, 35, 950–952. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Gene | DDR Pathway | Percentage (%) of Germline Mutations in Tested Prostate Cancer Patients | |
|---|---|---|---|
| Nicolosi et al. [75] | Pritchard et al. [61] | ||
| ATM | Homologous Recombination | 2.03 | 1.59 |
| ATR | Homologous Recombination | Not tested | 0.29 |
| BRCA1 | Homologous Recombination | 1.25 | 0.87 |
| BRCA2 | 4.74 | 5.35 | |
| BARD1 | Homologous Recombination | 0.00 | 0.00 |
| BRIP1 | Homologous Recombination | 0.28 | 0.18 |
| CDKN2a | p16/cyclin-dependent kinase/retinoblastoma gene pathway | 0.13 | Not tested |
| CHEK2 | Homologous Recombination | 2.88 | 1.87 |
| FAM175A | Homologous Recombination | Not tested | 0.18 |
| GEN1 | Homologous Recombination | Not tested | 0.46 |
| MLH1 | Mismatch Repair | 0.06 | 0.00 |
| MSH2 | 0.69 | 0.14 | |
| MSH6 | 0.45 | 0.14 | |
| NBN | Homologous Recombination | 0.32 | 0.29 |
| PALB2 | Homologous Recombination | 0.56 | 0.43 |
| PMS2 | Mismatch Repair | 0.54 | 0.29 |
| RAD51C | Homologous Recombination | 0.21 | 0.14 |
| RAD51D | Homologous Recombination | 0.15 | 0.43 |
| Clinical Trial | Phase | Study PARPi | Strategy | Primary Endpoint |
|---|---|---|---|---|
| TOPARP-A [67] | II | Olaparib | Olaparib in mCRPC | RR, PSA, CTC |
| TOPARP-B [120] | II | Olaparib | Olaparib in mCRPC with gene analysis | RR, PSA, CTC |
| PROFOUND [121] | III | Olaparib | Olaparib vs ARSi with gene analysis | rPFS |
| TRITON2 [122,123] | II | Rucaparib | Rucaparib in mCRPC | ORR |
| GALAHAD [124] | II | Niraparib | Niraparib in mCRPC | ORR |
| TALAPRO-1 [119] | II | Talazoparib | Talazoparib in mCRPC | ORR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, S.; Rachmat, R.; Enyioma, S.; Ghose, A.; Revythis, A.; Boussios, S. BRCA Mutations in Prostate Cancer: Assessment, Implications and Treatment Considerations. Int. J. Mol. Sci. 2021, 22, 12628. https://doi.org/10.3390/ijms222312628
Shah S, Rachmat R, Enyioma S, Ghose A, Revythis A, Boussios S. BRCA Mutations in Prostate Cancer: Assessment, Implications and Treatment Considerations. International Journal of Molecular Sciences. 2021; 22(23):12628. https://doi.org/10.3390/ijms222312628
Chicago/Turabian StyleShah, Sidrah, Rachelle Rachmat, Synthia Enyioma, Aruni Ghose, Antonios Revythis, and Stergios Boussios. 2021. "BRCA Mutations in Prostate Cancer: Assessment, Implications and Treatment Considerations" International Journal of Molecular Sciences 22, no. 23: 12628. https://doi.org/10.3390/ijms222312628