
Empagliflozin Ameliorates Free Fatty Acid Induced-Lipotoxicity in Renal Proximal Tubular Cells via the PPARγ/CD36 Pathway in Obese Mice

 , , ,
, , ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
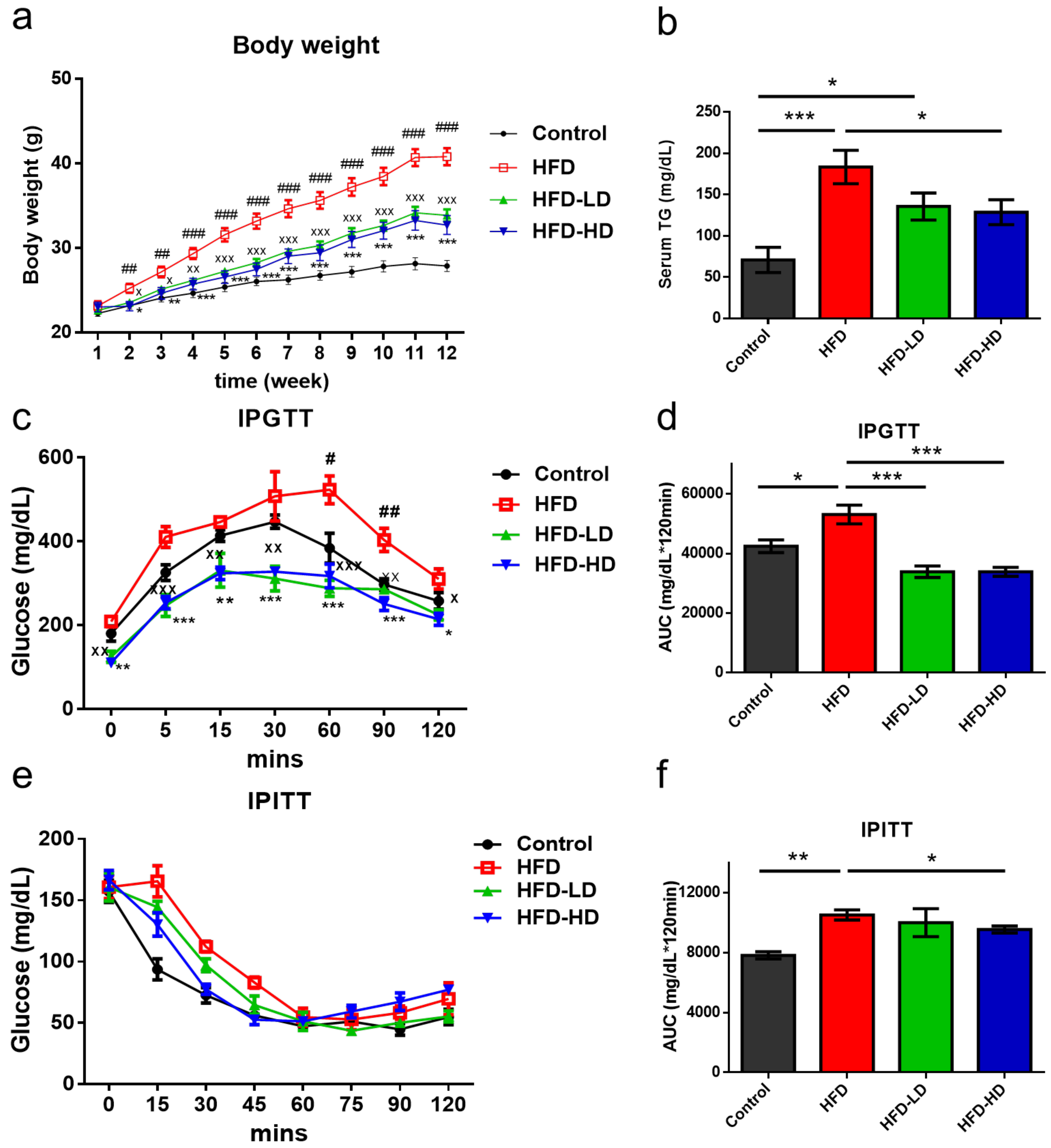
2.1. Empagliflozin Attenuated HFD-Induced Body Weight Gain and Insulin Resistance
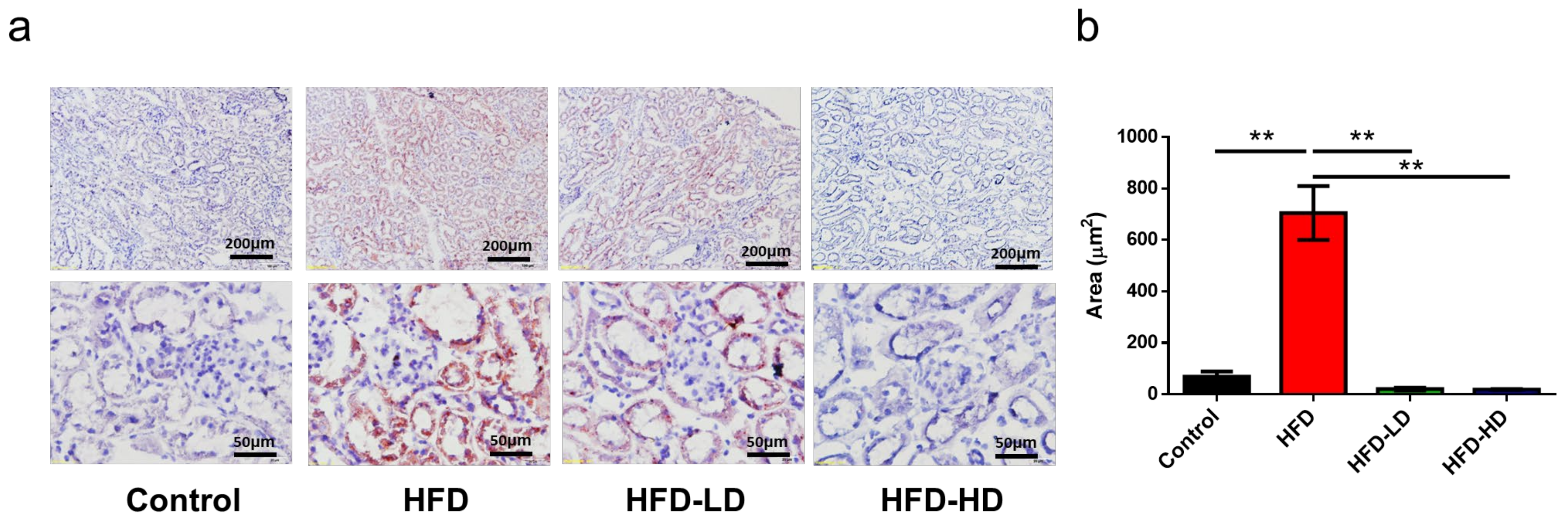
2.2. Empagliflozin Attenuated HFD-Induced Lipid Accumulation in Mouse Kidneys
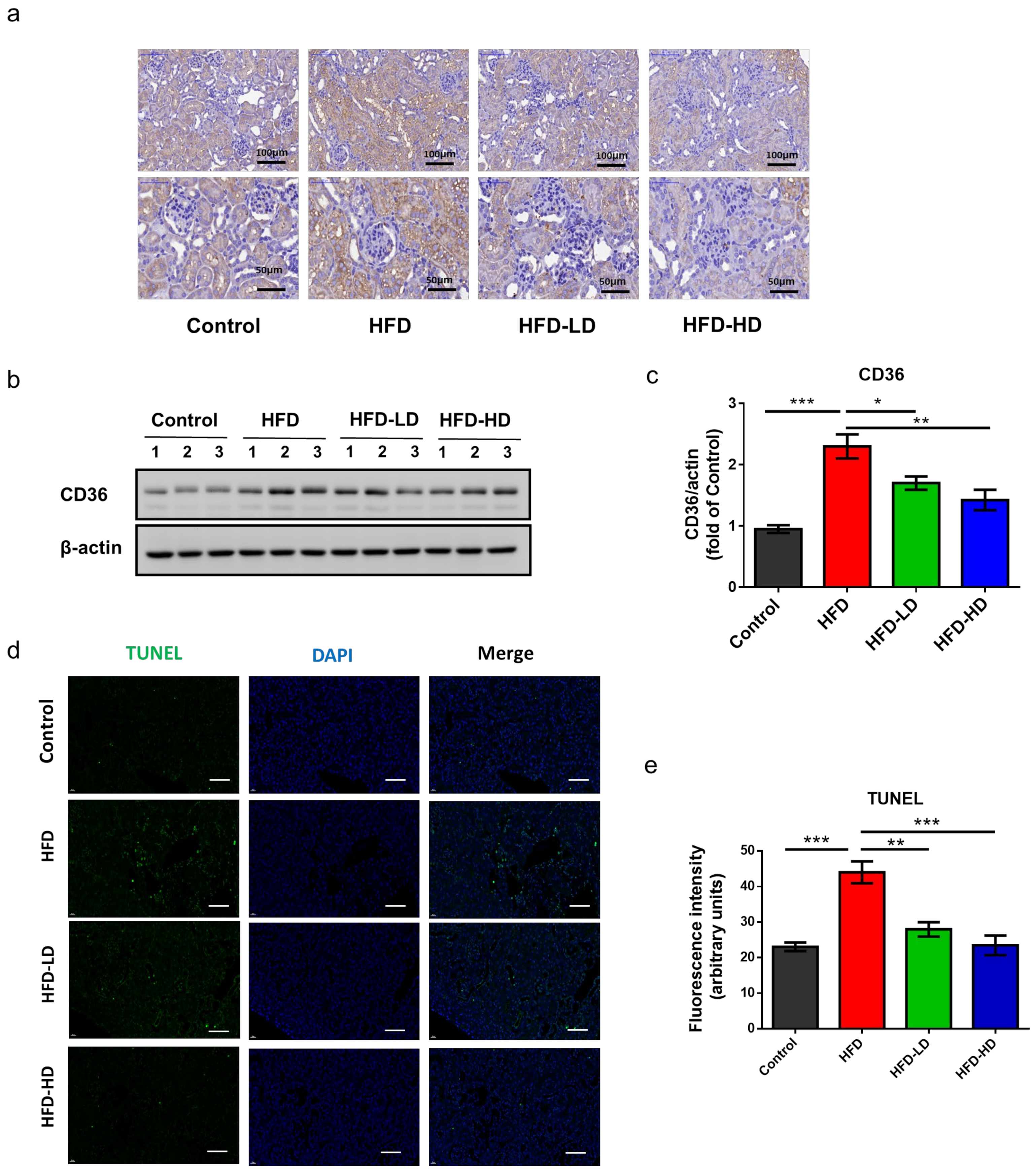
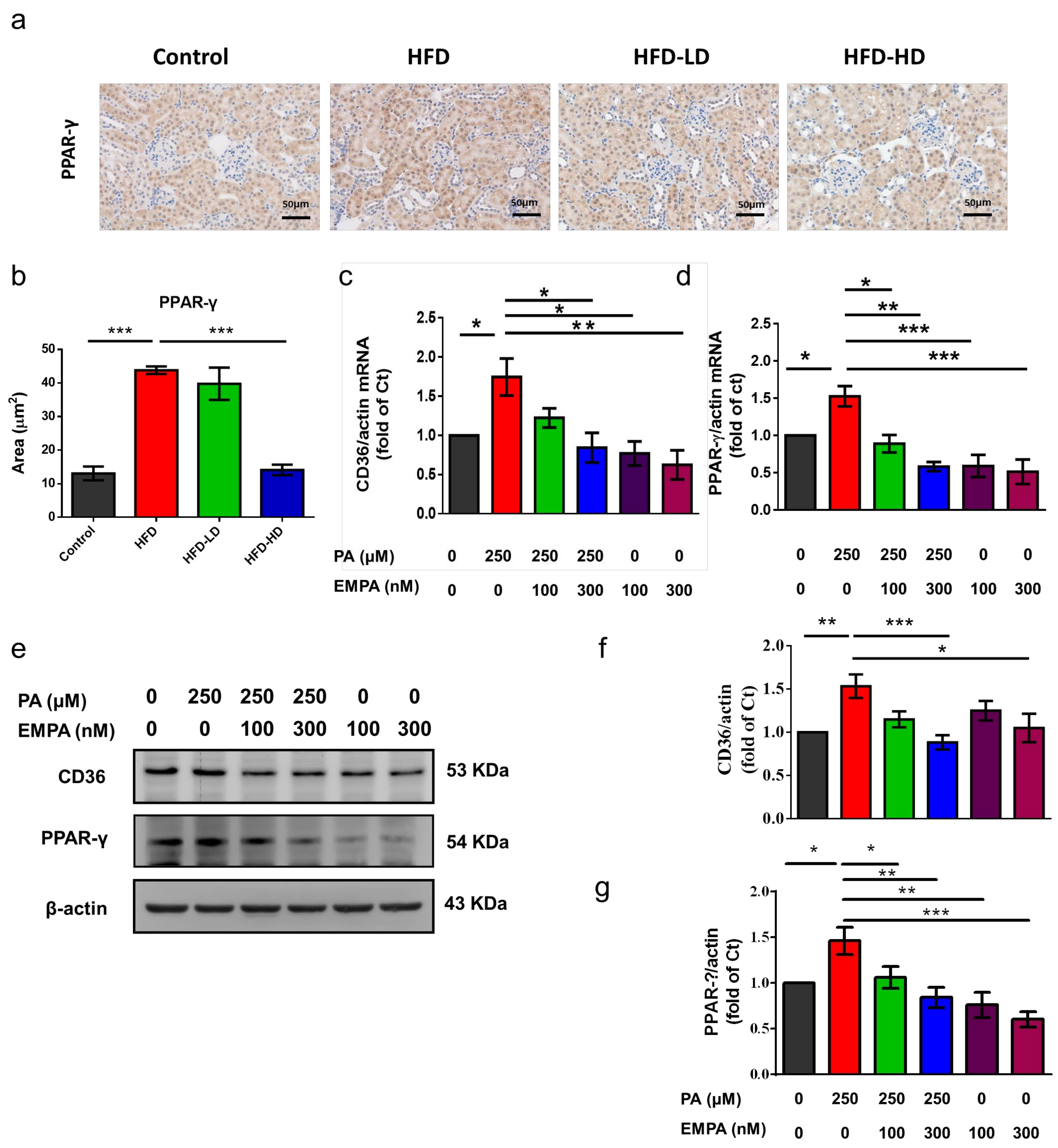
2.3. Empagliflozin Attenuated HFD-Induced CD36 Expression and Cell Apoptosis in Mouse Kidneys
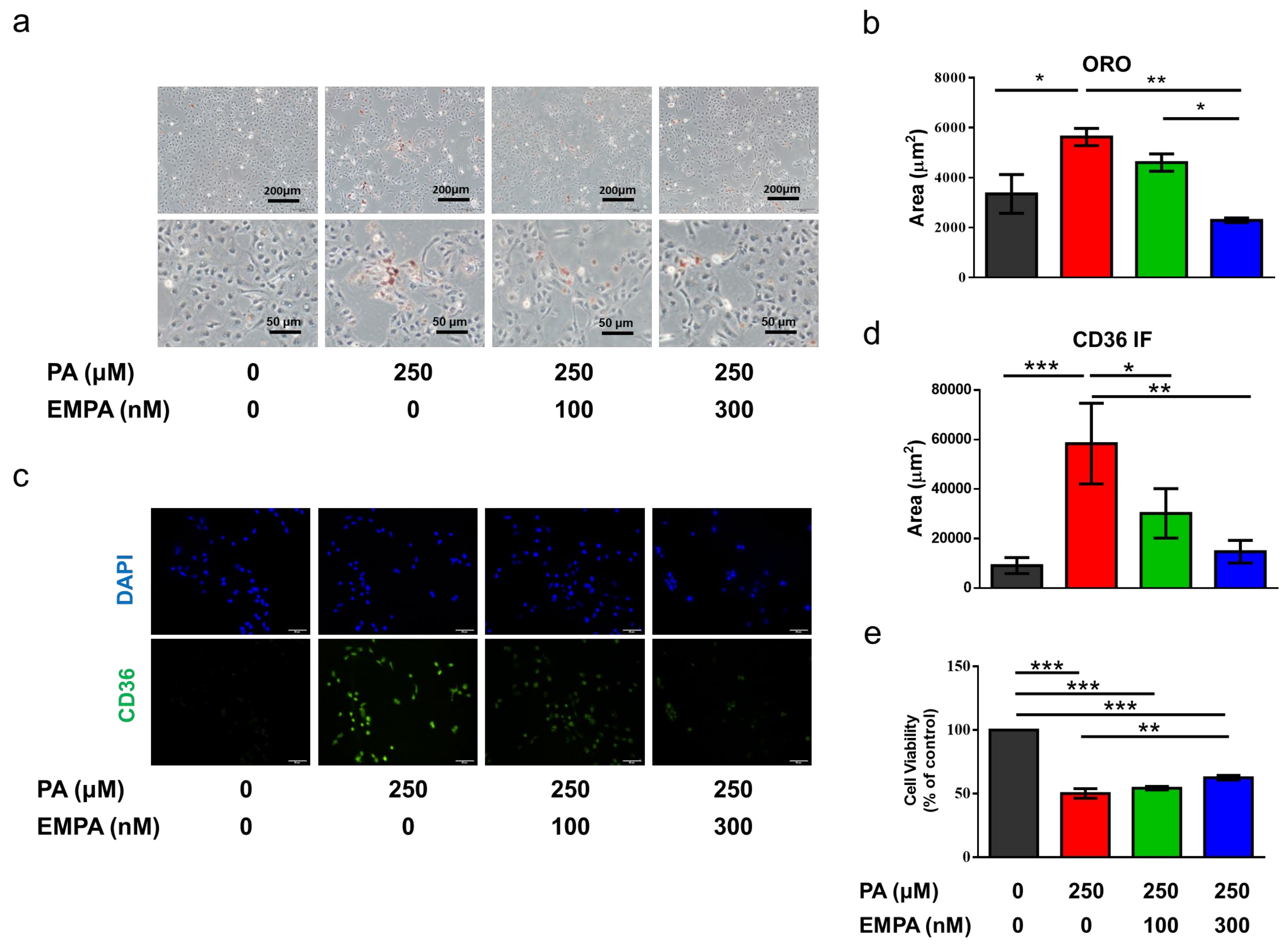
2.4. Empagliflozin Decreased Palmitic Acid-Induced CD36 Expression and Cell Death in a Proximal Tubular Cell Line (HK-2)
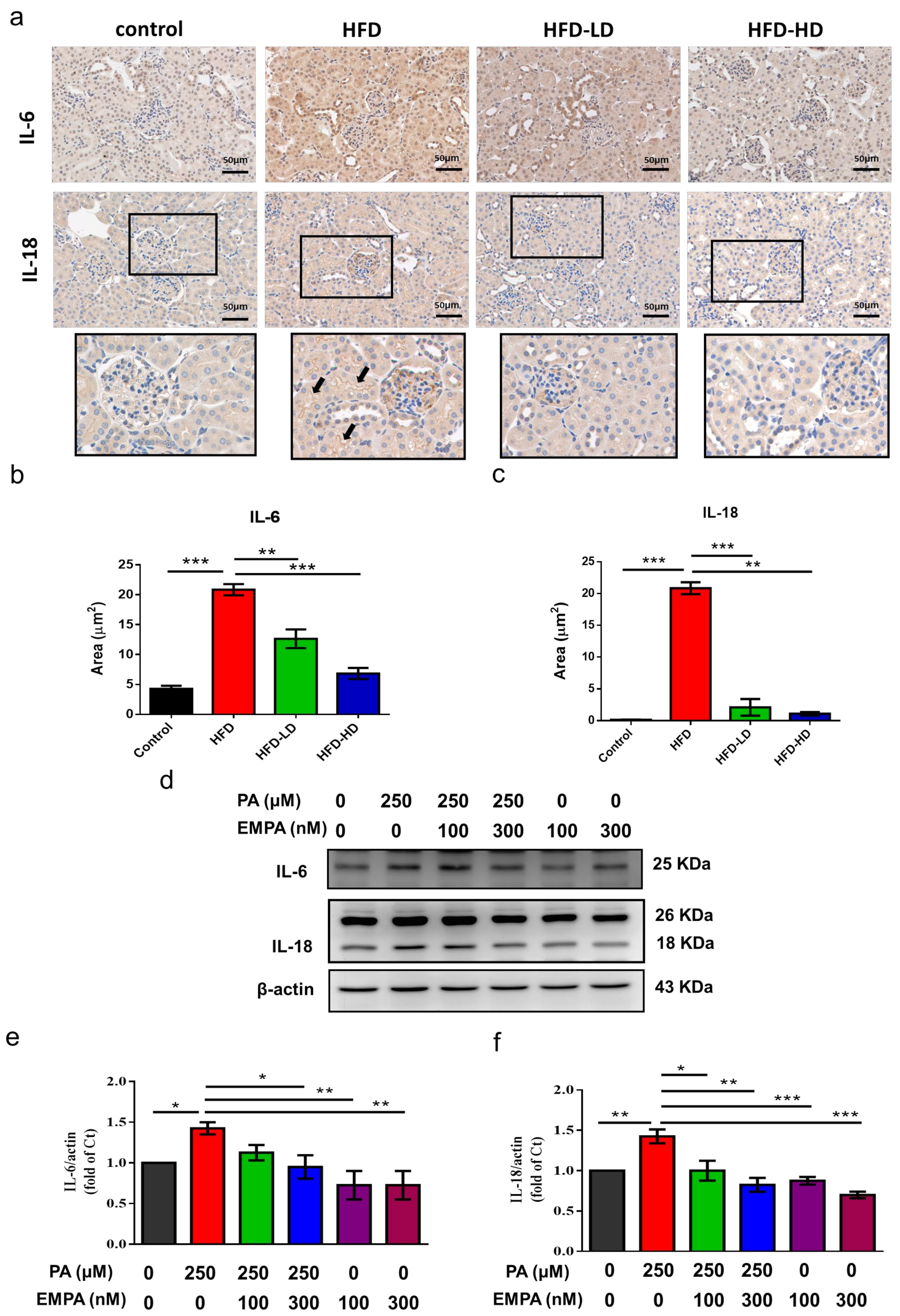
2.5. Empagliflozin Abolished HFD- and PA-Induced Inflammation in Proximal Tubular Cells
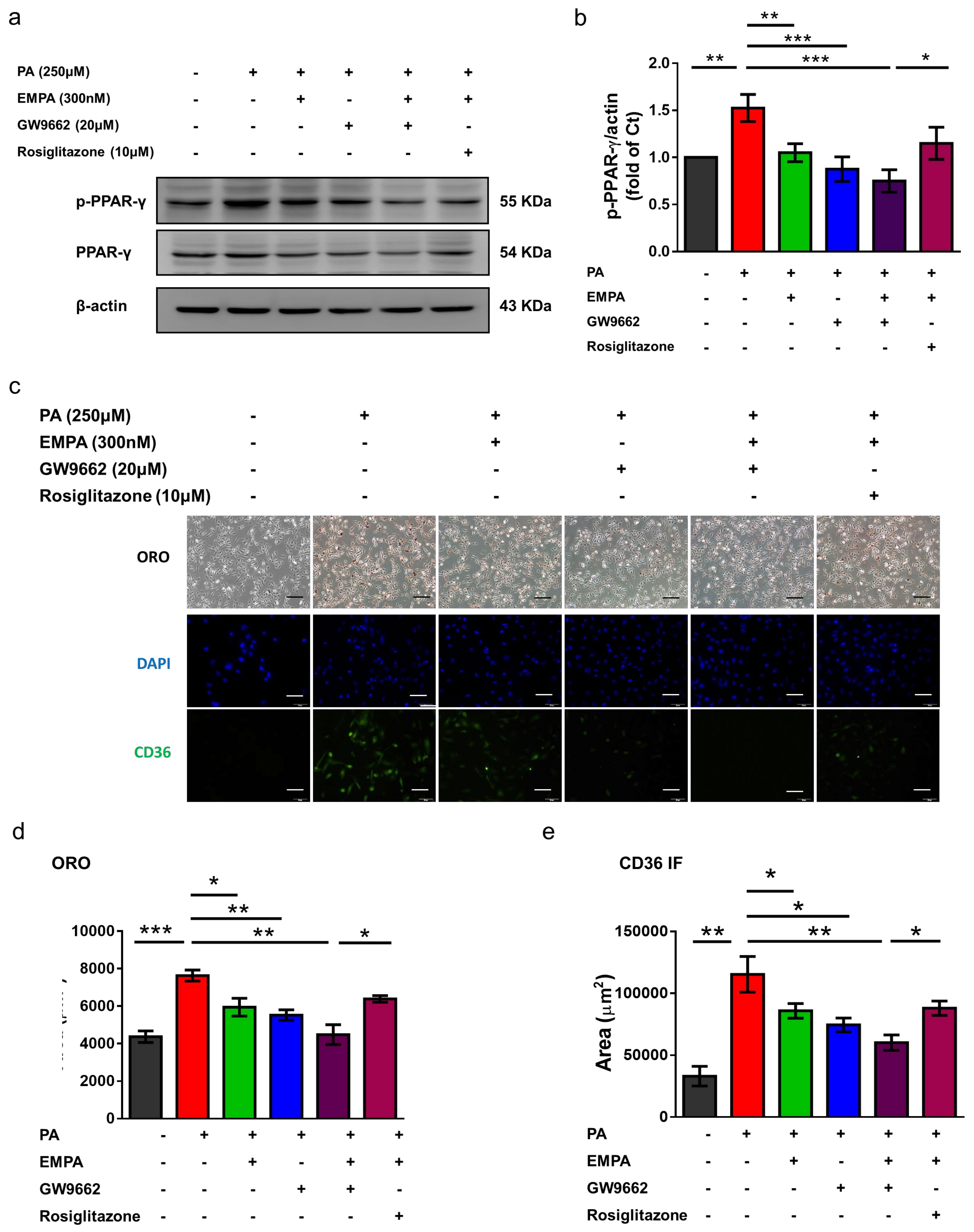
2.6. Empagliflozin Ameliorated Peroxisome Proliferator-Activated Receptor (PPAR)-γ Expression and Phosphorylation in HFD-Treated Kidneys and PA-Treated HK-2 Cells
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Cell Cultures
4.3. Cell Viability
4.4. Western Blotting
4.5. Immunohistochemistry and Immunofluorescent Staining
4.6. Quantitative Reverse-Transcription Polymerase Chain Reaction
4.7. Oil Red O Staining
4.8. Terminal Deoxynucleotide Transferase-Mediated dUTP Nick End Labeling Assay
4.9. Measurement of Metabolic Parameters
- (1)
- Fasting blood glucose levels were determined using a glucometer (Accu-Chek® Active, Roche, Mannheim, Germany) in a blood sample collected from the tail vein after 6 h of fasting.
- (2)
- IPGTT was performed at week 12 after fasting for 16 h. Glucose (2 g/kg BW) was intraperitoneally administered, and blood was collected from the tail tip of mice. Glucose concentration was measured using a glucometer, as described previously, at time point 0 (prior to glucose administration). In addition, blood glucose levels were assessed at 5, 15, 30, 60, 90, and 120 min after intraperitoneal injection.
- (3)
- For performing the IPITT, food was withdrawn for 4 h, and then mice were intraperitoneally administered insulin (0.75 U/kg) at time 0. Blood sugar was assessed at 15, 30, 60, 90, and 120 min after intraperitoneal injection. [47]
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wisse, B.E. The Inflammatory Syndrome: The Role of Adipose Tissue Cytokines in Metabolic Disorders Linked to Obesity. J. Am. Soc. Nephrol. 2004, 15, 2792–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praga, M.; Morales, E. Obesity, proteinuria and progression of renal failure. Curr. Opin. Nephrol. Hypertens. 2006, 15, 481–486. [Google Scholar] [CrossRef]
- Arner, P.; Rydén, M. Fatty Acids, Obesity and Insulin Resistance. Obes. Facts 2015, 8, 147–155. [Google Scholar] [CrossRef]
- Reaven, G.M.; Hollenbeck, C.; Jeng, C.-Y.; Wu, M.S.; Chen, Y.-D.I. Measurement of Plasma Glucose, Free Fatty Acid, Lactate, and Insulin for 24 h in Patients With NIDDM. Diabetes 1988, 37, 1020–1024. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, H.; Yanagita, M. Targeting the fatty acid transport protein CD36, a class B scavenger receptor, in the treatment of renal disease. Kidney Int. 2016, 89, 740–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mwaikambo, B.R.; Yang, C.; Chemtob, S.; Hardy, P. Hypoxia Up-regulates CD36 Expression and Function via Hypoxia-inducible Factor-1- and Phosphatidylinositol 3-Kinase-dependent Mechanisms. J. Biol. Chem. 2009, 284, 26695–26707. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.C.; Luiken, J.J.F.P. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Iwao, Y.; Nakajou, K.; Nagai, R.; Kitamura, K.; Anraku, M.; Maruyama, T.; Otagiri, M. CD36 is one of important receptors promoting renal tubular injury by advanced oxidation protein products. Am. J. Physiol. Physiol. 2008, 295, F1871–F1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubhayasekera, S.J.K.A.; Staaf, J.; Forslund, A.; Bergsten, P.; Bergquist, J. Free fatty acid determination in plasma by GC-MS after conversion to Weinreb amides. Anal. Bioanal. Chem. 2013, 405, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Ding, G.; Zhao, M.; Bai, M.; Yang, L.; Ni, J.; Wang, R.; Jia, Z.; Huang, S.; Zhang, A. NLRP3 Inflammasome Mediates Albumin-induced Renal Tubular Injury through Impaired Mitochondrial Function. J. Biol. Chem. 2014, 289, 25101–25111. [Google Scholar] [CrossRef] [Green Version]
- Baines, R.J.; Chana, R.S.; Hall, M.; Febbraio, M.; Kennedy, D.; Brunskill, N.J. CD36 mediates proximal tubular binding and uptake of albumin and is upregulated in proteinuric nephropathies. Am. J. Physiol. Physiol. 2012, 303, F1006–F1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, H.-J.; Muruve, D.A. The Inflammasomes in Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 1007–1018. [Google Scholar] [CrossRef]
- Susztak, K.; Ciccone, E.; McCue, P.; Sharma, K.; Böttinger, E.P. Multiple Metabolic Hits Converge on CD36 as Novel Mediator of Tubular Epithelial Apoptosis in Diabetic Nephropathy. PLoS Med. 2005, 2, e45. [Google Scholar] [CrossRef] [Green Version]
- Son, N.-H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.-Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinol. 2016, 157, 570–585. [Google Scholar] [CrossRef] [Green Version]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.-H.; Lambers-Heerspink, H.; De Zeeuw, D. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1799–1802. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.; McMurray, J.J.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Luo, Y.; Wang, X.; Orlicky, D.J.; Myakala, K.; Yang, P.; Levi, M. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Renal and Liver Disease in Western Diet Induced Obesity Mice. Int. J. Mol. Sci. 2018, 19, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef]
- Sato, O.; Kuriki, C.; Fukui, Y.; Motojima, K. Dual Promoter Structure of Mouse and Human Fatty Acid Translocase/CD36 Genes and Unique Transcriptional Activation by Peroxisome Proliferator-activated Receptor α and γ Ligands. J. Biol. Chem. 2002, 277, 15703–15711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef] [Green Version]
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2006, 26, 968–976. [Google Scholar] [CrossRef]
- Bolinder, J.; Ljunggren, Ö.; Kullberg, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sugg, J.; Parikh, S. Effects of Dapagliflozin on Body Weight, Total Fat Mass, and Regional Adipose Tissue Distribution in Patients with Type 2 Diabetes Mellitus with Inadequate Glycemic Control on Metformin. J. Clin. Endocrinol. Metab. 2012, 97, 1020–1031. [Google Scholar] [CrossRef] [Green Version]
- Bolinder, J.; Ljunggren, Ö.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sjöström, C.D.; Sugg, J.; Parikh, S. Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes. Metab. 2014, 16, 159–169. [Google Scholar] [CrossRef]
- Monami, M.; Nardini, C.; Mannucci, E. Efficacy and safety of sodium glucose co-transport-2 inhibitors in type 2 diabetes: A meta-analysis of randomized clinical trials. Diabetes Obes. Metab. 2014, 16, 457–466. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Maione, M.; Lai, V.; Lee, A.; Fagan, N.M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; et al. Renal Hemodynamic Effect of Sodium-Glucose Cotransporter 2 Inhibition in Patients With Type 1 Diabetes Mellitus. Circulation 2014, 129, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Ruan, X.Z.; Varghese, Z.; Moorhead, J.F. An update on the lipid nephrotoxicity hypothesis. Nat. Rev. Nephrol. 2009, 5, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, K.; Takata, T.; Sugihara, T.; Matono, T.; Koda, M.; Kanda, T.; Taniguchi, S.; Ida, A.; Mae, Y.; Yamamoto, M.; et al. Ipragliflozin Ameliorates Endoplasmic Reticulum Stress and Apoptosis through Preventing Ectopic Lipid Deposition in Renal Tubules. Int. J. Mol. Sci. 2019, 21, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahl, S.; Gancheva, S.; Straßburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 2019, 43, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.-C.; Yang, J.-L.; Lee, W.-C.; Chen, J.-B.; Lee, C.-T.; Wang, P.-W.; Vaghese, Z.; Chen, W.-Y. Palmitate aggravates proteinuria-induced cell death and inflammation via CD36-inflammasome axis in the proximal tubular cells of obese mice. Am. J. Physiol. Physiol. 2018, 315, F1720–F1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, W.; Huang, H.-Z.; Tan, L.-T.; Wan, J.-M.; Gui, H.-B.; Zhao, L.; Ruan, X.-Z.; Chen, X.-M.; Du, X.-G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.C.; Leung, J.C.; Lai, K.N. Diabetic Tubulopathy: An Emerging Entity. Nephrol. Public Health Worldw. 2011, 170, 124–134. [Google Scholar] [CrossRef]
- Miyauchi, K.; Takiyama, Y.; Honjyo, J.; Tateno, M.; Haneda, M. Upregulated IL-18 expression in type 2 diabetic subjects with nephropathy: TGF-β1 enhanced IL-18 expression in human renal proximal tubular epithelial cells. Diabetes Res. Clin. Pr. 2009, 83, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Shikata, K.; Hiramatsu, M.; Nakatou, T.; Kitamura, T.; Wada, J.; Itoshima, T.; Makino, H. Serum Interleukin-18 Levels Are Associated With Nephropathy and Atherosclerosis in Japanese Patients With Type 2 Diabetes. Diabetes Care 2005, 28, 2890–2895. [Google Scholar] [CrossRef] [Green Version]
- Bhat, O.M.; Kumar, P.U.; Giridharan, N.; Kaul, D.; Kumar, M.M.; Dhawan, V. Interleukin-18-induced atherosclerosis involves CD36 and NF-κB crosstalk in Apo E−/− mice. J. Cardiol. 2015, 66, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Brasier, A.R. The nuclear factor- B-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Jaikumkao, K.; Pongchaidecha, A.; Chueakula, N.; Thongnak, L.; Wanchai, K.; Chatsudthipong, V.; Chattipakorn, N.; Lungkaphin, A. Dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, slows the progression of renal complications through the suppression of renal inflammation, endoplasmic reticulum stress and apoptosis in prediabetic rats. Diabetes Obes. Metab. 2018, 20, 2617–2626. [Google Scholar] [CrossRef]
- Jedidi, I.; Couturier, M.; Thérond, P.; Gardès-Albert, M.; Legrand, A.; Barouki, R.; Bonnefont-Rousselot, D.; Aggerbeck, M. Cholesteryl ester hydroperoxides increase macrophage CD36 gene expression via PPARα. Biochem. Biophys. Res. Commun. 2006, 351, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, X.Z.; Powis, S.H.; Fernando, R.; Mon, W.Y.; Wheeler, D.C.; Moorhead, J.F.; Varghese, Z. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: Evidence for a PPAR-γ–dependent mechanism. Kidney Int. 2005, 67, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.-J.; Lee, S.; Lee, K.-S.; Park, J.-H.; Jang, Y.; Lee, E.J.; Park, H.-Y. PPARγ activation induces CD36 expression and stimulates foam cell like changes in rVSMCs. Prostagland. Lipid Mediat. 2006, 80, 165–174. [Google Scholar] [CrossRef]
- Khan, S.; Cabral, P.D.; Schilling, W.P.; Schmidt, Z.W.; Uddin, A.N.; Gingras, A.; Madhavan, S.M.; Garvin, J.L.; Schelling, J.R. Kidney Proximal Tubule Lipoapoptosis Is Regulated by Fatty Acid Transporter-2 (FATP2). J. Am. Soc. Nephrol. 2017, 29, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.-W.; Kuo, H.-M.; Huang, H.-T.; Chang, A.Y.W.; Weng, S.-W.; Tai, M.-H.; Chuang, J.-H.; Chen, I.-Y.; Huang, S.-C.; Lin, T.-K.; et al. Biphasic Response of Mitochondrial Biogenesis to Oxidative Stress in Visceral Fat of Diet-Induced Obesity Mice. Antioxidants Redox Signal. 2014, 20, 2572–2588. [Google Scholar] [CrossRef]
- Mehlem, A.; Hagberg, C.; Muhl, L.; Eriksson, U.; Falkevall, A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 2013, 8, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-C.; Chou, C.-A.; Chen, W.-Y.; Yang, J.-L.; Lee, W.-C.; Chen, J.-B.; Lee, C.-T.; Li, L.-C. Empagliflozin Ameliorates Free Fatty Acid Induced-Lipotoxicity in Renal Proximal Tubular Cells via the PPARγ/CD36 Pathway in Obese Mice. Int. J. Mol. Sci. 2021, 22, 12408. https://doi.org/10.3390/ijms222212408
Huang C-C, Chou C-A, Chen W-Y, Yang J-L, Lee W-C, Chen J-B, Lee C-T, Li L-C. Empagliflozin Ameliorates Free Fatty Acid Induced-Lipotoxicity in Renal Proximal Tubular Cells via the PPARγ/CD36 Pathway in Obese Mice. International Journal of Molecular Sciences. 2021; 22(22):12408. https://doi.org/10.3390/ijms222212408
Chicago/Turabian StyleHuang, Chiang-Chi, Chia-An Chou, Wei-Yu Chen, Jenq-Lin Yang, Wen-Chin Lee, Jin-Bor Chen, Chien-Te Lee, and Lung-Chih Li. 2021. "Empagliflozin Ameliorates Free Fatty Acid Induced-Lipotoxicity in Renal Proximal Tubular Cells via the PPARγ/CD36 Pathway in Obese Mice" International Journal of Molecular Sciences 22, no. 22: 12408. https://doi.org/10.3390/ijms222212408