
A Multi-Perspective Proximity View on the Dynamic Head Region of the Ribosomal 40S Subunit

Department of Molecular Microbiology and Genetics, Institute of Microbiology and Genetics, Göttingen Center for Molecular Biosciences (GZMB), Georg-August-University Göttingen, 37077 Göttingen, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(21), 11653; https://doi.org/10.3390/ijms222111653
Submission received: 12 August 2021
/
Revised: 23 October 2021
/
Accepted: 25 October 2021
/
Published: 28 October 2021
(This article belongs to the Special Issue From Affinity to Proximity to Interaction: Mass Spectrometry Methods to Study Biomolecular Complexes)
Abstract
:A comparison of overlapping proximity captures at the head region of the ribosomal 40S subunit (hr40S) in Saccharomyces cerevisiae from four adjacent perspectives, namely Asc1/RACK1, Rps2/uS5, Rps3/uS3, and Rps20/uS10, corroborates dynamic co-localization of proteins that control activity and fate of both ribosomes and mRNA. Co-locating factors that associate with the hr40S are involved in (i) (de)ubiquitination of ribosomal proteins (Hel2, Bre5-Ubp3), (ii) clamping of inactive ribosomal subunits (Stm1), (iii) mRNA surveillance and vesicular transport (Smy2, Syh1), (iv) degradation of mRNA (endo- and exonucleases Ypl199c and Xrn1, respectively), (v) autophagy (Psp2, Vps30, Ykt6), and (vi) kinase signaling (Ste20). Additionally, they must be harmonized with translation initiation factors (eIF3, cap-binding protein Cdc33, eIF2A) and mRNA-binding/ribosome-charging proteins (Scp160, Sro9). The Rps/uS-BioID perspectives revealed substantial Asc1/RACK1-dependent hr40S configuration indicating a function of the β-propeller in context-specific spatial organization of this microenvironment. Toward resolving context-specific constellations, a Split-TurboID analysis emphasized the ubiquitin-associated factors Def1 and Lsm12 as neighbors of Bre5 at hr40S. These shuttling proteins indicate a common regulatory axis for the fate of polymerizing machineries for the biosynthesis of proteins in the cytoplasm and RNA/DNA in the nucleus.
1. Introduction
Comprehension of the cellular integration of ribosomes requires an understanding of dynamic interactions between ribosome and non-ribosomal components as well as between two proximal ribosomes (e.g., collided or aggregated ones). These interactions may be temporary and dynamic in regard to specific cellular contexts. An exposed ribosomal contact point is the head region of the 40S ribosomal subunit (hr40S). In recent years, different proximity labeling techniques have emerged and are continuously being optimized, which are well suited for the identification of non-ribosomal and ribosomal components entering within short distance of this site [1]. The biotin IDentification (BioID) approach makes use of a promiscuous biotin ligase that is fused to a protein of interest and causes in vivo labeling of proximal proteins with biotin [2]. Biotinylated proteins are enriched from cell lysates using affinity matrices such as Streptavidin or Strep-Tactin® and can be identified using targeted validation like Western blot experiments or unbiased approaches like liquid chromatography-mass spectrometry (LC-MS). This methodology is especially convenient for studying dynamic contact sites of large protein or ribonucleoprotein complexes. For instance, factors involved in the supply or regulation of biosynthetic machineries associate context specifically to these sites, and due to their weak and/or transient binding, are generally difficult to identify with pull-down experiments. The BioID methodology does not rely on sustained protein binding within cell lysates, but rather on cellular proximity during culture growth. In previous work, we have established a quantitative BioID approach with stable isotope labeling with amino acids in cell culture (SILAC-BioID) to start identifying proteins that co-localize with the WD40-repeat and β-propeller protein Asc1 in S. cerevisiae, the conserved homolog of RACK1 in higher eukaryotes [3]. Asc1/RACK1 resides at the hr40S close to the mRNA exit tunnel and is linked to the mRNA entry channel via binding to the extended C-terminal arm of Rps3 [4]. Asc1/RACK1 is exposed to the cytoplasm and faces regulators of the initiation of mRNA translation and signal transduction components [3,5,6,7]. It provides resistance against and viability in challenging growth conditions such as nutrient depletion, osmotic stress, temperature shock or cell wall burden and is required for multicellular development in higher fungi [8,9,10]. In metazoa, RACK1 is crucial for embryogenesis [11,12], and aberrant expression levels of RACK1 are associated with poor clinical outcome of different types of cancers [13]. RACK1 is involved in cellular signaling and contributes to fundamental cellular processes such as cell proliferation, cell migration, apoptosis, and angiogenesis [13].
We previously identified proteins that had not been known for being close to ribosomes, specifically at the hr40S, such as Mbf1 and Def1, the E3-ubiquitin RING ligase Hel2, and the deubiquitination complex Bre5-Ubp3 [3]. In the meantime, it has been shown that Mbf1, in concert with Rps3 and Asc1, prevents ribosomal frameshifting at inhibitory CGA–CGA codon pairs [14]. Moreover, Mbf1 and its mammalian homolog EDF1 are present at colliding ribosomes near the mRNA entry channel upon stall-inducing conditions [15]. Def1 is involved in the degradation of stalled RNA polymerase II (RNAPII) within the nucleus [16]. Co-purification experiments showed that Def1 and RNAPII also interact with Bre5-Ubp3, and that Ubp3 mediates deubiquitination of RNAPII [17].
Since Asc1 is considered a scaffold protein that organizes protein microenvironments and might also appear in a ribosome-unbound state, we wanted to (i) validate Asc1-proximity of proteins in the hr40S microenvironment and (ii) study the impact of Asc1-depletion on their association with the hr40S, and with that on the integrity of this ribosomal microenvironment. Toward this end, we recently performed a BioID experiment from a slightly shifted perspective using Rps2/uS5-BirA* instead of Asc1-BirA* [18]. This confirmed, e.g., the ribosomal localization of Def1 and also indicated Asc1-dependence of Bre5-Ubp3 association with the hr40S [18]. Here, we consolidated the overall picture with two further ribosomal perspectives at the hr40S, namely that of Rps3/uS3 and Rps20/uS10 (Figure 1A). We established a new control for the hr40S BioID experiments, namely Rpl5/uL18-BirA*. Rpl5 is a protein of the 60S ribosomal subunit that on the one hand is part of the common machinery, but on the other hand is located at a distal site within the complex. We therefore call it a distal control.
Overall, our multi-perspective proximity analysis describes a range of proteins occurring at the head region close to the center of initiation of translation. Several factors have an impact on ubiquitin-mediated ribosome disposition linked to cellular signaling. The multitude of ribosomes within one cell and the high number of cells taken for a BioID experiment, lead to a cross-section of identified proteins that localize within this dynamic microenvironment with context-specific hr40S constellations. For the dissection of different protein constellations in ongoing studies, we established here the Split-TurboID approach for the first time for S. cerevisiae and identified proteins that are specific for an hr40S constellation that contains Bre5.
2. Results
2.1. Same Complex but Distal Site: A New Reference Point for hr40S SILAC-BioID Experiments
Appropriate controls serving as relative quantification reference points have to be chosen for the faithful identification of co-localizing proteins with proximity label/MS. With that, false positive evaluation of naturally biotinylated proteins or proteins labeled by stray activity, e.g., of an untethered BirA*-fusion protein moiety, can be avoided. In previous SILAC-BioID experiments, we used a wild-type yeast strain as well as a strain expressing a free BirA* ligase as negative controls. However, the latter one can cause substantial background biotinylation that might mask true neighbors of the bait-BirA* fusion protein [18]. This can be especially detrimental when the cellular abundance of the BirA*-bait protein is significantly less than that of free BirA*. In this case, the proximity-dependent biotinylation through the bait-BirA* fusion protein can be less than the unspecific background biotinylation through free BirA*. Alternatively, additional controls can be established with BirA* fused to proteins that are (i) expected to be functionally and regionally unrelated to the bait protein or (ii) members of the same complex as the bait protein but localizing apart at a distal site. Therefore, we tested BirA* fusions to the C-termini of the following proteins: (i) fructose 1,6-bisphosphate aldolase Fba1, an enzyme involved in glycolysis and gluconeogenesis, and (ii) Rpl5/uL18 and Rpl25/uL23, two proteins of the large 60S ribosomal subunit (Figure 1A). These two ribosomal proteins (RPs) appeared promising since comparable GFP fusion chimera have earlier been proven functional and applicable with respect to the baits’ localization and function [19]. The bait-BirA* fusion proteins were expressed in yeast cells bearing the respective endogenous wild-type alleles of the bait proteins. The abundance ratios of BirA*-fused and endogenous RPs were representatively analyzed for Rps3 with Western blot experiments (Figure 1B). The levels of endogenous untagged Rps3 decreased relative to the additional expression of plasmid-borne Rps3-BirA*, attributable to the excess ribosomal protein quality control (ERISQ) [20]. The expression of the RP-BirA* fusion proteins does not compromise general growth on standard yeast minimal medium neither in the presence nor in the absence of additional biotin. Wild-type-like resistance against the mRNA translation inhibitor cycloheximide also verified natural ribosome resilience when RP-BirA* fusion proteins were expressed (Figure 1C). Asc1-deficient asc1− cells became only marginally more sensitive to cycloheximide when they were expressing the RP-BirA* fusion proteins. We tested for the expression of the RP-BirA* fusion proteins and their biotinylation activities (Figure 1D). Stable expression of all BirA* fusion proteins was confirmed with Western blot experiments using a BirA-specific antibody. Depletion of Asc1 neither affected the abundance of the fusion proteins nor their overall biotinylation activity. As mentioned before, a high dosage of biotin causes strong overall protein biotinylation for cells expressing free BirA* (Figure 1E, [3,18]). The biotinylation activity of Fba1-BirA* was comparably strong to that of free BirA*, probably due to the high cellular protein abundance of Fba1 (Figure 1D,E, [21]). By contrast, only a slight increase in biotinylation activity was observed for Rpl25-BirA*. For Rps3-BirA* and Rpl5-BirA*, a substantial and similar increase in overall protein biotinylation was observed. Since biotin labeling by these two RP-BirA* fusion proteins seemed to be most similar among the tested constructs, Rpl5-BirA* was chosen as a distal reference control for SILAC quantification in further proximity mappings at the hr40S and is subsequently referred to as distal control.
2.2. Proximity Labeling with Biotin at the hr40S from the Rps3 Perspective
For an Rps3-BirA* SILAC-BioID experiment, an RPS3-birA* fusion gene-bearing strain was grown with light and an RPL5-birA*-bearing strain (the newly established distal control) with medium isotope-labeled SILAC amino acids. Additionally, a heavy isotope-labeled asc1− strain bearing RPS3-birA* was included to analyze the Asc1-dependence of the captured Rps3 microenvironment (Figure 2A). After separate culture growth, similar numbers of cells of the three cultures were pooled and lysed in one batch under denaturing conditions (4% SDS). Aliquots were taken from the separate cultures for cell lysis to confirm stable expression and biotinylation activity of bait-BirA* fusion proteins by Western blot experiments (Figure 2B). Biotinylated proteins from the lysate of the cell pool were enriched with Strep-Tactin® gravity flow columns, digested with trypsin, and the resulting peptides were analyzed by LC-MS (Figure 2A). Three independent biological replicates were analyzed, and the MS data were searched against an S. cerevisiae-specific protein database (derived from UniProt) for peptide/protein identification and relative quantification by using the MaxQuant software [22]. The quantitative search result data were further analyzed with the Perseus software [23] according to the workflow summarized in Table S1. SILAC ratios were logarithmized and proteins with valid enrichment quantification values for all three replicates were further considered (360 proteins). The BioID input samples (total proteomes) were additionally analyzed with LC-MS to account for possible expression variances within the different yeast strains. The BioID-enrichment ratios were then normalized to these proteome ratios, leading to proteome-corrected BioID-enrichment values. Based on low cellular expression, some proteins with BioID-enrichment data were not identified/quantified from the proteome input samples. For these proteins, data analysis proceeded on the basis of the enrichment ratios only. A one-sample t-test was performed for the proteome-corrected enrichment ratios of the ASC1 + RPS3-birA*/ASC1 + RPL5-birA* comparison with a p-value threshold of 0.05. Additionally, proteins were filtered for at least 1.5-fold enrichment (log2 SILAC ratios ≥ 0.585). A total of 42 proteins passed these thresholds and were thus considered as occurring proximal to Rps3 (Figure 3A, highlighted in green in the zoomed in box; Table S2).
Among these proteins were seven ribosomal proteins (Rps1a/b, Rps2, Rps17a/b, Rps20, Rps26a/b, Rpl8a/b, and Rpl24a), the ribosome clamping factor Stm1, translation initiation factors (Cdc33, Tif1, Tif3, Tif35, eIF2A), the poly(A)-binding protein Pab1, the Pab1-binding protein Pbp1, mRNA-binding proteins (Sro9, Scp160, and Slf1), and the Rps3-chaperone Yar1, indicating that the native Rps3 microenvironment was captured. Other proteins identified from previous BioID experiments with Asc1-BirA* and Rps2-BirA* recurred with Rps3-BirA* here, namely, Def1, Lsm12, Mbf1, and the two paralogs Smy2 and Syh1, and Vps30 [3,18]. Vps30, a subunit of the phosphatidylinositol 3-kinase (PI3K) complexes I and II, was identified in the Rps2-BirA* microenvironment only in the absence of Asc1 [18]. However, here we observed Vps30 proximal to Rps3-BirA* also in the presence of Asc1. The PI3K complexes I and II are involved in autophagosome formation and vacuolar protein sorting [24].
Also, Psp2, a protein that has recently been described to be involved in autophagy regulation in yeast, was identified within the Rps3-BirA* microenvironment. Upon nitrogen starvation, Psp2 binds 5′UTRs of mRNAs encoding the autophagy factors Atg1 and Atg13, and by this promotes the initiation of their translation through interaction with components of the eIF4F complex [25]. Further, the R-SNARE Ykt6 required for autophagosome-vacuole fusion [26,27] and the small GTPase Arl1 involved in autophagosome formation and fusion with the vacuole [28] were found proximal to Rps3-BirA*. Some further captured proteins are involved in signal transduction, e.g., the upstream mitogen-activated protein 4 kinase (MAP4K) Ste20 and the GTP-binding protein Ras2.
Beyond that, we also identified 274 proteins with only two out of three valid quantification values. Among them were an additional 19 proteins with at least 1.5-fold enrichment from the Rps3-BirA* samples (Figure 3A; Table S3). These were, e.g., the ubiquitin ligase Hel2, the deubiquitinase Ubp3 and its co-factor Bre5, and the translation initiation factor eIF3a (Rpg1) that have also been found proximal to the hr40S in our previous BioID experiments [3,18]. Ypl199c is another protein of this group. The Smr domain containing proteins Ypl199c and Cue2 are homologs of the Caenorhabditis elegans endonuclease NONU-1, and they have recently been shown to redundantly support nonstop mRNA decay [29]. To our knowledge, this is the first study indicating the localization of the putative endonuclease Ypl199c to the hr40S in S. cerevisiae.
2.3. The Asc1-Dependent Microenvironment of Rps3-BirA*
To reveal Asc1-dependent differences at the hr40S, we then quantitatively compared the Rps3-BirA* proximal proteins in the presence and absence of Asc1 by using ASC1 wild-type and asc1− strains (Figure 3B). Again, proteins with either three or two enrichment values were taken into account, and an averaged log2 difference of ≥ 0.585 or ≤ −0.585 was set as a threshold for Asc1-dependent Rps3-BirA* proximity (Table S1). A total of 19 proteins showed decreased and ten proteins showed increased capture in the absence of Asc1 (Figure 3B; Tables S4 and S5). These data confirmed a previously observed Asc1-dependence in the hr40S localization of Bre5-Ubp3 [18], a complex known to deubiquitinate Rps3 [30]. In addition, the E3-ubiquitin RING ligase Hel2 that ubiquitinates Rps3 at collided ribosomes and the putative endonuclease Ypl199c were less captured from the asc1− strain. Reduced capture of the aforementioned autophagy factors Psp2, Arl1, and Ykt6 might indicate an impact of Asc1 on autophagy-related processes.
2.4. Another Step Ahead: The hr40S Proxiome from the Rps20-BirA* Perspective
For the results described thus far, Rpl5-BirA* served as a good control, so we held on to it for another SILAC-BioID experiment at the hr40S with Rps20/uS10-BirA* (Figure 1A). We observed stable expression of Rps20-BirA* and no detrimental effects on colony growth and translational stress tolerance (Figure 1C and Figure 2B). The workflow of the SILAC-BioID experiment and the data analysis was essentially identical to that described before (Figure 2A and Table S1; for minor variations see materials and methods). A total of 401 proteins with quantification values from all three replicates were identified and a further 338 proteins with two quantification values. Regarding the proteins with three quantification values, 14 of them were specifically enriched with Rps20-BirA*, including the bait protein itself (≥1.5-fold enrichment, p-value threshold of 0.05; Figure 4A; Table S6). Eight additional proteins appeared that had only two enrichment ratio values, but at least 1.5-fold enrichment (Table S7). Seven of all these proteins were known from previous SILAC-BioID experiments as being proximal to the hr40S. Furthermore, the known Rps20 co-localizing [NU+] prion formation protein New1 was identified. New1 is very similar to the translation elongation factor eEF3 with regard to its domain architecture as well as its binding to the 80S ribosome [31]. New1 is important for translation termination and/or ribosome recycling at stop codons that are preceded by lysine and arginine codons [31].
Thioredoxin peroxidase Tsa1 was another protein within the proximity of Rps20-BirA*. Tsa1 has a described, but thus far unspecified, ribosomal localization and appears to serve as an antioxidant for translating ribosomes [32]. The enzyme Spe3 which catalyzes the synthesis of the polyamine spermidine was also identified. Polyamines bind nucleic acids and promote the efficiency and fidelity of translation [33]. Spermidine is a substrate for the essential hypusine modification of eIF5A, a translation factor required for the synthesis of proteins containing polyproline sequences and for the termination of translation [33]. The absence of Asc1 only caused minor changes within the Rps20-BirA* microenvironment: Sro9, Lsm12, and Set5 showed increased capture (Figure 4B; Tables S8 and S9) and for Set5, no proteome values were obtained for the input normalization, indicating that the enrichment could also have been due to increased cellular protein abundance in the absence of Asc1.
2.5. Toward the Bre5-Specific hr40S Microenvironment with Split-TurboID
Thus far, our BioID survey has provided a cross section of a dynamic hr40S microenvironment where the ribosomal proteins Asc1, Rps2, Rps3, and Rps20 encounter a variety of non-ribosomal proteins, many of them putatively present there within a specific ribosomal context. Toward resolving context-specific protein proximities at the hr40S, we extended our analysis to Split-BioID experiments [34,35,36]. For a Split-BioID experiment, a biotin ligase is split into two catalytically inactive halves that are genetically fused to two bait proteins (Figure 5A). Upon co-localization of these bait proteins, a functional biotin ligase is reconstituted that biotinylates proteins within their common proximal microenvironment. Here, this methodology was adapted to S. cerevisiae for the first time with split halves of the BirA* variant TurboID, which has a stronger catalytic activity than BirA* [34]. Thus, short incubation times with biotin are sufficient to obtain pronounced protein biotinylation. To test whether Split-TurboID is generally applicable in yeast, we initially fused the N- and C-terminal TurboID halves (NTb and CTb) to the C-termini of Asc1 and Rps2. Both proteins are highly abundant in yeast cells, their C-termini are proximal to each other and should therefore induce biotinylation activity through reconstitution of Split-TurboID. Additionally, we tested the free NTb and CTb halves. To confirm their expression with Western blot experiments using a myc-specific antibody, all constructs also contained a C-terminal myc tag; with Streptavidin-HRP we then monitored biotinylated proteins from cell lysates (Figure S1): Both combinations, Asc1-CTb•Rps2-NTb and Asc1-NTb•Rps2-CTb, resulted in substantial biotinylation activity indicating the reconstitution of a complemented TurboID. We also observed that the CTb on its own possesses low residual biotinylation activity. Moreover, free NTb and CTb halves reassemble to form a functional biotin ligase if expressed simultaneously. When one Tb-half was fused to either Asc1 or Rps2 and the other one was expressed individually without bait, substantial biotinylation was observed. Overexpression of Split-TurboID halves can lead to unspecific reconstitution of TurboID and should be avoided [37].
Previous BioID experiments with Asc1-, Rps2-, and Rps3-BirA* established Bre5, co-factor of the deubiquitinase Ubp3, as a protein that localizes to the hr40S. Bre5-Ubp3 plays a key role in ribophagy and stress granule formation [38], but also acts in the nucleus [17]. The complex also regulates anterograde and retrograde transport between the endoplasmic reticulum and the Golgi apparatus [39]. Thus, Bre5 is supposed to come across many different proteins in different cellular contexts. Here, we performed an hr40S-specific Bre5-Split-TurboID experiment to characterize its ribosomal microenvironment. Expression levels of Bre5 are far below that of RPs [21]. We expressed Bre5-CTb from its genomic locus with its natural promoter to avoid overexpression and combined it with plasmid-borne Rps2-NTb. Bre5-CTb and Rps2-NTb are supposed to form a functional biotin ligase when both bait proteins co-localize at the hr40S (Figure 5B). To account for the mild background biotinylation activity of CTb, a strain expressing Bre5-CTb only (complemented with an empty vector, EV) was used as a negative control (BRE5-CTb•EV). Again, we considered to use Rpl5 for a distal control, and thus included a Bre5-CTb strain transformed by a plasmid expressing Rpl5-NTb (BRE5-CTb•RPL5-NTb) in the experiment (Figure 5B).
Expression and functionality of Bre5-CTb were confirmed by Western blot experiments using a BirA-specific antibody and by drop dilution growth assays on medium containing cycloheximide, respectively (Figure 5C). We performed a Split-TurboID experiment followed by LC-MS analysis essentially as described before for BioID experiments, including SILAC-labeling for relative enrichment quantification of proteins (Figure 5D). Here, we used yeast strains prototrophic for lysine and arginine and performed 2nSILAC [40]. We evaluated the labeling efficiency by determining the percentage of labeled peptides (based on annotated fragmentation spectra) in samples derived from the separate SILAC-labeled cultures (Tables S10 and S11). Over 98% of the peptides were correctly labeled in each sample, validating the 2nSILAC approach for the purpose of Split-TurboID experiments.
Taking advantage of the strong biotinylation activity of TurboID, the incubation time of cell cultures with biotin was reduced from overnight to 3 h. Western blot experiments with cell lysates from the separate cell cultures showed stable expression of Rps2-NTb and Rpl5-NTb (Figure 5D). Additionally, overall biotinylation activity was evaluated using Streptavidin-HRP. Co-expression of Bre5-CTb and Rps2-NTb resulted in an obvious increase in biotinylated proteins. For the distal control BRE5-CTb•RPL5-NTb, a slightly increased biotinylation was observed in comparison to the BRE5-CTb•EV control.
All LC-MS data of the Split-TurboID experiment were analyzed similarly as described before for BioID experiments with regard to initial filtering steps, proteome-correction, and statistical validation (for details see Table S12). Using the Bre5-CTb•EV strain as a negative control, 14 proteins were found as significantly captured from the Bre5-Rps2 microenvironment (Figure 6A; Tables S13 and S15), and 11 proteins with the distal control Bre5-CTb•Rpl5-NTb (Figure 6B; Tables S14 and S15). Seven of the proteins had a common overlap found with both controls and with that form the high confidence core of the Bre5-CTb•Rps2-NTb Split-TurboID experiment (Figure 6, proteins highlighted in green; Table S15). These seven proteins are the deubiquitinase Ubp3, the ubiquitin-associated factors Def1 and Lsm12 [17,41], Pbp1, the RNA helicase Ded1, Lsg1 (factor releasing nonsense-mediated mRNA decay factor 3 from the 60S subunit, Nmd3 [42]), and the mRNA-binding protein Sro9. These results on the one hand confirm hr40S localization of proteins identified with the Asc1- and Rps-BioID experiments, and on the other hand verify transitional hr40S localization of the nucleus-active RNA polymerase II degradation factor Def1 and of the Pbp1-Pbp4-interacting protein Lsm12 [16,43].
3. Discussion
Proximity of proteins not only occurs through direct physical binding, but also through transient common localization at or near larger structural components or machineries. Accurately localized bait-biotin ligases within the hr40S microenvironment covalently label proximal proteins in cell culture that are captured and identified afterwards from detergent-solubilized cell extracts. By taking different labeling positions within the hr40S, we have taken four pictures from this specific microenvironment from slightly shifted perspectives, namely from a biotin ligase fused to Asc1/RACK1, Rps2/uS5, Rps3/uS3, or Rps20/uS10 (Figure 7). Three non-ribosomal proteins appeared as common proximal factors of all four bait proteins, namely the mRNA-binding proteins Scp160 and Sro9, and the ribosome clamping factor Stm1. Common to these proteins is that they provide for mRNA tunnel occupation of the ribosomal 40S subunit, either with mRNA (through Scp160 and Sro9) or on its own (Stm1). The largest intersection set between two perspectives derived from Asc1-BirA* and Rps3-BirA*. This set contains 14 additional non-ribosomal proteins. Among them are the mRNA cap-binding protein Cdc33 and the a-subunit of eIF3 Rpg1, which reflects an mRNA 5′UTR-orientation of both Asc1 and the C-terminus of Rps3 at the hr40S. Slf1 is a paralog of the afore mentioned mRNA-binding protein Sro9. Ubiquitination, ubiquitin binding, and deubiquitination processes within that microenvironment are indicated by the proximity of the E3 RING ubiquitin ligase Hel2, the CUE domain- and poly(Q)-containing protein Def1, and the deubiquitination complex Bre5-Ubp3, respectively. The GYF-domain containing paralogs Smy2 and Syh1, and the mRNA 5′-3′ exonuclease Xrn1 hint to a broader translation-associated context of mRNA surveillance within that area. Mbf1, in concert with Asc1 and Rps3, has been described to prevent translational frameshifting at inhibitory CGA-CGA codon repeats [14]. The known interacting proteins Pbp1 and Lsm12, together with Pbp4 and Dhh1, are related to stress granules [44].
We also observed changes at the hr40S caused by the absence of Asc1, e.g., Hel2 and Bre5-Ubp3 were captured less (Figure 7). There are two possibilities for Asc1-dependent changes in protein captures with BioID: (i) Changes in the abundance of proteins at the hr40S or (ii) altered accessibility of proteins for biotinylation. The latter might be the case for Rps3 itself and for Rps26a/b in the Rps3-BioID experiment. The lysine residues within the C-terminal arm of Rps3 become increasingly accessible in the absence of Asc1 (Figure 1A), an observation that we had already made from the Rps2 perspective [18]. Shielding through Asc1 might also explain why Vps30, a subunit of the PI3K complexes I and II, was captured from the Rps2-BirA* perspective only in the absence of Asc1 [18], whereas it was captured independently of Asc1 from the Rps3-BirA* perspective.
The large number of proteins at the hr40S represents a cross-section of various microenvironments of context-specific ribosomes. Contexts can be specified by the presence of non-ribosomal proteins that co-localize at the hr40S only upon certain circumstances, e.g., early during ribosome biogenesis or later upon ribosome collision as a consequence of ribosome stalling. A first pioneering Split-TurboID experiment with Bre5 and the constitutive hr40S protein Rps2 provides a promising basis for studying distinct ribosomal microenvironments. In the following sections, we discuss the identified proteins within their, thus far, known contexts with regard to ribosome homeostasis and Asc1-dependence.
3.1. Regulation of eIF2α Phosphorylation by Gcn2 Might Require an Asc1-Dependent Positioning of Hel2 at hr40S
Asc1/RACK1 has an essential role in the recognition of translational stalling of ribosomes, e.g., on aberrant mRNAs during translation [45,46,47,48]. To prevent the synthesis of aberrant and toxic polypeptides and to take care of stuck polysomes, stalled ribosomes are recognized by the ribosome-associated protein quality control pathway (RQC). This pathway subjects the nascent polypeptide chain to proteasomal degradation [45]. Furthermore, ribosomal subunits need to be released and the mRNA degraded by endo- and exonucleolytic activities [49]. Ribosome stalling can induce collisions with trailing ribosomes and the formation of disomes [50,51]. The interface of such disomes is mainly formed by two 40S subunits and causes direct contact between the Asc1 molecules from the leading and the colliding ribosomes [50]. RPs within the concerned area are marked through ubiquitination. The E3-ubiquitin RING ligase Hel2 modifies Rps3/uS3 and Rps20/uS10 in S. cerevisiae, and its mammalian homolog ZNF598 ubiquitinates eS10 and uS10 [48,51,52,53,54]. Ubiquitination of RPs is followed by ribosomal subunit separation, the degradation of the associated nascent polypeptide chain and the aberrant mRNA, and by a recycling of ribosome subunits [50,51,54]. In mammalian cells, deubiquitination of RPs by the conserved Bre5-Ubp3 homologous deubiquitination complex G3BP1-family-USP10 is an important late step during 40S subunit recycling to prevent its lysosomal degradation [55]. In yeast, Bre5-Ubp3 also antagonizes the Hel2-mediated ubiquitination of Rps3 [30]. Our recent Asc1-BioID study revealed proximity of Asc1 to Hel2 and Bre5-Ubp3 [3]. With our new Rps3-BioID results, we confirmed an Asc1-dependent localization of Bre5-Ubp3 at the hr40S, which we had already observed earlier by studying the Rps2 perspective (Figure 7, [18]). The Rps3-BioID data further imply displacement of Hel2 in the absence of Asc1. An impaired ribosome localization of Hel2 has previously been reported for Asc1-deficient cells with polysome gradient experiments [56]. Thus, our findings underscore the importance of Asc1 for balancing ubiquitination and deubiquitination at the hr40S.
Recent studies have shown that ribosome collisions not only trigger translational quality control pathways, but also lead to broader cellular stress responses when they become more abundant and persistent [57,58,59]. The action of Hel2 during RQC prevents the activation of the integrated stress response (ISR) under low stress conditions when ribosome collisions are rare [59]. Under stress conditions and upon increased frequency of collisions, however, the eIF2α kinase Gcn2 is activated especially by ribosomes with an empty tRNA acceptor site [59]. eIF2α is a subunit of the heterotrimeric eIF2 complex. Phosphorylation of eIF2α by Gcn2 at S51 prevents formation of the ternary complex eIF2·GTP·Met-tRNAi, which is required for initiation of translation, and thus attenuates global translation [60,61]. Deletion of HEL2 increases cellular eIF2α phosphorylation [59]. Previously, we observed elevated levels of eIF2α phosphorylation for an ∆asc1 strain independent of applied stress conditions [9]. The impaired hr40S localization of Hel2 in Asc1-deficient cells and thus impairment of RQC might abrogate Hel2’s inhibitory effect on premature activation of Gcn2 under non-stress conditions leading to increased levels of phosphorylated eIF2α.
Similar to eIF2 the initiation factor eIF2A promotes Met-tRNAi binding to the 40S subunit, yet, it is not required for global translation initiation and appears to be rather involved in specific initiation events, such as re-initiation, internal initiation, or initiation at non-AUG codons [62]. Here, we observed increased capture of eIF2A from the Rps3 microenvironment in the absence of Asc1. The observed increase in the phosphorylation of eIF2α in ∆asc1 cells and the correlated activated ISR might cause the need for an alternative way to initiate translation that might be mediated by eIF2A.
3.2. The GYF Domain Proteins Smy2/Syh1, the Putative Endonuclease Ypl199c, and the 5′-3′ Exonuclease Xrn1 Localize to the hr40S
In this study, we also observed displacement of Smy2 at the hr40S in the absence of Asc1. Moreover, the capture of the Smy2 paralog Syh1 was Asc1-dependent in previous BioID experiments with Rps2-BirA* (Figure 7, [18]). Smy2 and Syh1 are potential homologs of mammalian GIGYF2, an inhibitor of translation [63,64]. Recent studies revealed that re-initiation of translation during RQC is prevented through the action of GIGYF2 and mRNA cap-binding protein 4EHP [65]. Their recruitment to faulty mRNAs seems to be mediated by different factors, including the Hel2 homolog ZNF598 [65] and the Mbf1 homolog EDF1 [15]. 4EHP is an eIF4E1 ortholog and inhibits initiation of translation through competition with eIF4E for cap-binding [66,67]. S. cerevisiae contains no obvious 4EHP homolog, and Smy2 and Syh1 seem to mediate mRNA decay rather than repression of translation during RQC [65]. However, Smy2 binds the eIF4E-binding protein Eap1, a translational repressor that competes with eIF4G for binding to eIF4E [68]. Smy2 and Eap1 together with Scp160 and Asc1 form the SESA network to regulate translation of POM34 mRNA that encodes an integral component of the nuclear pore complex [68].
Here, for the first time, the yeast endonuclease Ypl199c was found proximal to Rps3. Ypl199c and Cue2 are two yeast endonucleases with homology to the C. elegans protein NONU-1 and have recently been described to have an evolutionarily conserved function in mRNA cleavage during nonstop and no-go mRNA decay [29,69]. In the absence of Asc1, Ypl199c enriched less from the Rps3-microenvironment. This might explain the previously reported importance of Asc1 for endonucleolytic cleavage of mRNA during nonstop decay [70]. Endonucleolytically cleaved mRNA is further degraded by the 5′-3′ exonuclease Xrn1 and the exosome [71]. Xrn1 localizes close to Asc1 [3,72] and was also captured here within the proximity of Rps3.
3.3. The hr40S Might Be Directly Linked to MAP Kinase Signaling through the MAP4K Ste20
We identified the MAP4K Ste20 as being proximal to Rps3 and thus, most likely, to translating ribosomes. In metazoa, an important sensor for colliding ribosomes is the MAP3K ZAKα [57,58]. The MAP3K associates with translating ribosomes and auto-phosphorylates itself when stable collisions become increasingly abundant and the quality control pathway gets overburdened [58]. Depending on the degree of ribosome collisions, ZAKα is required for the activation of GCN2 and stress-activated kinases p38 and JNK to reduce translation initiation and to induce apoptosis, respectively [58]. Similar to ZAKα, Ste20 might communicate the translation status to stress-activated kinases. Co-localization of Asc1 and Ste20 has been described earlier [6], and deletion of the ASC1 gene causes increased phosphorylation of Ste20 at three different sites [73]. Phosphorylation of the downstream targets of Ste20-comprising MAPK cascades, the MAPKs Kss1 and the p38 homolog Hog1, increases and decreases upon ASC1 deletion, respectively [6,73].
Overall, the hr40S seems to be sensitive to and dependent on the activity status of (poly)ribosomes and/or the context of their stalling, and thus might directly contribute to the adequate progressive, preservative or degradative processes. This encompasses cellular signaling by (de)ubiquitination and kinase-mediated protein phosphorylation, together regulating protein processing, structure remodeling, and protein trafficking. With that, ribosomes coupled to any specific cellular process might stall context-specifically (e.g., upon substrate depletion, stress, faulty mRNA) and simultaneously trigger the release of ribosome-bound factors destined for specific cellular target sites, such as mRNA binding proteins and Def1 for the nucleus. hr40S residence of Def1 was corroborated by the Bre5-CTb•Rps2-NTb Split-TurboID experiment, indicating the possibility that ribosome stalling or collision might trigger its ribosomal release and consecutive nuclear transfer. Indeed, it has been shown previously that ubiquitin-dependent chopping of a glutamine-rich C-terminal moiety of Def1 is essential for its nuclear accumulation [74]. Ongoing studies will address the hypothesis that signal and origin of the nuclear transfer of Def1 emerge from the hr40S, possibly coordinated by the scaffold protein Asc1.
4. Materials and Methods
4.1. Plasmid Construction
Plasmids used in this study are listed in Table 1. All new plasmids in this study were generated using the GeneArt™ Seamless Cloning and Assembly Kit (#10378809, Fisher Scientific GmbH, Schwerte, Germany), except for pME5058, which was generated with the In-Fusion HD Cloning Kit (#639650, Clontech Laboratories, Inc., Mountain View, CA, USA). To obtain plasmids pME4800 and pME5059-5061, the plasmid pME4478 was used as a template to generate the linearized vector backbone by PCR, and inserts were amplified from genomic S. cerevisiae DNA (RH2817), introducing the linker sequence and 15 bp overhangs complementary to the ends of the vector backbone. For the construction of pME5058, the insert contained 20 bp overhangs. Plasmids pME4984 (NTb-myc) and pME4985 (CTb-myc) are based on plasmids pME2785 and pME2787. The NTb insert was amplified from pME4480 and the CTb insert from a codon optimized template (provided by Hans Dieter Schmitt) [34,75]. For the NTb insert, a codon exchange was introduced via the PCR primer to obtain the amino acid exchange Q65P. In the next step, pME4984 (NTb-myc) and pME4985 (CTb-myc) were used to amplify plasmid backbones for the construction of plasmids pME4986-4989 and pME5312. Inserts were amplified from genomic DNA and linker sequences were introduced via the oligonucleotides used for PCR. For plasmids pME4986 and pME4987, the MET25 promoter was replaced by the native ASC1 promoter [76]. Proteins were fused via four glycine-serine-serine (GSS) repeats as a linker to the BirA* protein, NTb, or CTb. The Rps3 linker contained two additional GSS repeats. Plasmid pME5307 was based on plasmid pFA6a-VN-TRP1 [77]. The coding sequence for the N-terminal fragment of Venus (VN) was replaced by the CTb-myc sequence and a linker sequence coding for the amino acid sequence GRRIPGLGSAGSAAGSGE was introduced.
4.2. Yeast Strains and Growth Conditions
Yeast strains used in this study are of the genetic ∑1278b background and listed in Table 2. Yeasts cells were cultivated in liquid yeast nitrogen base medium (YNB; 0.15% YNB, 0.5% ammonium sulfate, 2% glucose). L-arginine (20 mg/L), L-lysine HCl (30 mg/L), L-tryptophan (20 mg/L), and L-proline (200 mg/L) were added as required. To obtain strain RH3902 for the expression of Bre5-CTb-myc, the transformation cassette was amplified from plasmid pME5307. Primers used for PCR contained overhangs identical to the target site for homologous recombination. The transformation cassette was inserted into the genome of strain RH2817.
4.3. Western Blot Analysis
Cell lysis was performed as described in Schmitt and Valerius (2019) [18] according to Kushnirov (2000) [79]. For each sample, an equal number of yeast cells (calculated according to the OD600 of the culture, e.g., 6.25 mL of culture with OD600 of 0.8) was harvested by centrifugation. Pelleted cells were washed with water and incubated in 0.1 M NaOH for 5 min at room temperature. NaOH was removed and yeast cells were resuspended in 100 µL of 1:4 diluted SDS-PAGE loading buffer (0.25 M Tris pH 6.8, 30% glycerol, 15% β-mercaptoethanol, 7% SDS, 0.3% bromphenol blue). Samples were incubated at 95 °C for 3 min and centrifuged at 13,000 rpm for 5 min. Proteins of 6 µL of the supernatant were separated by SDS-PAGE, blotted onto a nitrocellulose membrane and subsequently stained with Ponceau S (0.2% Ponceau S, 3% trichloroacetic acid). After blocking of the membranes with 5% milk powder in tris buffered saline (TBS; 20 mM Tris, 150 mM NaCl), the membranes were incubated with a BirA antibody (#orb230654, biorbyt), c-myc antibody (#9E10, Santa Cruz SC40), or Rps3 antibody (provided by Heike Krebber, [3]), followed by incubation with the peroxidase-coupled goat anti-rabbit antibody. For the detection of biotinylated proteins, membranes were blocked overnight with 1% BSA in phosphate buffered saline (PBS, 8 mM Na2HPO4, 2 mM NaH2PO4, 150 mM NaCl) and incubated with PierceTM High Sensitivity Streptavidin-HRP (#21130, Thermo Fisher Scientific, Waltham, Massachusetts, USA) diluted 1:2000 in the blocking buffer with 0.1% Tween 20) for 1 h at room temperature. The FUSION-SL-4 (Peqlab Biotechnology GmbH, Erlangen, Germany) was used for the detection of chemiluminescent signals.
4.4. Proximity Labeling MS Experiments
Yeast cells were grown in 200 mL (Rps3-BioID experiment) or 100 mL (Rps20-BioID and Split-TurboID experiment) YNB medium containing 10 µM D-biotin to mid-log phase. For the Split-TurboID experiment, biotin was added when the cells reached an OD600 of 0.25 and cells were harvested after 3 h incubation. The media additionally contained differentially labeled lysine and arginine for SILAC (see Figure 2A and Figure 5D for labeling of the strains). 13C6-L-arginine HCl, 13C615N4-L-arginine HCl, 4,4,5,5-D4-L-lysine HCl, 13C6-L-lysine HCl were used for the labeling of the strains with medium and heavy amino acids. For the Split-TurboID experiment, L-proline was additionally added. After harvesting the cells, equal amounts of cells from each culture were mixed according to the OD600 of the main cultures. Cells were washed with washing buffer 1 (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2) and lysed in 1.8 mL breaking buffer (wash buffer 1 with 1 cOmpleteTM EDTA-free protease inhibitor cocktail tablet (#5056489001, Merck) per 50 mL, 0.5 mM DTT and 0.5 mM PMSF) using glass beads, and SDS was added to a final w/v of 4%. For the enrichment of biotinylated proteins in the Rps3-BioID experiments, Strep-Tactin® Sepharose® gravity flow columns with 0.2 mL bed volume were used (#2-1202-550, IBA Lifesciences GmbH, Göttingen, Germany) as described in Schmitt and Valerius (2019) [18]. For the enrichment of biotinylated proteins in the Rps20-BioID and the Split-TurboID experiments, 200–300 µL Strep-Tactin® Superflow resin (#2-1206-025, IBA Lifesciences GmbH) were used instead. Due to the smaller culture volume, cells were lysed in only 0.9 mL breaking buffer. The Strep-Tactin® beads were washed twice with 750 µL washing buffer 2 (0.1 M Tris-Cl, 150 mM NaCl, 1 mM EDTA, #2-1003-100, IBA Lifesciences GmbH) for equilibration. The cell lysate was added to the equilibrated beads and incubated for 30 min with gentle rotation. The samples were washed three times with 1 mL washing buffer 2 containing 0.4% w/v SDS. Biotinylated proteins were eluted through incubation of the beads with 150 µL BXT buffer (0.1 M Tris-Cl, 150 mM NaCl, 1 mM EDTA, 50 mM biotin #2-1042-025, IBA Lifesciences GmbH) for 10 min. The elution step was repeated once, and proteins were isolated from the eluate by chloroform-methanol extraction [80]. Protein pellets were reconstituted in 20 µL 8 M urea/2 M thiourea and subjected to SDS-PAGE followed by incubation in fixing solution (40% ethanol, 10% acetic acid) for at least 1 h. As input control samples, 50 µg of proteins from the whole cell lysate were separated by SDS-PAGE. Complete gel lanes were divided in ten fractions/gel slices, the proteins digested within the gel with trypsin, and peptides were extracted [81]. To evaluate the labeling efficiency for the Split-TurboID experiment, protein lysates were prepared as described in 4.3 using aliquots of the still separate cell cultures. A total of 6 µL of each cell lysate was used for SDS-PAGE. One gel slice was cut out from the gel lane and used for in-gel digest of proteins with trypsin. Peptides were purified using C18 (#2215, 3M) stop and go extraction (stage) tips [82,83]. Peptides were dried, dissolved in 20 µL sample buffer (2% acetonitrile, 0.1% formic acid), and analyzed through LC-MS using an UltiMate 3000 RSLCnano system coupled to either an Orbitrap Velos Pro hybrid ion trap-Orbitrap or a Q Exactive HF mass spectrometer (all Thermo Fisher Scientific). For the first replicate of the Rps20-BioID, the samples from the eluate fractions were measured twice. SILAC ratios for identified proteins were later on averaged for these two technical replicates. LC-MS analysis was performed as described before [18]. LC-MS method programming and data acquisition were performed with the software XCalibur 2.2 (Thermo Fisher Scientific). MS/MS2 data were searched against an S. cerevisiae-specific protein database (UniProt Proteome ID UP000002311) additionally containing the amino acid sequence of Escherichia coli BirA (UniProtKB: P06709) or of NTb and CTb [34] using the software MaxQuant 1.6.12.0 [22]. With respect to the SILAC labeling of the yeast cultures, Arg6 and Lys4 alongside Arg10 and Lys8 were defined as the medium and heavy labels, respectively. SILAC ratios for BirA* were later not further considered due to the different molecular weight of the BirA* fusion proteins, and thus, their distribution in different fractions. Also, SILAC ratios for NTb and CTb were removed. Biotinylation of lysine was set as variable modification in addition to oxidation of methionine and N-terminal acetylation. Re-quantification was enabled and a minimum of one ratio count was required for protein quantification using unique and razor peptides. For all other parameters, the default settings were used. MaxQuant output data were analyzed using the Perseus software 1.6.0.7 [23].
4.5. Phenotypic Growth Test
Yeast cells were cultivated to mid-log phase in YNB medium and diluted to an OD600 of 0.1. Three tenfold dilutions were prepared, and 20 µL of each dilution were spotted on the agar plates containing either 10 µM biotin or 0.05 µg/mL cycloheximide. A plate without additional supplements was used for the growth control. Plates were incubated at 30 °C for 3 to 4 days.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ijms222111653/s1.
Author Contributions
Conceptualization, K.S. and O.V.; Methodology, K.S. and O.V.; Validation, K.S. and O.V.; Formal analysis, K.S.; Investigation, K.S., A.-A.K. and O.V.; Writing—original draft preparation, K.S. and O.V.; Writing—review and editing, K.S., A.-A.K. and O.V.; Visualization, K.S. and O.V.; Funding acquisition, O.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) grants VA 352/2-2, DFG-GZ: A 630 (ZUK41/1), DFG-GZ: INST 186/1230-1 FUGG. Publication was supported by the Open Access Publication Funds of the Göttingen University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Acknowledgments
We thank Gerhard H. Braus for his general support and critical review of the manuscript. We thank Anna Lao Dudek for technical assistance and proofreading and Nadine Opitz for generating the Rps3-BirA* plasmid. We thank our graduate students Olga Steinbrenner, Georg Aschenbrandt, Judith Aylin Weyergraf, and Lucas Sebastian Hollstein for their contribution. We thank Heike Krebber (Georg-August-University Göttingen) for providing the Rps3-antibody and Hans Dieter Schmitt (Max Planck Institute for Biophysical Chemistry, Göttingen) for providing a codon usage-optimized CTb DNA template for S. cerevisiae.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Samavarchi-Tehrani, P.; Samson, R.; Gingras, A.C. Proximity Dependent Biotinylation: Key Enzymes and Adaptation to Proteomics Approaches. Mol. Cell Proteom. 2020, 19, 757–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opitz, N.; Schmitt, K.; Hofer-Pretz, V.; Neumann, B.; Krebber, H.; Braus, G.H.; Valerius, O. Capturing the Asc1p/Receptor for Activated C Kinase 1 (RACK1) Microenvironment at the Head Region of the 40S Ribosome with Quantitative BioID in Yeast. Mol. Cell Proteom. 2017, 16, 2199–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef] [Green Version]
- Kouba, T.; Rutkai, E.; Karásková, M.; Valášek, L. The eIF3c/NIP1 PCI domain interacts with RNA and RACK1/ASC1 and promotes assembly of translation preinitiation complexes. Nucleic Acids Res. 2012, 40, 2683–2699. [Google Scholar] [CrossRef] [Green Version]
- Zeller, C.E.; Parnell, S.C.; Dohlman, H.G. The RACK1 ortholog Asc1 functions as a G-protein beta subunit coupled to glucose responsiveness in yeast. J. Biol. Chem. 2007, 282, 25168–25176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Li, Y.; Rushing, B.R.; Harris, S.E.; McRitchie, S.L.; Jones, J.C.; Dominguez, D.; Sumner, S.J.; Dohlman, H.G. Multi-omics analysis of glucose-mediated signaling by a moonlighting Gbeta protein Asc1/RACK1. PLoS Genet. 2021, 17, e1009640. [Google Scholar] [CrossRef] [PubMed]
- Rachfall, N.; Schmitt, K.; Bandau, S.; Smolinski, N.; Ehrenreich, A.; Valerius, O.; Braus, G.H. RACK1/Asc1p, a ribosomal node in cellular signaling. Mol. Cell Proteom. 2013, 12, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Valerius, O.; Kleinschmidt, M.; Rachfall, N.; Schulze, F.; López Marín, S.; Hoppert, M.; Streckfuss-Bömeke, K.; Fischer, C.; Braus, G.H. The Saccharomyces homolog of mammalian RACK1, Cpc2/Asc1p, is required for FLO11-dependent adhesive growth and dimorphism. Mol. Cell Proteom. 2007, 6, 1968–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, B.; Wanke, C.; Lapaglia, S.K.; Braus, G.H. c-Jun and RACK1 homologues regulate a control point for sexual development in Aspergillus nidulans. Mol. Microbiol. 2000, 37, 28–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadrmas, J.L.; Smith, M.A.; Pronovost, S.M.; Beckerle, M.C. Characterization of RACK1 function in Drosophila development. Dev. Dyn. 2007, 236, 2207–2215. [Google Scholar] [CrossRef] [PubMed]
- Volta, V.; Beugnet, A.; Gallo, S.; Magri, L.; Brina, D.; Pesce, E.; Calamita, P.; Sanvito, F.; Biffo, S. RACK1 depletion in a mouse model causes lethality, pigmentation deficits and reduction in protein synthesis efficiency. Cell Mol. Life Sci. 2012, 70, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Xie, D. RACK1, a versatile hub in cancer. Oncogene 2015, 34, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.; Yang, Q.; Grayhack, E.J. Multi-protein bridging factor 1(Mbf1), Rps3 and Asc1 prevent stalled ribosomes from frameshifting. Elife 2018, 7, e39637. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.K.; Ordureau, A.; Best, K.; Saba, J.A.; Zinshteyn, B.; Sundaramoorthy, E.; Fulzele, A.; Garshott, D.M.; Denk, T.; Thoms, M.; et al. EDF1 coordinates cellular responses to ribosome collisions. Elife 2020, 9, e58828. [Google Scholar] [CrossRef] [PubMed]
- Woudstra, E.C.; Gilbert, C.; Fellows, J.; Jansen, L.; Brouwer, J.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature 2002, 415, 929–933. [Google Scholar] [CrossRef]
- Kvint, K.; Uhler, J.P.; Taschner, M.J.; Sigurdsson, S.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. Reversal of RNA polymerase II ubiquitylation by the ubiquitin protease Ubp3. Mol. Cell 2008, 30, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.; Valerius, O. yRACK1/Asc1 proxiOMICs-Towards Illuminating Ships Passing in the Night. Cells 2019, 8, 1384. [Google Scholar] [CrossRef] [Green Version]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 2008, 10, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.K.; Porras-Yakushi, T.R.; Reitsma, J.M.; Huber, F.M.; Sweredoski, M.J.; Hoelz, A.; Hess, S.; Deshaies, R.J. A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. Elife 2016, 5, e19105. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Baryshnikova, A.; Brown, G.W. Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst 2018, 6, 192–205.e193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Reidick, C.; Boutouja, F.; Platta, H.W. The class III phosphatidylinositol 3-kinase Vps34 in Saccharomyces cerevisiae. Biol. Chem. 2017, 398, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Liu, X.; Ariosa, A.; Huang, H.; Jin, M.; Karbstein, K.; Klionsky, D.J. Psp2, a novel regulator of autophagy that promotes autophagy-related protein translation. Cell Res. 2019, 29, 994–1008. [Google Scholar] [CrossRef]
- Bas, L.; Papinski, D.; Licheva, M.; Torggler, R.; Rohringer, S.; Schuschnig, M.; Kraft, C. Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome-vacuole fusion. J. Cell Biol. 2018, 217, 3656–3669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Reggiori, F.; Ungermann, C. A novel in vitro assay reveals SNARE topology and the role of Ykt6 in autophagosome fusion with vacuoles. J. Cell Biol. 2018, 217, 3670–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Rosenwald, A.G. Autophagy in Saccharomyces cerevisiae requires the monomeric GTP-binding proteins, Arl1 and Ypt6. Autophagy 2016, 12, 1721–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, M.L.; Burroughs, A.M.; Monem, P.C.; Egelhofer, T.A.; Pule, M.N.; Aravind, L.; Arribere, J.A. NONU-1 Encodes a Conserved Endonuclease Required for mRNA Translation Surveillance. Cell Rep. 2020, 30, 4321–4331.e4324. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, H.D.; Yang, H.W.; Kim, H.J.; Jang, C.Y.; Kim, J. Modulating cellular balance of Rps3 mono-ubiquitination by both Hel2 E3 ligase and Ubp3 deubiquitinase regulates protein quality control. Exp. Mol. Med. 2017, 49, e390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasari, V.; Pochopien, A.A.; Margus, T.; Murina, V.; Turnbull, K.; Zhou, Y.; Nissan, T.; Graf, M.; Nováček, J.; Atkinson, G.C.; et al. A role for the Saccharomyces cerevisiae ABCF protein New1 in translation termination/recycling. Nucleic Acids Res. 2019, 47, 8807–8820. [Google Scholar] [CrossRef] [PubMed]
- Trotter, E.W.; Rand, J.D.; Vickerstaff, J.; Grant, C.M. The yeast Tsa1 peroxiredoxin is a ribosome-associated antioxidant. Biochem. J. 2008, 412, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Dever, T.E.; Ivanov, I.P. Roles of polyamines in translation. J. Biol. Chem. 2018, 293, 18719–18729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.F.; Branon, T.C.; Rajeev, S.; Svinkina, T.; Udeshi, N.D.; Thoudam, T.; Kwak, C.; Rhee, H.W.; Lee, I.K.; Carr, S.A.; et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl. Acad. Sci. USA 2020, 117, 12143–12154. [Google Scholar] [CrossRef] [PubMed]
- De Munter, S.; Görnemann, J.; Derua, R.; Lesage, B.; Qian, J.; Heroes, E.; Waelkens, E.; Van Eynde, A.; Beullens, M.; Bollen, M. Split-BioID: A proximity biotinylation assay for dimerization-dependent protein interactions. FEBS Lett. 2017, 591, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopp, I.M.; Amaya Ramirez, C.C.; Debeljak, J.; Kreibich, E.; Skribbe, M.; Wild, K.; Béthune, J. Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat. Commun. 2017, 8, 15690. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.F.; Branon, T.C.; Udeshi, N.D.; Myers, S.A.; Carr, S.A.; Ting, A.Y. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc. 2020, 15, 3971–3999. [Google Scholar] [CrossRef]
- Nostramo, R.; Varia, S.N.; Zhang, B.; Emerson, M.M.; Herman, P.K. The Catalytic Activity of the Ubp3 Deubiquitinating Protease Is Required for Efficient Stress Granule Assembly in Saccharomyces cerevisiae. Mol. Cell Biol. 2015, 36, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.; Stutz, F.; Dargemont, C. Deubiquitination, a new player in Golgi to endoplasmic reticulum retrograde transport. J. Biol. Chem. 2003, 278, 51989–51992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenmaier, S.; Stiller, S.B.; Morgenstern, M.; Lübbert, P.; Oeljeklaus, S.; Wiedemann, N.; Warscheid, B. Complete Native Stable Isotope Labeling by Amino Acids of Saccharomyces cerevisiae for Global Proteomic Analysis. Anal. Chem. 2018, 90, 10501–10509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, R.; Shi, L.; Wu, C.; Qiao, W.; Liu, L.; Wu, J. Lsm12 Mediates Deubiquitination of DNA Polymerase eta To Help Saccharomyces cerevisiae Resist Oxidative Stress. Appl. Environ. Microbiol. 2019, 85, e01988-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedges, J.; West, M.; Johnson, A.W. Release of the export adapter, Nmd3p, from the 60S ribosomal subunit requires Rpl10p and the cytoplasmic GTPase Lsg1p. EMBO J. 2005, 24, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischer, T.C.; Weaver, C.M.; McAfee, K.J.; Jennings, J.L.; Link, A.J. Systematic identification and functional screens of uncharacterized proteins associated with eukaryotic ribosomal complexes. Genes Dev. 2006, 20, 1294–1307. [Google Scholar] [CrossRef]
- Swisher, K.D.; Parker, R. Localization to, and effects of Pbp1, Pbp4, Lsm12, Dhh1, and Pab1 on stress granules in Saccharomyces cerevisiae. PLoS ONE 2010, 5, e10006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandman, O.; Stewart-Ornstein, J.; Wong, D.; Larson, A.; Williams, C.C.; Li, G.W.; Zhou, S.; King, D.; Shen, P.S.; Weibezahn, J.; et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 2012, 151, 1042–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroha, K.; Akamatsu, M.; Dimitrova, L.; Ito, T.; Kato, Y.; Shirahige, K.; Inada, T. Receptor for activated C kinase 1 stimulates nascent polypeptide-dependent translation arrest. EMBO Rep. 2010, 11, 956–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitron, C.S.; Park, J.H.; Brandman, O. Asc1, Hel2, and Slh1 couple translation arrest to nascent chain degradation. RNA 2017, 23, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaramoorthy, E.; Leonard, M.; Mak, R.; Liao, J.; Fulzele, A.; Bennett, E.J. ZNF598 and RACK1 Regulate Mammalian Ribosome-Associated Quality Control Function by Mediating Regulatory 40S Ribosomal Ubiquitylation. Mol. Cell 2017, 65, 751–760.e754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, C.J.; Eyler, D.E.; Green, R. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science 2010, 330, 369–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, K.; Tesina, P.; Matsuo, Y.; Sugiyama, T.; Cheng, J.; Saeki, Y.; Tanaka, K.; Becker, T.; Beckmann, R.; Inada, T. Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 2019, 38, e100276. [Google Scholar] [CrossRef]
- Juszkiewicz, S.; Chandrasekaran, V.; Lin, Z.; Kraatz, S.; Ramakrishnan, V.; Hegde, R.S. ZNF598 Is a Quality Control Sensor of Collided Ribosomes. Mol. Cell 2018, 72, 469–481.e467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garzia, A.; Jafarnejad, S.M.; Meyer, C.; Chapat, C.; Gogakos, T.; Morozov, P.; Amiri, M.; Shapiro, M.; Molina, H.; Tuschl, T.; et al. The E3 ubiquitin ligase and RNA-binding protein ZNF598 orchestrates ribosome quality control of premature polyadenylated mRNAs. Nat. Commun. 2017, 8, 16056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, Y.; Ikeuchi, K.; Saeki, Y.; Iwasaki, S.; Schmidt, C.; Udagawa, T.; Sato, F.; Tsuchiya, H.; Becker, T.; Tanaka, K.; et al. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat. Commun. 2017, 8, 159. [Google Scholar] [CrossRef] [Green Version]
- Simms, C.L.; Yan, L.L.; Zaher, H.S. Ribosome Collision Is Critical for Quality Control during No-Go Decay. Mol. Cell 2017, 68, 361–373.e365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Garzia, A.; Morozov, P.; Molina, H.; Tuschl, T. The G3BP1-Family-USP10 Deubiquitinase Complex Rescues Ubiquitinated 40S Subunits of Ribosomes Stalled in Translation from Lysosomal Degradation. Mol. Cell 2020, 77, 1193–1205.e1195. [Google Scholar] [CrossRef] [PubMed]
- Winz, M.L.; Peil, L.; Turowski, T.W.; Rappsilber, J.; Tollervey, D. Molecular interactions between Hel2 and RNA supporting ribosome-associated quality control. Nat. Commun. 2019, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Vind, A.C.; Snieckute, G.; Blasius, M.; Tiedje, C.; Krogh, N.; Bekker-Jensen, D.B.; Andersen, K.L.; Nordgaard, C.; Tollenaere, M.A.X.; Lund, A.H.; et al. ZAKalpha Recognizes Stalled Ribosomes through Partially Redundant Sensor Domains. Mol. Cell 2020, 78, 700–713.e707. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Peterson, A.; Zinshteyn, B.; Regot, S.; Green, R. Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 2020, 182, 404–416.e414. [Google Scholar] [CrossRef]
- Yan, L.L.; Zaher, H.S. Ribosome quality control antagonizes the activation of the integrated stress response on colliding ribosomes. Mol. Cell 2021, 81, 614–628.e614. [Google Scholar] [CrossRef]
- Dever, T.E.; Feng, L.; Wek, R.C.; Cigan, A.M.; Donahue, T.F.; Hinnebusch, A.G. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 1992, 68, 585–596. [Google Scholar] [CrossRef]
- Dever, T.E.; Yang, W.; Aström, S.; Byström, A.S.; Hinnebusch, A.G. Modulation of tRNA(iMet), eIF-2, and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2.GTP.Met-tRNA(iMet) ternary complexes. Mol. Cell Biol. 1995, 15, 6351–6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komar, A.A.; Merrick, W.C. A Retrospective on eIF2A-and Not the Alpha Subunit of eIF2. Int. J. Mol. Sci. 2020, 21, 2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ash, M.R.; Faelber, K.; Kosslick, D.; Albert, G.I.; Roske, Y.; Kofler, M.; Schuemann, M.; Krause, E.; Freund, C. Conserved beta-hairpin recognition by the GYF domains of Smy2 and GIGYF2 in mRNA surveillance and vesicular transport complexes. Structure 2010, 18, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Ler, L.W.; Fabian, M.R.; Siddiqui, N.; Mullin, M.; Henderson, V.C.; Alain, T.; Fonseca, B.D.; Karashchuk, G.; Bennett, C.F.; et al. A novel 4EHP-GIGYF2 translational repressor complex is essential for mammalian development. Mol. Cell Biol. 2012, 32, 3585–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, K.L.; Dickson, K.; Cogan, J.Z.; Replogle, J.M.; Schoof, M.; D’Orazio, K.N.; Sinha, N.K.; Hussmann, J.A.; Jost, M.; Frost, A.; et al. GIGYF2 and 4EHP Inhibit Translation Initiation of Defective Messenger RNAs to Assist Ribosome-Associated Quality Control. Mol. Cell 2020, 79, 950–962.e956. [Google Scholar] [CrossRef]
- Rom, E.; Kim, H.C.; Gingras, A.C.; Marcotrigiano, J.; Favre, D.; Olsen, H.; Burley, S.K.; Sonenberg, N. Cloning and characterization of 4EHP, a novel mammalian eIF4E-related cap-binding protein. J. Biol. Chem. 1998, 273, 13104–13109. [Google Scholar] [CrossRef] [Green Version]
- Zuberek, J.; Kubacka, D.; Jablonowska, A.; Jemielity, J.; Stepinski, J.; Sonenberg, N.; Darzynkiewicz, E. Weak binding affinity of human 4EHP for mRNA cap analogs. RNA 2007, 13, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Sezen, B.; Seedorf, M.; Schiebel, E. The SESA network links duplication of the yeast centrosome with the protein translation machinery. Genes Dev. 2009, 23, 1559–1570. [Google Scholar] [CrossRef] [Green Version]
- D’Orazio, K.N.; Wu, C.C.; Sinha, N.; Loll-Krippleber, R.; Brown, G.W.; Green, R. The endonuclease Cue2 cleaves mRNAs at stalled ribosomes during No Go Decay. Elife 2019, 8, e49117. [Google Scholar] [CrossRef]
- Ikeuchi, K.; Inada, T. Ribosome-associated Asc1/RACK1 is required for endonucleolytic cleavage induced by stalled ribosome at the 3’ end of nonstop mRNA. Sci. Rep. 2016, 6, 28234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamyshev, A.L.; Karamysheva, Z.N. Lost in Translation: Ribosome-Associated mRNA and Protein Quality Controls. Front. Genet. 2018, 9, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesina, P.; Heckel, E.; Cheng, J.; Fromont-Racine, M.; Buschauer, R.; Kater, L.; Beatrix, B.; Berninghausen, O.; Jacquier, A.; Becker, T.; et al. Structure of the 80S ribosome-Xrn1 nuclease complex. Nat. Struct Mol. Biol. 2019, 26, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.; Smolinski, N.; Neumann, P.; Schmaul, S.; Hofer-Pretz, V.; Braus, G.H.; Valerius, O. Asc1p/RACK1 Connects Ribosomes to Eukaryotic Phosphosignaling. Mol. Cell Biol. 2017, 37, e00279-16. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.D.; Harreman, M.; Taschner, M.; Reid, J.; Walker, J.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. Proteasome-mediated processing of Def1, a critical step in the cellular response to transcription stress. Cell 2013, 154, 983–995. [Google Scholar] [CrossRef] [Green Version]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, M.; Schulz, R.; Braus, G.H. The yeast CPC2/ASC1 gene is regulated by the transcription factors Fhl1p and Ifh1p. Curr. Genet. 2006, 49, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.K.; Huh, W.K. Bimolecular fluorescence complementation analysis system for in vivo detection of protein-protein interaction in Saccharomyces cerevisiae. Yeast 2007, 24, 767–775. [Google Scholar] [CrossRef]
- Mumberg, D.; Müller, R.; Funk, M. Regulatable promoters of Saccharomyces cerevisiae: Comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994, 22, 5767–5768. [Google Scholar] [CrossRef]
- Kushnirov, V.V. Rapid and reliable protein extraction from yeast. Yeast 2000, 16, 857–860. [Google Scholar] [CrossRef]
- Wessel, D.; Flügge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
BirA* fusion protein expression and monitoring of biotinylation activity. (A) Structure of the S. cerevisiae 80S ribosome with Asc1 (red), Rps3 (blue), Rps20 (orange), Rpl5 (cyan), and Rpl25 (pink) highlighted. rRNA and proteins of the 40S subunit are colored in light gray and rRNA and proteins of the 60S subunit in dark gray. Since the proteins of interest were C-terminally fused to BirA*, the (approximate) position of each C-terminus is indicated with a red-rimmed circle filled with the respective color. For Rps3, the C-terminal amino acids R226-A240, which are structurally not resolved, are indicated with an arrow. Crystal structure data from PDB entry 4V88 [4] were visualized using the PyMOL molecular graphics system. (B) Protein extracts of ASC1 wild-type cells expressing either free BirA* or Rps3-BirA* were separated by SDS polyacrylamide gel electrophoresis (PAGE) and proteins were blotted onto a nitrocellulose membrane. An Rps3-specific antibody [3] was used to detect native endogenous Rps3 and the Rps3-BirA* fusion protein. Respective signals are labeled and highlighted with an arrow. Strains were cultivated in the presence and absence of 10 µM biotin. (C) The growth of ASC1 wild-type and asc1− strains (RH2817 and RH3510, respectively) expressing RP-BirA* fusion proteins was monitored on YNB medium with 10 µM biotin or 0.05 µg/mL cycloheximide (CHX) in comparison to strains with the empty vector (EV). A YNB plate without additional supplements served as a control. Tenfold serial dilutions of the cell cultures were spotted on the plates, and the cells were incubated for 3 d without and 4 d with CHX, respectively. (D,E) S. cerevisiae ASC1 wild-type and asc1− strains (RH3493 and RH3520) expressing plasmid-borne free BirA*, Rps3-BirA*, Rpl5-BirA*, Rpl25-BirA*, or Fba1-BirA* were cultivated in liquid medium and used for the preparation of protein extracts. Streptavidin-HRP was used for visualization of biotinylated proteins and a BirA-specific antibody for the detection of both free BirA* and the fusion proteins. Ponceau staining served as a loading control. (D) Free BirA* and the fusion proteins were expressed in ASC1 wild-type (+) and asc1− strains (−), and biotin (final concentration 10 µM) was added to all cultures. (E) ASC1 wild-type cells expressing the indicated proteins were cultivated in the presence (+) or absence (−) of 10 µM biotin in the medium.
Figure 1.
BirA* fusion protein expression and monitoring of biotinylation activity. (A) Structure of the S. cerevisiae 80S ribosome with Asc1 (red), Rps3 (blue), Rps20 (orange), Rpl5 (cyan), and Rpl25 (pink) highlighted. rRNA and proteins of the 40S subunit are colored in light gray and rRNA and proteins of the 60S subunit in dark gray. Since the proteins of interest were C-terminally fused to BirA*, the (approximate) position of each C-terminus is indicated with a red-rimmed circle filled with the respective color. For Rps3, the C-terminal amino acids R226-A240, which are structurally not resolved, are indicated with an arrow. Crystal structure data from PDB entry 4V88 [4] were visualized using the PyMOL molecular graphics system. (B) Protein extracts of ASC1 wild-type cells expressing either free BirA* or Rps3-BirA* were separated by SDS polyacrylamide gel electrophoresis (PAGE) and proteins were blotted onto a nitrocellulose membrane. An Rps3-specific antibody [3] was used to detect native endogenous Rps3 and the Rps3-BirA* fusion protein. Respective signals are labeled and highlighted with an arrow. Strains were cultivated in the presence and absence of 10 µM biotin. (C) The growth of ASC1 wild-type and asc1− strains (RH2817 and RH3510, respectively) expressing RP-BirA* fusion proteins was monitored on YNB medium with 10 µM biotin or 0.05 µg/mL cycloheximide (CHX) in comparison to strains with the empty vector (EV). A YNB plate without additional supplements served as a control. Tenfold serial dilutions of the cell cultures were spotted on the plates, and the cells were incubated for 3 d without and 4 d with CHX, respectively. (D,E) S. cerevisiae ASC1 wild-type and asc1− strains (RH3493 and RH3520) expressing plasmid-borne free BirA*, Rps3-BirA*, Rpl5-BirA*, Rpl25-BirA*, or Fba1-BirA* were cultivated in liquid medium and used for the preparation of protein extracts. Streptavidin-HRP was used for visualization of biotinylated proteins and a BirA-specific antibody for the detection of both free BirA* and the fusion proteins. Ponceau staining served as a loading control. (D) Free BirA* and the fusion proteins were expressed in ASC1 wild-type (+) and asc1− strains (−), and biotin (final concentration 10 µM) was added to all cultures. (E) ASC1 wild-type cells expressing the indicated proteins were cultivated in the presence (+) or absence (−) of 10 µM biotin in the medium.

Figure 2.
Workflow of the Rps3 and Rps20 proximity labeling MS experiments. (A) For the Rps3-BioID experiment, ASC1 wild-type and asc1− strains (RH3493 and RH3520, respectively) expressing plasmid-borne Rps3-BirA* were cultivated with light and heavy SILAC amino acids, respectively. As a negative control, an ASC1 wild-type strain expressing Rpl5-BirA* was labeled with the medium SILAC amino acids. Equivalently, the Rps20-BioID experiment was performed using strains expressing Rps20-BirA* instead of Rps3-BirA*. Cells were cultivated in the presence of the respective SILAC amino acids and with 10 µM biotin. Cells of the individual cultures were harvested in the exponential growth phase and combined. An aliquot of the still separate cell cultures was taken for cell lysis for the Western blot experiments depicted in (B). The protein extracts of the pooled cells were used to enrich biotinylated proteins via affinity purification. Proteins were subjected to SDS-PAGE followed by in-gel digestion with trypsin. Peptides were analyzed with LC-MS, and data analysis was performed using the MaxQuant and Perseus software. (B) Cell lysates of the indicated strains were separated by SDS-PAGE and proteins transferred onto nitrocellulose membranes to detect RP-BirA* fusion proteins and biotinylated proteins using a BirA-specific antibody and Streptavidin-HRP, respectively. One of three replicates is shown representatively.
Figure 2.
Workflow of the Rps3 and Rps20 proximity labeling MS experiments. (A) For the Rps3-BioID experiment, ASC1 wild-type and asc1− strains (RH3493 and RH3520, respectively) expressing plasmid-borne Rps3-BirA* were cultivated with light and heavy SILAC amino acids, respectively. As a negative control, an ASC1 wild-type strain expressing Rpl5-BirA* was labeled with the medium SILAC amino acids. Equivalently, the Rps20-BioID experiment was performed using strains expressing Rps20-BirA* instead of Rps3-BirA*. Cells were cultivated in the presence of the respective SILAC amino acids and with 10 µM biotin. Cells of the individual cultures were harvested in the exponential growth phase and combined. An aliquot of the still separate cell cultures was taken for cell lysis for the Western blot experiments depicted in (B). The protein extracts of the pooled cells were used to enrich biotinylated proteins via affinity purification. Proteins were subjected to SDS-PAGE followed by in-gel digestion with trypsin. Peptides were analyzed with LC-MS, and data analysis was performed using the MaxQuant and Perseus software. (B) Cell lysates of the indicated strains were separated by SDS-PAGE and proteins transferred onto nitrocellulose membranes to detect RP-BirA* fusion proteins and biotinylated proteins using a BirA-specific antibody and Streptavidin-HRP, respectively. One of three replicates is shown representatively.

Figure 3.
Proteins occurring proximal to Rps3 and their Asc1-dependence. Volcano plots show the averaged log2 SILAC ratios for (A) ASC1 + RPS3-birA* (light SILAC) to ASC1 + RPL5-birA* (medium SILAC) and (B) asc1− + RPS3-birA* (heavy SILAC) to ASC1 + RPS3-birA* (light SILAC) on the x-axes and the -log10 (p-value) of the t-test on the y-axes. Gray dotted threshold lines indicate a p-value of 0.05 (horizontal) and a log2 SILAC ratio of 0.585 or −0.585 (vertical). Proteins with two instead of three quantification values (w/o p-values) that passed the threshold of two values ≥ 0.585 are depicted below the horizontal 0-value line with their averaged log2 SILAC ratios (L/M in A and H/L in B). Not applicable (N/A) on the y-axis indicates that no t-test was performed for these proteins and thus no p-value is available. For proteins highlighted with an asterisks (*), no information about their protein abundance was obtained from the proteome data. For all other proteins that are highlighted in red or green, at least one quantification value was obtained from the proteome analysis. Gray squares represent proteins not passing the set criteria for proximity (A) or Asc1-dependence (B). For details see Tables S1–S5.
Figure 3.
Proteins occurring proximal to Rps3 and their Asc1-dependence. Volcano plots show the averaged log2 SILAC ratios for (A) ASC1 + RPS3-birA* (light SILAC) to ASC1 + RPL5-birA* (medium SILAC) and (B) asc1− + RPS3-birA* (heavy SILAC) to ASC1 + RPS3-birA* (light SILAC) on the x-axes and the -log10 (p-value) of the t-test on the y-axes. Gray dotted threshold lines indicate a p-value of 0.05 (horizontal) and a log2 SILAC ratio of 0.585 or −0.585 (vertical). Proteins with two instead of three quantification values (w/o p-values) that passed the threshold of two values ≥ 0.585 are depicted below the horizontal 0-value line with their averaged log2 SILAC ratios (L/M in A and H/L in B). Not applicable (N/A) on the y-axis indicates that no t-test was performed for these proteins and thus no p-value is available. For proteins highlighted with an asterisks (*), no information about their protein abundance was obtained from the proteome data. For all other proteins that are highlighted in red or green, at least one quantification value was obtained from the proteome analysis. Gray squares represent proteins not passing the set criteria for proximity (A) or Asc1-dependence (B). For details see Tables S1–S5.

Figure 4.
Proteins occurring proximal to Rps20 and their Asc1-dependence. Volcano plots show the averaged log2 SILAC ratios for (A) ASC1 RPS20-birA* (light SILAC) to ASC1 RPL5-birA* (medium SILAC) and (B) asc1− RPS20-birA* (heavy SILAC) to ASC1 RPS20-birA* (light SILAC) on the x-axes and the -log10 (p-value) of the t-test on the y-axes. Gray dotted threshold lines indicate a p-value of 0.05 (horizontal) and a log2 SILAC ratio of 0.585 or −0.585 (vertical). Proteins with two instead of three quantification values (w/o p-values) that passed the threshold of two values ≥ 0.585 are depicted below the horizontal 0-value line with their averaged log2 SILAC ratios (L/M in (A) and H/L in (B)). Not applicable (N/A) on the y-axis indicates that no t-test was performed for these proteins and thus no p-value is available. For proteins highlighted with an asterisks (*), no information about their protein abundance was obtained from the proteome data. For all other proteins that are highlighted in red or green, at least one quantification value was obtained from the proteome analysis. Gray squares represent proteins not passing the set criteria for proximity (A) or Asc1-dependence (B). For details see Tables S1 and S6–S9.
Figure 4.
Proteins occurring proximal to Rps20 and their Asc1-dependence. Volcano plots show the averaged log2 SILAC ratios for (A) ASC1 RPS20-birA* (light SILAC) to ASC1 RPL5-birA* (medium SILAC) and (B) asc1− RPS20-birA* (heavy SILAC) to ASC1 RPS20-birA* (light SILAC) on the x-axes and the -log10 (p-value) of the t-test on the y-axes. Gray dotted threshold lines indicate a p-value of 0.05 (horizontal) and a log2 SILAC ratio of 0.585 or −0.585 (vertical). Proteins with two instead of three quantification values (w/o p-values) that passed the threshold of two values ≥ 0.585 are depicted below the horizontal 0-value line with their averaged log2 SILAC ratios (L/M in (A) and H/L in (B)). Not applicable (N/A) on the y-axis indicates that no t-test was performed for these proteins and thus no p-value is available. For proteins highlighted with an asterisks (*), no information about their protein abundance was obtained from the proteome data. For all other proteins that are highlighted in red or green, at least one quantification value was obtained from the proteome analysis. Gray squares represent proteins not passing the set criteria for proximity (A) or Asc1-dependence (B). For details see Tables S1 and S6–S9.

Figure 5.
Split-TurboID experiment to analyze the Bre5 microenvironment during co-localization with Rps2. (A) Principle of Split-TurboID. Upon co-localization of the bait proteins, the Split-TurboID halves reconstitute to a functional biotin ligase. The enzyme catalyzes the formation of biotinyl-AMP that reacts with primary amines of proteins in the proximity (dubbed x and y). (B) Bre5-CTb and Rps2-NTb were co-expressed to biotinylate proteins in their common microenvironment. As a negative control, Bre5-CTb is expressed in the absence of any NTb to account for mild biotinylation activity of CTb. As a distal control, Bre5-CTb was co-expressed with Rpl5-NTb. They are supposed to locate at distal sites at the ribosome and residual biotinylation activity must be either unspecific or originate at a microenvironment apart from the hr40S. (C) Expression of Bre5-CTb in the presence and absence of additional biotin (+B/−B) was validated in a Western blot experiment using a BirA-specific antibody. A drop dilution assay confirmed the functionality of the Bre5-CTb fusion protein. Tenfold serial dilutions of the wild-type, the ∆bre5 and the BRE5-CTb strain were spotted on YNB plates containing 0.05 µg/mL cycloheximide (CHX). YNB plates served as a growth control. Plates were incubated for 3 d at 30 °C. (D) The Bre5-Rps2 Split-TurboID workflow: Strains were cultivated with SILAC amino acids as indicated until the cultures reached an OD600 of 0.25. After addition of biotin, incubation was continued for a further 3 h. Cells were harvested and combined. An aliquot of the still separate cell cultures was taken for cell lysis to obtain samples for the depicted Western blot experiments. Stable expression of Rps2-NTb and Rpl5-NTb was verified using an antibody against the myc-tag of the Tb-halves. Bre5-CTb is not visible according to its significantly lower cellular abundance compared to the ribosomal proteins. Cellular biotinylation activity was evaluated using Streptavidin-HRP. The cell lysates were further used to evaluate the efficiency of protein labeling with 2nSILAC by LC-MS analysis. The remaining cells of the main cultures were pooled for preparation of cell lysates, and the samples were further processed as described for the BioID workflow in Figure 2A.
Figure 5.
Split-TurboID experiment to analyze the Bre5 microenvironment during co-localization with Rps2. (A) Principle of Split-TurboID. Upon co-localization of the bait proteins, the Split-TurboID halves reconstitute to a functional biotin ligase. The enzyme catalyzes the formation of biotinyl-AMP that reacts with primary amines of proteins in the proximity (dubbed x and y). (B) Bre5-CTb and Rps2-NTb were co-expressed to biotinylate proteins in their common microenvironment. As a negative control, Bre5-CTb is expressed in the absence of any NTb to account for mild biotinylation activity of CTb. As a distal control, Bre5-CTb was co-expressed with Rpl5-NTb. They are supposed to locate at distal sites at the ribosome and residual biotinylation activity must be either unspecific or originate at a microenvironment apart from the hr40S. (C) Expression of Bre5-CTb in the presence and absence of additional biotin (+B/−B) was validated in a Western blot experiment using a BirA-specific antibody. A drop dilution assay confirmed the functionality of the Bre5-CTb fusion protein. Tenfold serial dilutions of the wild-type, the ∆bre5 and the BRE5-CTb strain were spotted on YNB plates containing 0.05 µg/mL cycloheximide (CHX). YNB plates served as a growth control. Plates were incubated for 3 d at 30 °C. (D) The Bre5-Rps2 Split-TurboID workflow: Strains were cultivated with SILAC amino acids as indicated until the cultures reached an OD600 of 0.25. After addition of biotin, incubation was continued for a further 3 h. Cells were harvested and combined. An aliquot of the still separate cell cultures was taken for cell lysis to obtain samples for the depicted Western blot experiments. Stable expression of Rps2-NTb and Rpl5-NTb was verified using an antibody against the myc-tag of the Tb-halves. Bre5-CTb is not visible according to its significantly lower cellular abundance compared to the ribosomal proteins. Cellular biotinylation activity was evaluated using Streptavidin-HRP. The cell lysates were further used to evaluate the efficiency of protein labeling with 2nSILAC by LC-MS analysis. The remaining cells of the main cultures were pooled for preparation of cell lysates, and the samples were further processed as described for the BioID workflow in Figure 2A.

Figure 6.
Proteins within a common microenvironment of Bre5 and Rps2. Volcano plots illustrate the averaged log2 SILAC ratios for (A) BRE5-CTb•RPS2-NTb (light SILAC) to BRE5-CTb•EV (medium SILAC) and (B) BRE5-CTb•RPS2-NTb (light SILAC) to BRE5-CTb•RPL5-NTb (heavy SILAC) on the x-axes and the -log10(p-value) of the t-test on the y-axes. A p-value of 0.05 (horizontal) and a log2 SILAC ratio of 0.585 or −0.585 (vertical) are indicated with gray dotted threshold lines. Proteins found as significantly enriched from the Bre5-Rps2 microenvironment with both controls are highlighted in green. Proteins in orange passed these thresholds for only one of the comparisons. For all labeled proteins, eluate ratios were corrected on total proteome ratios (input normalization). Gray squares represent proteins not passing the set criteria for proximity. For details see Tables S12–S15.
Figure 6.
Proteins within a common microenvironment of Bre5 and Rps2. Volcano plots illustrate the averaged log2 SILAC ratios for (A) BRE5-CTb•RPS2-NTb (light SILAC) to BRE5-CTb•EV (medium SILAC) and (B) BRE5-CTb•RPS2-NTb (light SILAC) to BRE5-CTb•RPL5-NTb (heavy SILAC) on the x-axes and the -log10(p-value) of the t-test on the y-axes. A p-value of 0.05 (horizontal) and a log2 SILAC ratio of 0.585 or −0.585 (vertical) are indicated with gray dotted threshold lines. Proteins found as significantly enriched from the Bre5-Rps2 microenvironment with both controls are highlighted in green. Proteins in orange passed these thresholds for only one of the comparisons. For all labeled proteins, eluate ratios were corrected on total proteome ratios (input normalization). Gray squares represent proteins not passing the set criteria for proximity. For details see Tables S12–S15.

Figure 7.
Four adjacent and overlapping perspectives at the hr40S: Comparison of microenvironments captured with Asc1-, Rps2-, Rps3-, and Rps20-BioID. The identified proteins co-localizing with the four different RPs are depicted within a Venn diagram. Proteins that were additionally identified in the common microenvironment of Bre5 and Rps2 with the Bre5-CTb•Rps2-NTb Split-TurboID are underlined.
Figure 7.
Four adjacent and overlapping perspectives at the hr40S: Comparison of microenvironments captured with Asc1-, Rps2-, Rps3-, and Rps20-BioID. The identified proteins co-localizing with the four different RPs are depicted within a Venn diagram. Proteins that were additionally identified in the common microenvironment of Bre5 and Rps2 with the Bre5-CTb•Rps2-NTb Split-TurboID are underlined.


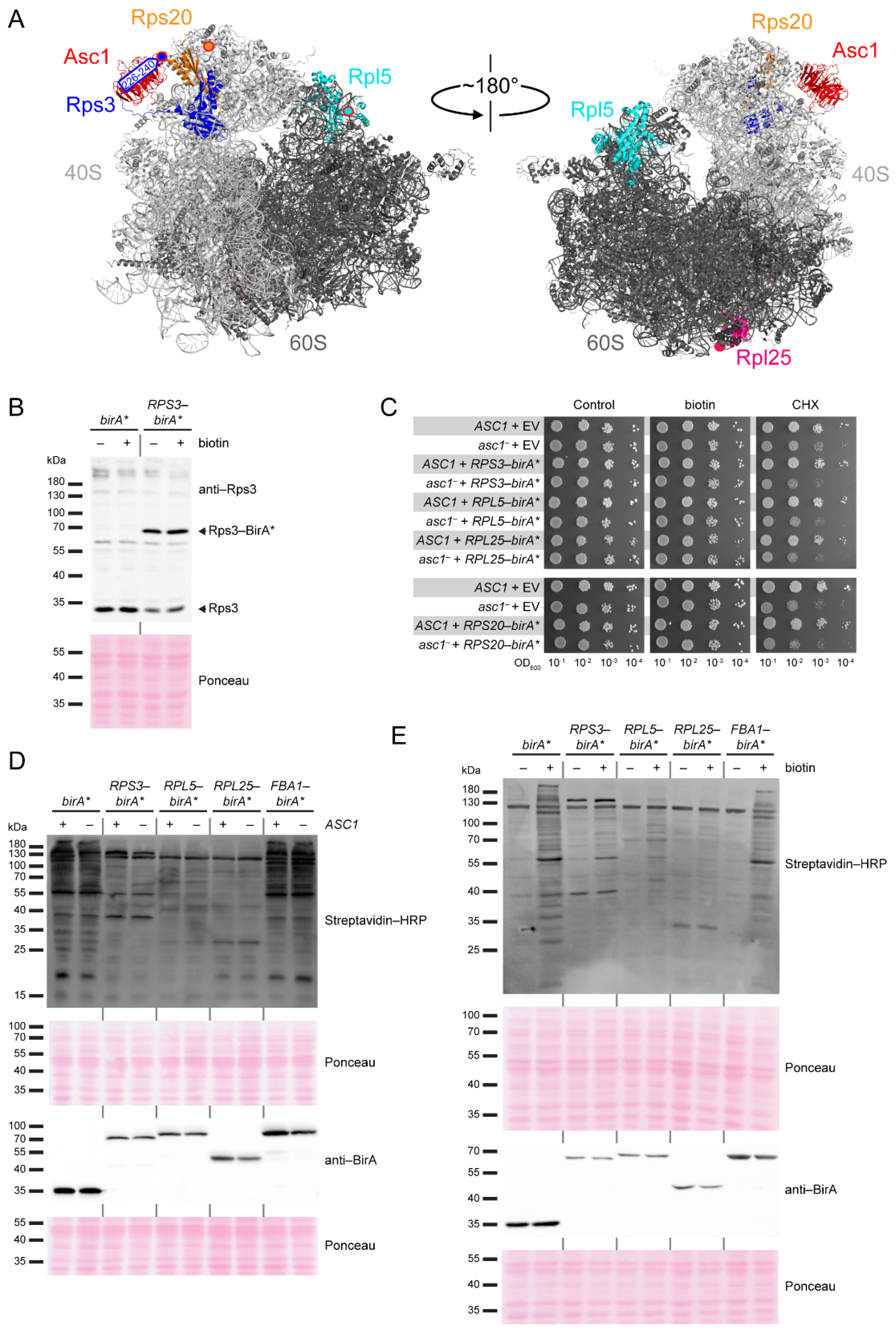{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Plasmids used in this study.
| Plasmid Name | Description | Reference |
|---|---|---|
| pME2785 | MET25Prom, CYC1Term, TRP1, 2 μm | [78] |
| pME2787 | MET25Prom, CYC1Term, URA3, 2 μm | [78] |
| pME4480 | MET25Prom, CYC1Term, URA3, 2 μm, birA* | [3] |
| pME4478 | MET25Prom, CYC1Term, URA3, 2 μm, ASC1-birA* | [3] |
| pME4800 | MET25Prom, CYC1Term, URA3, 2 μm, RPS20-birA* | This study |
| pME5058 | MET25Prom, CYC1Term, URA3, 2 μm, RPS3-birA* | This study |
| pME5059 | MET25Prom, CYC1Term, URA3, 2 μm, FBA1-birA* | This study |
| pME5060 | MET25Prom, CYC1Term, URA3, 2 μm, RPL5-birA* | This study |
| pME5061 | MET25Prom, CYC1Term, URA3, 2 μm, RPL25-birA* | This study |
| pME4984 | MET25Prom, CYC1Term, URA3, 2 μm, NTb-myc | This study |
| pME4985 | MET25Prom, CYC1Term, TRP1, 2 μm, CTb-myc | This study |
| pME4986 | natPromASC1, CYC1Term, URA3, 2 μm, ASC1-NTb-myc | This study |
| pME4987 | natPromASC1, CYC1Term, TRP1, 2 μm, ASC1-CTb-myc | This study |
| pME4988 | MET25Prom, CYC1Term, URA3, 2 μm, RPS2-NTb-myc | This study |
| pME4989 | MET25Prom, CYC1Term, TRP1, 2 μm, RPS2-CTb-myc | This study |
| pFA6a-VN-TRP1 | pFA6a, VN, TRP1, ADH1Term | [77] |
| pME5307 | pFA6a, CTb-myc, TRP1, ADH1Term | This study |
| pME5312 | MET25Prom, CYC1Term, URA3, 2 μm, RPL5-NTb-myc | This study |
Table 2.
S. cerevisiae strains used in this study.
| Strain Name | Description | Reference |
|---|---|---|
| RH2817 | MATα, ura3-52, trp1::hisG | [9] |
| RH3510 | MATα, ura3-52, trp1::hisG, asc1-loxP SNR24 | [8] |
| RH3493 | MATα, ura3-52, trp1::hisG, Δarg4::loxP, Δlys1::loxP | [73] |
| RH3520 | MATα, ura3-52, trp1::hisG, asc1-loxP SNR24, ∆arg4::loxP, ∆lys1::loxP | [73] |
| RH3789 | MATα, ura3-52, trp1::hisG, Δbre5::kanMX4 | [18] |
| RH3902 | MATα, ura3-52, trp1::hisG, BRE5-CTb-myc::TRP1 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schmitt, K.; Kraft, A.-A.; Valerius, O. A Multi-Perspective Proximity View on the Dynamic Head Region of the Ribosomal 40S Subunit. Int. J. Mol. Sci. 2021, 22, 11653. https://doi.org/10.3390/ijms222111653
AMA Style
Schmitt K, Kraft A-A, Valerius O. A Multi-Perspective Proximity View on the Dynamic Head Region of the Ribosomal 40S Subunit. International Journal of Molecular Sciences. 2021; 22(21):11653. https://doi.org/10.3390/ijms222111653
Chicago/Turabian StyleSchmitt, Kerstin, Alina-Andrea Kraft, and Oliver Valerius. 2021. "A Multi-Perspective Proximity View on the Dynamic Head Region of the Ribosomal 40S Subunit" International Journal of Molecular Sciences 22, no. 21: 11653. https://doi.org/10.3390/ijms222111653
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.