
Therapy Prospects for Mitochondrial DNA Maintenance Disorders

 , , , , , , and
, , , , , , and
Abstract
:1. Mitochondria and Mitochondrial Diseases
2. Mitochondrial DNA Depletion and Multiple Deletions Syndromes (MDDS)
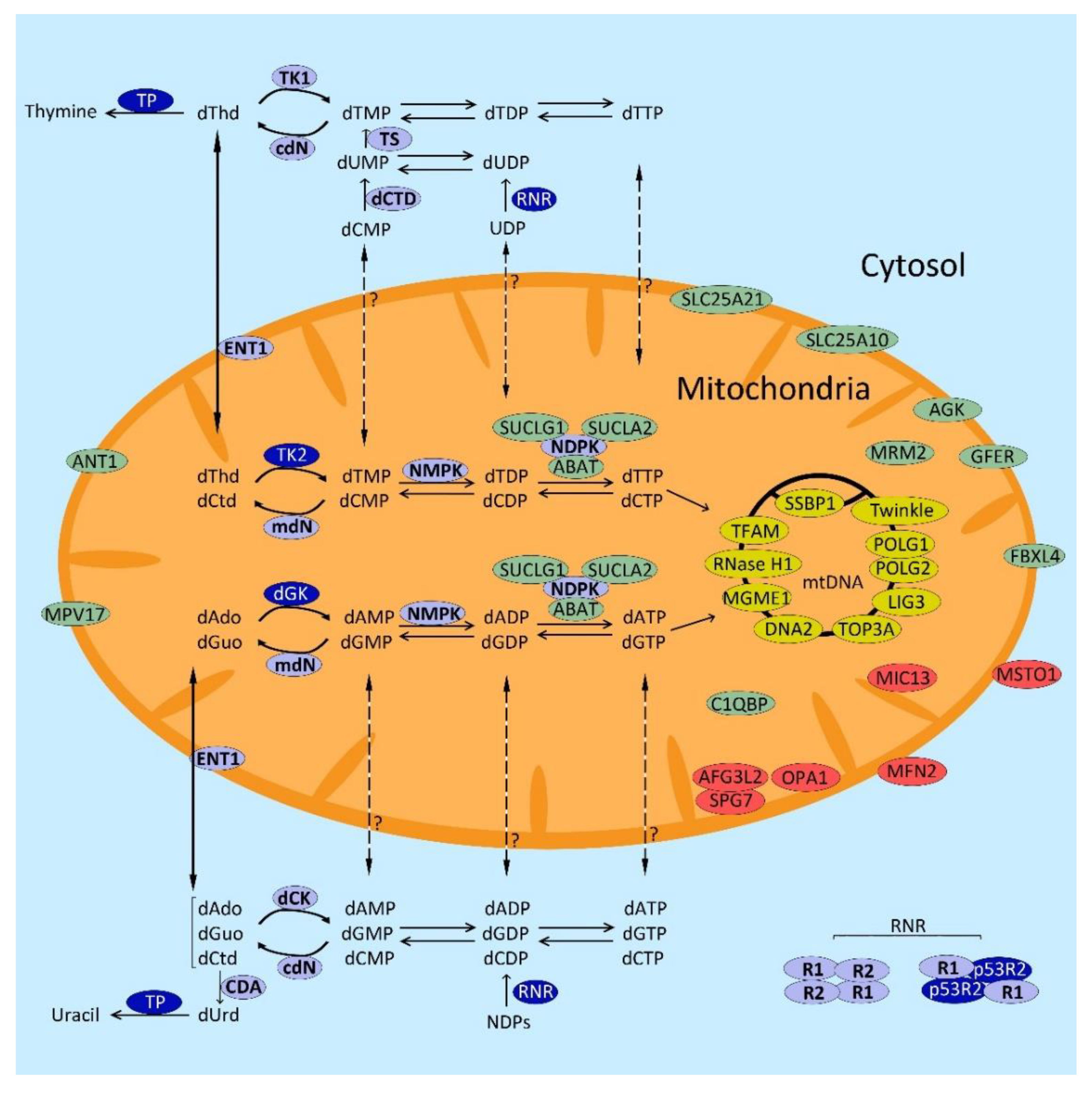
2.1. Genes Encoding Proteins of the mtDNA Replication Machinery
2.2. Genes Involved in dNTP Metabolism
2.2.1. dNTP Anabolism
De Novo Pathway
Salvage Pathway
2.2.2. dNTP Catabolism
2.2.3. Nucleoside/Nucleotide Transporters
2.3. Genes Involved in Mitochondrial Dynamics
2.4. Genes Involved in mtDNA Maintenance through Unknown Mechanisms
3. Non-Targeted Therapies for Mitochondrial DNA Maintenance Disorders
3.1. Symptomatic Treatments and Other Non-Targeted Treatments Applied to MDDS Patients
3.2. Non-Targeted Experimental Approaches
4. Targeted Therapies for MDDS
4.1. Direct Scavenging of Toxic Metabolites
4.2. Enzyme Replacement
4.3. Allogeneic Hematopoietic Stem Cell Transplantation (AHSCT)
4.4. Liver Transplantation
4.5. Administration of Deoxyribonucleosides (dNs)
4.6. Gene Therapy
4.7. Improving Mitochondrial Shape and Other Approaches
5. Prospects and Specific Barriers
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than Just a Powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [Green Version]
- Hajnóczky, G.; Csordás, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Roy, S.S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Breda, C.N.D.S.; Davanzo, G.G.; Basso, P.J.; Câmara, N.O.S.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 225–274, IN1–IN6. [Google Scholar] [CrossRef]
- Gray, M.W. Lynn Margulis and the endosymbiont hypothesis: 50 years later. Mol. Biol. Cell 2017, 28, 1285–1287. [Google Scholar] [CrossRef]
- García-Rodríguez, L.J. Appendix Basic Properties of Mitochondria. Echinoderms Part B 2007, 80, 809–812. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- DiMauro, S. Mitochondrial DNA Medicine. Biosci. Rep. 2007, 27, 5–9. [Google Scholar] [CrossRef]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Ronchi, D.; Comi, G.P. Genes and Pathways Involved in Adult Onset Disorders Featuring Muscle Mitochondrial DNA Instability. Int. J. Mol. Sci. 2015, 16, 18054–18076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suomalainen, A.; Isohanni, P. Mitochondrial DNA depletion syndromes—Many genes, common mechanisms. Neuromuscul. Disord. 2010, 20, 429–437. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Craigen, W.J.; Wong, L.J.C.; Scaglia, F. Mitochondrial DNA maintenance defects overview. In Gene Reviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Garone, C.; Tadesse, S.; Hirano, M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain 2011, 134, 3326–3332. [Google Scholar] [CrossRef] [Green Version]
- Van Goethem, G.; Dermaut, B.; Lofgren, A.; Martin, J.J.; van Broeckhoven, C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001, 28, 211–212. [Google Scholar] [CrossRef]
- Longley, M.J.; Clark, S.; Yu Wai Man, C.; Hudson, G.; Durham, S.E.; Taylor, R.W.; Nightingale, S.; Turnbull, D.M.; Copeland, W.C.; Chinnery, P.F. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am. J. Hum. Genet. 2006, 78, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Spelbrink, J.N.; Li, F.-Y.; Tiranti, V.; Nikali, K.; Yuan, Q.-P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornblum, C.; Nicholls, T.J.; Haack, T.B.; Scholer, S.; Peeva, V.; Danhauser, K.; Hallmann, K.; Zsurka, G.; Rorbach, J.; Iuso, A.; et al. Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat. Genet. 2013, 45, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Ronchi, D.; di Fonzo, A.; Lin, W.; Bordoni, A.; Liu, C.; Fassone, E.; Pagliarani, S.; Rizzuti, M.; Zheng, L.; Filosto, M.; et al. Mutations in DNA2 Link Progressive Myopathy to Mitochondrial DNA Instability. Am. J. Hum. Genet. 2013, 92, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Reyes, A.; Melchionda, L.; Nasca, A.; Carrara, F.; Lamantea, E.; Zanolini, A.; Lamperti, C.; Fang, M.; Zhang, J.; Ronchi, D.; et al. RNASEH1 Mutations Impair mtDNA Replication and Cause Adult-Onset Mitochondrial Encephalomyopathy. Am. J. Hum. Genet. 2015, 97, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Stiles, A.R.; Simon, M.; Stover, A.; Eftekharian, S.; Khanlou, N.; Wang, H.L.; Magaki, S.; Lee, H.; Partynski, K.; Dorrani, N.; et al. Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol. Genet. Metab. 2016, 119, 91–99. [Google Scholar] [CrossRef]
- Martin, C.A.; Sarlos, K.; Logan, C.V.; Thakur, R.S.; Parry, D.A.; Bizard, A.H.; Leitch, A.; Cleal, L.; Ali, N.S.; Al-Owain, M.A.; et al. Mutations in TOP3A Cause a Bloom Syndrome-Like Disorder. Am. J. Hum. Genet. 2018, 103, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Jurkute, N.; Leu, C.; Pogoda, H.; Arno, G.; Robson, A.G.; Nürnberg, G.; Altmüller, J.; Thiele, H.; Motameny, S.; Toliat, M.R.; et al. SSBP1 mutations in dominant optic atrophy with variable retinal degeneration. Ann. Neurol. 2019, 86, 368–383. [Google Scholar] [CrossRef]
- Bonora, E.; Chakrabarty, S.; Kellaris, G.; Tsutsumi, M.; Bianco, F.; Bergamini, C.; Ullah, F.; Isidori, F.; Liparulo, I.; Diquigiovanni, C.; et al. Biallelic variants in LIG3 cause a novel mitochondrial neurogastrointestinal encephalomyopathy. Brain 2021, awab056. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Spinazzola, A.; Hirano, M. Thymidine Phosphorylase Gene Mutations in MNGIE, a Human Mitochondrial Disorder. Science 1999, 283, 689–692. [Google Scholar] [CrossRef]
- Saada, A.; Shaag, A.; Mandel, H.; Nevo, Y.; Eriksson, S.; Elpeleg, O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat. Genet. 2001, 29, 342–344. [Google Scholar] [CrossRef]
- Mandel, H.; Szargel, R.; Labay, V.; Elpeleg, O.; Saada, A.; Shalata, A.; Anbinder, Y.; Berkowitz, D.; Hartman, C.; Barak, M.; et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat. Genet. 2001, 29, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chretien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Amati-Bonneau, P.; Blakely, E.L.; Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.; Ahlqvist, K.; Suomalainen, A.; Reynier, P.; et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain 2008, 131, 329–337. [Google Scholar] [CrossRef]
- Rouzier, C.; Bannwarth, S.; Chaussenot, A.; Chevrollier, A.; Verschueren, A.; Bonello-Palot, N.; Fragaki, K.; Cano, A.; Pouget, J.; Pellissier, J.F.; et al. The MFN2 gene is re-sponsible for mitochondrial DNA instability and optic atrophy ’plus’ phenotype. Brain 2012, 135, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Wedding, I.M.; Koht, J.; Tran, G.T.; Misceo, D.; Selmer, K.K.; Holmgren, A.; Frengen, E.; Bindoff, L.; Tallaksen, C.M.; Tzoulis, C. Spastic paraplegia type 7 is associated with multiple mitochondrial DNA deletions. PLoS ONE 2014, 9, e86340. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.; Pfeffer, G.; Griffin, H.R.; Blakely, E.L.; Kurzawa-Akanbi, M.; Gabriel, J.; Sitarz, K.; Roberts, M.; Schoser, B.; Pyle, A.; et al. Clonal Expansion of Secondary Mitochondrial DNA Deletions Associated with Spinocerebellar Ataxia Type. JAMA Neurol. 2015, 72, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Nasca, A.; Scotton, C.; Zaharieva, I.; Neri, M.; Selvatici, R.; Magnusson, O.T.; Gal, A.; Weaver, D.; Rossi, R.; Armaroli, A.; et al. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum. Mutat. 2017, 38, 970–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, B.E.; Whaley, K.G.; Bove, K.E.; Labilloy, A.; Lombardo, R.C.; Hopkin, R.J.; Leslie, N.D.; Prada, C.; Assouline, Z.; Barcia, G.; et al. Expanding and Underscoring the Hepato-Encephalopathic Phenotype of QIL1/MIC13. Hepatology 2019, 70, 1066–1070. [Google Scholar] [CrossRef]
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttälä, A.; Zeviani, M.; Comi, G.; Keränen, S.; Peltonen, L.; Suomalainen-Wartiovaara, A. Role of Adenine Nucleotide Translocator 1 in mtDNA Maintenance. Science 2000, 289, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, P.; Calvo, S.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006, 38, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; King, M.S.; Smith, A.C.; Oláhová, M.; Bansagi, B.; Roos, A.; Eyassu, F.; Borchers, C.; Ramesh, V.; Lochmuller, H.; et al. Mitochondrial oxodicarboxylate carrier deficiency is associated with mitochondrial DNA depletion and spinal muscular atrophy–like disease. Genet. Med. 2018, 20, 1224–1235. [Google Scholar] [CrossRef] [Green Version]
- Punzi, G.; Porcelli, V.; Ruggiu, M.; Hossain, F.; Menga, A.; Scarcia, P.; Castegna, A.; Gorgoglione, R.; Pierri, C.L.; Laera, L.; et al. SLC25A10 biallelic mutations in intractable epileptic encephalopathy with complex I deficiency. Hum. Mol. Genet. 2018, 27, 499–504. [Google Scholar] [CrossRef]
- Elpeleg, O.; Miller, C.; Hershkovitz, E.; Bitner-Glindzicz, M.; Bondi-Rubinstein, G.; Rahman, S.; Pagnamenta, A.; Eshhar, S.; Saada, A. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am. J. Hum. Genet. 2005, 76, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Ostergaard, E.; Christensen, E.; Kristensen, E.; Mogensen, B.; Duno, M.; Shoubridge, E.A.; Wibrand, F. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am. J. Hum. Genet. 2007, 81, 383–387. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012, 4, 118ra10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fonzo, A.; Ronchi, D.; Lodi, T.; Fassone, E.; Tigano, M.; Lamperti, C.; Corti, S.; Bordoni, A.; Fortunato, F.; Nizzardo, M.; et al. The Mitochondrial Disulfide Relay System Protein GFER Is Mutated in Autosomal-Recessive Myopathy with Cataract and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet. 2009, 84, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besse, A.; Wu, P.; Bruni, F.; Donti, T.; Graham, B.H.; Craigen, W.J.; McFarland, R.; Moretti, P.; Lalani, S.; Scott, K.L.; et al. The GABA transaminase, ABAT, is essential for mitochondrial nucleoside metabolism. Cell Metab. 2015, 21, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnen, P.E.; Yarham, J.W.; Besse, A.; Wu, P.; Faqeih, E.A.; Al-Asmari, A.M.; Saleh, M.A.; Eyaid, W.; Hadeel, A.; He, L.; et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am. J. Hum. Genet. 2013, 93, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Garone, C.; D’Souza, A.R.; Dallabona, C.; Lodi, T.; Rebelo-Guiomar, P.; Rorbach, J.; Donati, M.A.; Procopio, E.; Montomoli, M.; Guerrini, R.; et al. Defective mitochondrial rRNA methyltransferase MRM2 causes MELAS-like clinical syndrome. Hum. Mol. Genet. 2017, 26, 4257–4266. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, R.G.; Olahova, M.; Kishita, Y.; Garone, C.; Kremer, L.S.; Yagi, M.; Uchiumi, T.; Jourdain, A.A.; Thompson, K.; D’Souza, A.R.; et al. Biallelic C1QBP Mutations Cause Severe Neonatal-, Childhood-, or Later-Onset Cardiomyopathy Associated with Combined Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2017, 101, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.T.; Pontes, C.D.B.; Ciesielski, G.L. Roles of the mitochondrial replisome in mitochondrial DNA deletion formation. Genet. Mol. Biol. 2020, 43, e20190069. [Google Scholar] [CrossRef] [Green Version]
- Young, M.J.; Copeland, W.C. Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 2016, 38, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Lecrenier, N.; van der Bruggen, P.; Foury, F. Mitochondrial DNA polymerases from yeast to man: A new family of polymerases. Gene 1997, 185, 147–152. [Google Scholar] [CrossRef]
- Rahman, S.; Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef]
- Del Bo, R.; Bordoni, A.; Sciacco, M.; di Fonzo, A.; Galbiati, S.; Crimi, M.; Bresolin, N.; Comi, G.P. Remarkable infidelity of polymerase γA associated with mutations in POLG1 exonuclease domain. Neurology 2003, 61, 903–908. [Google Scholar] [CrossRef]
- Hikmat, O.; Naess, K.; Engvall, M.; Klingenberg, C.; Rasmussen, M.; Tallaksen, C.M.; Brodtkorb, E.; Ostergaard, E.; Coo, I.F.M.; Pias-Peleteiro, L.; et al. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J. Inherit. Metab. Dis. 2020, 43, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Young, M.J.; Longley, M.J.; Li, F.-Y.; Kasiviswanathan, R.; Wong, L.-J.; Copeland, W.C. Biochemical analysis of human POLG2 variants associated with mitochondrial disease. Hum. Mol. Genet. 2011, 20, 3052–3066. [Google Scholar] [CrossRef] [PubMed]
- Varma, H.; Faust, P.L.; Iglesias, A.D.; Lagana, S.M.; Wou, K.; Hirano, M.; DiMauro, S.; Mansukani, M.M.; Hoff, K.E.; Nagy, P.L.; et al. Whole exome sequencing identifies a homozygous POLG2 missense variant in an infant with fulminant hepatic failure and mitochondrial DNA depletion. Eur. J. Med. Genet. 2016, 59, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.E.; DeBalsi, K.L.; Sanchez-Quintero, M.J.; Longley, M.J.; Hirano, M.; Naini, A.B.; Copeland, W.C. Characterization of the human homozygous R182W POLG2 mutation in mitochondrial DNA depletion syndrome. PLoS ONE 2018, 13, e0203198. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Gaspari, M.; Falkenberg, M. TWINKLE Has 5′ → 3′ DNA Helicase Activity and Is Specifically Stimulated by Mitochondrial Single-stranded DNA-binding Protein. J. Biol. Chem. 2003, 278, 48627–48632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffart, S.; Cooper, H.M.; Tyynismaa, H.; Wanrooij, S.; Suomalainen, A.; Spelbrink, J.N. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 2009, 18, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikali, K.; Suomalainen, A.; Saharinen, J.; Kuokkanen, M.; Spelbrink, J.N.; Lonnqvist, T.; Peltonen, L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum. Mol. Genet. 2005, 14, 2981–2990. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, M.D.; García-Martínez, A.; Corral-Juan, M.; Pérez-Álvarez, Á.I.; Plasencia, A.M.; Villamar, M.; Moreno-Pelayo, M.A.; Matilla-Dueñas, A.; Menéndez-González, M.; del Castillo, I. Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: Overlap of TWNK-related recessive disorders. J. Transl. Med. 2019, 17, 290. [Google Scholar] [CrossRef] [PubMed]
- Morino, H.; Pierce, S.B.; Matsuda, Y.; Walsh, T.; Ohsawa, R.; Newby, M.; Hiraki-Kamon, K.; Kuramochi, M.; Lee, M.K.; Klevit, R.E.; et al. Mutations in Twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology 2014, 83, 2054–2061. [Google Scholar] [CrossRef] [Green Version]
- Wanrooij, S.; Luoma, P.; van Goethem, G.; van Broeckhoven, C.; Suomalainen, A.; Spelbrink, J.N. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004, 32, 3053–3064. [Google Scholar] [CrossRef] [Green Version]
- Nishigaki, Y.; Marti, R.; Copeland, W.C.; Hirano, M. Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. J. Clin. Investig. 2003, 111, 1913–1921. [Google Scholar] [CrossRef] [Green Version]
- Del Dotto, V.; Ullah, F.; di Meo, I.; Magini, P.; Gusic, M.; Maresca, A.; Caporali, L.; Palombo, F.; Tagliavini, F.; Baugh, E.H.; et al. SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder. J. Clin. Investig. 2020, 130, 108–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, T.J.; Nadalutti, C.A.; Motori, E.; Sommerville, E.W.; Gorman, G.; Basu, S.; Hoberg, E.; Turnbull, D.M.; Chinnery, P.F.; Larsson, N.-G.; et al. Topoisomerase 3α Is Required for Decatenation and Segregation of Human mtDNA. Mol. Cell 2018, 69, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Wang, L. Mitochondrial purine and pyrimidine metabolism and beyond. Nucleosides Nucleotides Nucleic Acids 2016, 35, 578–594. [Google Scholar] [CrossRef]
- Ferraro, P.; Nicolosi, L.; Bernardi, P.; Reichard, P.; Bianchi, V. Mitochondrial deoxynucleotide pool sizes in mouse liver and evidence for a transport mechanism for thymidine monophosphate. Proc. Natl. Acad. Sci. USA 2006, 103, 18586–18591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontarin, G.; Gallinaro, L.; Ferraro, P.; Reichard, P.; Bianchi, V. Origins of mitochondrial thymidine triphosphate: Dynamic relations to cytosolic pools. Proc. Natl. Acad. Sci. USA 2003, 100, 12159–12164. [Google Scholar] [CrossRef] [Green Version]
- Di Noia, M.A.; Todisco, S.; Cirigliano, A.; Rinaldi, T.; Agrimi, G.; Iacobazzi, V.; Palmieri, F. The human SLC25A33 and SLC25A36 genes of solute carrier family 25 encode two mitochondrial pyrimidine nucleotide transporters. J. Biol. Chem. 2014, 289, 33137–33148. [Google Scholar] [CrossRef] [Green Version]
- Floyd, S.; Favre, C.; Lasorsa, F.M.; Leahy, M.; Trigiante, G.; Stroebel, P.; Marx, A.; Loughran, G.; O’Callaghan, K.; Marobbio, C.M.; et al. The insulin-like growth factor-I-mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol. Biol. Cell 2007, 18, 3545–3555. [Google Scholar] [CrossRef] [Green Version]
- Franzolin, E.; Miazzi, C.; Frangini, M.; Palumbo, E.; Rampazzo, C.; Bianchi, V. The pyrimidine nucleotide carrier PNC1 and mitochondrial trafficking of thymidine phosphates in cultured human cells. Exp. Cell Res. 2012, 318, 2226–2236. [Google Scholar] [CrossRef] [PubMed]
- Shaibani, A.; Shchelochkov, O.A.; Zhang, S.; Katsonis, P.; Lichtarge, O.; Wong, L.J.; Shinawi, M. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch. Neurol. 2009, 66, 1028–1032. [Google Scholar] [CrossRef] [Green Version]
- Fratter, C.; Raman, P.; Alston, C.L.; Blakely, E.L.; Craig, K.; Smith, C.; Evans, J.; Seller, A.; Czermin, B.; Hanna, M.G.; et al. RRM2B mutations are frequent in familial PEO with multiple mtDNA deletions. Neurology 2011, 76, 2032–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horga, A.; Pitceathly, R.D.S.; Blake, J.C.; Woodward, C.E.; Zapater, P.; Fratter, C.; Mudanohwo, E.E.; Plant, G.T.; Houlden, H.; Sweeney, M.G.; et al. Peripheral neuropathy predicts nuclear gene defect in patients with mitochondrial ophthalmoplegia. Brain 2014, 137, 3200–3212. [Google Scholar] [CrossRef] [Green Version]
- Pitceathly, R.D.S.; Fassone, E.; Taanman, J.-W.; Sadowski, M.; Fratter, C.; Mudanohwo, E.E.; Woodward, C.E.; Sweeney, M.G.; Holton, J.L.; Hanna, M.G.; et al. Kearns-Sayre syndrome caused by defective R1/p53R2 assembly. J. Med. Genet. 2011, 48, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.S.; Smith, C.; Fratter, C.; Alston, C.L.; He, L.; Craig, K.; Blakely, E.L.; Evans, J.C.; Taylor, J.; Shabbir, Z.; et al. Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics. Brain 2012, 135, 3392–3403. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Ylikallio, E.; Patel, M.; Molnar, M.J.; Haller, R.G.; Suomalainen, A. A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions. Am. J. Hum. Genet. 2009, 85, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garone, C.; Taylor, R.W.; Nascimento, A.; Poulton, J.; Fratter, C.; Dominguez-Gonzalez, C.; Evans, J.C.; Loos, M.; Isohanni, P.; Suomalainen, A.; et al. Retrospective natural history of thymidine kinase 2 deficiency. J. Med. Genet. 2018, 55, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Caporali, L.; Bello, L.; Tagliavini, F.; La Morgia, C.; Maresca, A.; di Vito, L.; Liguori, R.; Valentino, M.L.; Cecchin, D.; Pegoraro, E.; et al. DGUOK recessive mutations in patients with CPEO, mitochondrial myopathy, parkinsonism and mtDNA deletions. Brain 2017, 141, e3. [Google Scholar] [CrossRef]
- Ronchi, D.; Garone, C.; Bordoni, A.; Rios, P.G.; Calvo, S.E.; Ripolone, M.; Ranieri, M.; Rizzuti, M.; Villa, L.; Magri, F.; et al. Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain 2012, 135, 3404–3415. [Google Scholar] [CrossRef] [Green Version]
- Rampazzo, C.; Gallinaro, L.; Milanesi, E.; Frigimelica, E.; Reichard, P.; Bianchi, V. A deoxyribonucleotidase in mitochondria: Involvement in regulation of dNTP pools and possible link to genetic disease. Proc. Natl. Acad. Sci. USA 2000, 97, 8239–8244. [Google Scholar] [CrossRef] [Green Version]
- Franzolin, E.; Pontarin, G.; Rampazzo, C.; Miazzi, C.; Ferraro, P.; Palumbo, E.; Reichard, P.; Bianchi, V. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 14272–14277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requena, C.E.; Perez-Moreno, G.; Ruiz-Perez, L.M.; Vidal, A.E.; Gonzalez-Pacanowska, D. The NTP pyro-phosphatase DCTPP1 contributes to the homoeostasis and cleansing of the dNTP pool in human cells. Biochem. J. 2014, 459, 171–180. [Google Scholar] [CrossRef]
- Martínez-Arribas, B.; Requena-Torres, C.E.; Pérez-Moreno, G.; Ruíz-Pérez, L.M.; Vidal, A.E.; González-Pacanowska, D. DCTPP1 prevents a mutator phenotype through the modulation of dCTP, dTTP and dUTP pools. Cell. Mol. Life Sci. 2019, 77, 1645–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, M.; Carelli, V.; de Giorgio, R.; Pironi, L.; Accarino, A.; Cenacchi, G.; D’Alessandro, R.; Filosto, M.; Marti, R.; Nonino, F.; et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Position paper on diagnosis, prognosis, and treatment by the MNGIE International Network. J. Inherit. Metab. Dis. 2021, 44, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, P.; Pontarin, G.; Crocco, L.; Fabris, S.; Reichard, P.; Bianchi, V. Mitochondrial deoxynucleotide pools in quiescent fibroblasts: A possible model for mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). J. Biol. Chem. 2005, 280, 24472–24480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vioque, E.G.; Torres-Torronteras, J.; Andreu, A.L.; Martí, R. Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE). PLoS Genet. 2011, 7, e1002035. [Google Scholar] [CrossRef]
- Pontarin, G.; Ferraro, P.; Valentino, M.L.; Hirano, M.; Reichard, P.; Bianchi, V. Mitochondrial DNA Depletion and Thymidine Phosphate Pool Dynamics in a Cellular Model of Mitochondrial Neurogastrointestinal Encephalomyopathy. J. Biol. Chem. 2006, 281, 22720–22728. [Google Scholar] [CrossRef] [Green Version]
- Spinazzola, A.; Marti, R.; Nishino, I.; Andreu, A.L.; Naini, A.; Tadesse, S.; Pela, I.; Zammarchi, E.; Donati, M.A.; Oliver, J.A.; et al. Altered Thymidine Metabolism Due to Defects of Thymidine Phosphorylase. J. Biol. Chem. 2002, 277, 4128–4133. [Google Scholar] [CrossRef] [Green Version]
- Lopez, L.C.; Akman, H.O.; García-Cazorla, Á.; Dorado, B.; Martí, R.; Nishino, I.; Tadesse, S.; Pizzorno, G.; Shungu, D.C.; Bonilla, E.; et al. Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase-deficient mice. Hum. Mol. Genet. 2008, 18, 714–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishigaki, Y.; Martiand, R.; Hirano, M. ND5 is a hot-spot for multiple atypical mitochondrial DNA deletions in mitochondrial neurogastrointestinal encephalomyopathy. Hum. Mol. Genet. 2003, 13, 91–101. [Google Scholar] [CrossRef]
- Cámara, Y.; Vioque, E.G.; Scarpelli, M.; Torres-Torronteras, J.; Caballero, A.; Hirano, M.; Martí, R. Administration of deoxyribonucleosides or inhibition of their catabolism as a pharmacological approach for mitochondrial DNA depletion syndrome. Hum. Mol. Genet. 2014, 23, 2459–2467. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Perez-Torras, S. Nucleoside transporter proteins as biomarkers of drug responsiveness and drug targets. Front. Pharmacol. 2015, 6, 13. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Urtasun, N.; Pérez-Torras, S. Intestinal Nucleoside Transporters: Function, Expression, and Regulation. Compr. Physiol. 2011, 8, 1003–1017. [Google Scholar] [CrossRef]
- Lai, Y.; Tse, C.-M.; Unadkat, J.D. Mitochondrial Expression of the Human Equilibrative Nucleoside Transporter 1 (hENT1) Results in Enhanced Mitochondrial Toxicity of Antiviral Drugs. J. Biol. Chem. 2004, 279, 4490–4497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, N.V.; Morris, M.R.; Cangul, H.; Gleeson, D.; Straatman-Iwanowska, A.; Davies, N.; Keenan, S.; Pasha, S.; Rahman, F.; Gentle, D.; et al. Mutations in SLC29A3, Encoding an Equilibrative Nucleoside Transporter ENT3, Cause a Familial Histiocytosis Syndrome (Faisalabad Histiocytosis) and Familial Rosai-Dorfman Disease. PLoS Genet. 2010, 6, e1000833. [Google Scholar] [CrossRef] [PubMed]
- Wevers, R.A.; Christensen, M.; Engelke, U.F.H.; Geuer, S.; Coene, K.; Kwast, J.T.; Lund, A.M.; Vissers, L.E.L.M. Functional disruption of pyrimidine nucleoside transporter CNT1 results in a novel inborn error of metabolism with high excretion of uridine and cytidine. J. Inherit. Metab. Dis. 2019, 42, 494–500. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Suleiman, J.; Almannai, M.; Scaglia, F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases. Mol. Genet. Metab. 2018, 125, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Yapa, N.M.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef] [PubMed]
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 2010, 21, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M.; Okano, Y. Human Misato regulates mitochondrial distribution and morphology. Exp. Cell Res. 2007, 313, 1393–1404. [Google Scholar] [CrossRef]
- Kishita, Y.; Shimura, M.; Kohda, M.; Akita, M.; Imai-Okazaki, A.; Yatsuka, Y.; Nakajima, Y.; Ito, T.; Ohtake, A.; Murayama, K.; et al. A novel homozygous variant in MICOS13/QIL1 causes hepato-encephalopathy with mitochondrial DNA depletion syndrome. Mol. Genet. Genom. Med. 2020, 8, e1427. [Google Scholar] [CrossRef]
- Martinelli, P.; La Mattina, V.; Bernacchia, A.; Magnoni, R.; Cerri, F.; Cox, G.; Quattrini, A.; Casari, G.; Rugarli, E.I. Genetic interaction between the m -AAA protease isoenzymes reveals novel roles in cerebellar degeneration. Hum. Mol. Genet. 2009, 18, 2001–2013. [Google Scholar] [CrossRef] [Green Version]
- Baderna, V.; Schultz, J.; Kearns, L.S.; Fahey, M.; Thompson, B.A.; Ruddle, J.B.; Huq, A.; Maltecca, F. A novel AFG3L2 mutation close to AAA domain leads to aberrant OMA1 and OPA1 processing in a family with optic atrophy. Acta Neuropathol. Commun. 2020, 8, 93. [Google Scholar] [CrossRef]
- Pfeffer, G.; Gorman, G.; Griffin, H.R.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137, 1323–1336. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef]
- Haghighi, A.; Haack, T.B.; Atiq, M.; Mottaghi, H.; Haghighi-Kakhki, H.; Bashir, R.A.; Ahting, U.; Feichtinger, R.G.; Mayr, J.A.; Rötig, A.; et al. Sengers syndrome: Six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients. Orphanet J. Rare Dis. 2014, 9, 119. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Epand, R.F.; Berno, B.; Pelosi, L.; Brandolin, G. Association of Phosphatidic Acid with the Bovine Mitochondrial ADP/ATP Carrier. Biochemistry 2009, 48, 12358–12364. [Google Scholar] [CrossRef]
- Kowluru, A.; Tannous, M.; Chen, H.Q. Localization and characterization of the mitochondrial isoform of the nucleoside diphosphate kinase in the pancreatic beta cell: Evidence for its complexation with mitochondrial succinyl-CoA synthetase. Arch. Bichem. Biophys. 2002, 398, 160–169. [Google Scholar] [CrossRef]
- Blázquez-Bermejo, C.; Carreño-Gago, L.; Molina-Granada, D.; Aguirre, J.; Ramón, J.; Torres-Torronteras, J.; Cabrera-Pérez, R.; Martin, M.Á.; Domínguez-González, C.; Cruz, X.; et al. Increased dNTP pools rescue mtDNA depletion in human POLG-deficient fibroblasts. FASEB J. 2019, 33, 7168–7179. [Google Scholar] [CrossRef]
- Dalla Rosa, I.; Camara, Y.; Durigon, R.; Moss, C.F.; Vidoni, S.; Akman, G.; Hunt, L.; Johnson, M.A.; Grocott, S.; Wang, L.; et al. MPV17 Loss Causes Deoxynucleotide Insufficiency and Slow DNA Replication in Mitochondrial. PLoS Genet. 2016, 12, e1005779. [Google Scholar] [CrossRef]
- Blázquez-Bermejo, C.; Molina-Granada, D.; Vila-Julià, F.; Jiménez-Heis, D.; Zhou, X.; Torres-Torronteras, J.; Karlsson, A.; Martí, R.; Cámara, Y. Age-related metabolic changes limit efficacy of deoxynucleoside-based therapy in thymidine kinase 2-deficient mice. EBioMedicine 2019, 46, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Nambot, S.; Gavrilov, D.; Thevenon, J.; Bruel, A.; Bainbridge, M.; Rio, M.; Goizet, C.; Rotig, A.; Jaeken, J.; Niu, N.; et al. Further delineation of a rare recessive encephalomyopathy linked to mutations in GFER thanks to data sharing of whole exome sequencing data. Clin. Genet. 2017, 92, 188–198. [Google Scholar] [CrossRef]
- Alsina, D.; Lytovchenko, O.; Schab, A.; Atanassov, I.; Schober, F.; Jiang, M.; Koolmeister, C.; Wedell, A.; Taylor, R.W.; Wredenberg, A.; et al. FBXL 4 deficiency increases mitochondrial removal by autophagy. EMBO Mol. Med. 2020, 12, 11659. [Google Scholar] [CrossRef]
- Marchet, S.; Legati, A.; Nasca, A.; di Meo, I.; Spagnolo, M.; Zanetti, N.; Lamantea, E.; Catania, A.; Lamperti, C.; Ghezzi, D. Homozygous mutations in C1QBP as cause of progressive external ophthalmoplegia (PEO) and mitochondrial myopathy with multiple mtDNA deletions. Hum. Mutat. 2020, 41, 1745–1750. [Google Scholar] [CrossRef]
- Camp, K.M.; Krotoski, D.; Parisi, M.A.; Gwinn, K.A.; Cohen, B.H.; Cox, C.S.; Enns, G.M.; Falk, M.J.; Goldstein, A.C.; Gopal-Srivastava, R.; et al. Nutritional interventions in primary mitochondrial disorders: Developing an evidence base. Mol. Genet. Metab. 2016, 119, 187–206. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S. Pathophysiology of mitochondrial disease causing epilepsy and status epilepticus. Epilepsy Behav. 2015, 49, 71–75. [Google Scholar] [CrossRef]
- Krahenbuhl, S.; Brandner, S.; Kleinle, S.; Liechti, S.; Straumann, D. Mitochondrial diseases represent a risk factor for valproate-induced fulminant liver failure. Liver Int. 2000, 20, 346–348. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA Depletion Syndromes: Review and Updates of Genetic Basis, Manifestations, and Therapeutic Options. Neurotherapeutics 2013, 10, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Kuszak, A.; Espey, M.G.; Falk, M.J.; Holmbeck, M.A.; Manfredi, G.; Shadel, G.S.; Vernon, H.J.; Zolkipli-Cunningham, Z. Nutritional Interventions for Mitochondrial OXPHOS Deficiencies: Mechanisms and Model Systems. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 163–191. [Google Scholar] [CrossRef]
- Santra, S.; Gilkerson, R.W.; Davidson, M.; Schon, E.A. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann. Neurol. 2004, 56, 662–669. [Google Scholar] [CrossRef]
- Ahola-Erkkilä, S.; Carroll, C.J.; Peltola-Mjösund, K.; Tulkki, V.; Mattila, I.; Seppänen-Laakso, T.; Oresic, M.; Tyynismaa, H.; Suomalainen, A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum. Mol. Genet. 2010, 19, 1974–1984. [Google Scholar] [CrossRef] [Green Version]
- Kaji, S.; Murayama, K.; Nagata, I.; Nagasaka, H.; Takayanagi, M.; Ohtake, A.; Iwasa, H.; Nishiyama, M.; Okazaki, Y.; Harashima, H.; et al. Fluctuating liver functions in siblings with MPV17 mutations and possible improvement associated with dietary and pharmaceutical treatments targeting respiratory chain complex II. Mol. Genet. Metab. 2009, 97, 292–296. [Google Scholar] [CrossRef]
- Hasselmann, O.; Blau, N.; Ramaekers, V.T.; Quadros, E.V.; Sequeira, J.; Weissert, M. Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol. Genet. Metab. 2010, 99, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.C.; Macdonald, J.R.; Mahoney, D.J.; Parise, G.; Beal, M.F.; Tarnopolsky, M.A. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve 2007, 35, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.A. Exercise as a Therapeutic Strategy for Primary Mitochondrial Cytopathies. J. Child. Neurol. 2014, 29, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [Green Version]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, J.; Purhonen, J.; Tegelberg, S.; Smolander, O.; Mörgelin, M.; Rozman, J.; Gailus-Durner, V.; Fuchs, H.; de Angelis, M.H.; Auvinen, P.; et al. Alternative oxidase-mediated respiration prevents lethal mitochondrial cardiomyopathy. EMBO Mol. Med. 2019, 11, e9456. [Google Scholar] [CrossRef]
- Siegmund, S.; Yang, H.; Sharma, R.; Javors, M.; Skinner, O.; Mootha, V.; Hirano, M.; Schon, E.A. Low-dose rapamycin extends lifespan in a mouse model of mtDNA depletion syndrome. Hum. Mol. Genet. 2017, 26, 4588–4605. [Google Scholar] [CrossRef] [Green Version]
- Sharma, H.; Singh, D.; Mahant, A.; Sohal, S.K.; Kesavan, A.K. Samiksha Development of mitochondrial replacement therapy: A review. Heliyon 2020, 6, 04643. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Turnbull, D.M.; Minczuk, M.; Gammage, P.A. Therapeutic Manipulation of mtDNA Heteroplasmy: A Shifting Perspective. Trends Mol. Med. 2020, 26, 698–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes, C.T.; Shanske, S.; Tritschler, H.J.; Aprille, J.R.; Andreetta, F.; Bonilla, E.; Schon, E.A.; DiMauro, S. mtDNA depletion with variable tissue expression: A novel genetic abnormality in mitochondrial diseases. Am. J. Hum. Genet. 1991, 48, 492–501. [Google Scholar] [PubMed]
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989, 339, 309–311. [Google Scholar] [CrossRef]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.D.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef]
- Roeben, B.; Marquetand, J.; Bender, B.; Billing, H.; Haack, T.B.; Sanchez-Albisua, I.; Schols, L.; Blom, H.J.; Synofzik, M. Hemodialysis in MNGIE transiently reduces serum and urine levels of thymidine and deoxyuridine, but not CSF levels and neurological function. Orphanet J. Rare Dis. 2017, 12, 135. [Google Scholar] [CrossRef]
- Yavuz, H.; Özel, A.; Christensen, M.; Christensen, E.; Schwartz, M.; Elmaci, M.; Vissing, J. Treatment of Mitochondrial Neurogastrointestinal Encephalomyopathy with Dialysis. Arch. Neurol. 2007, 64, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Pérez, R.; Torres-Torronteras, J.; Vila-Julià, F.; Ortega, F.; Cámara, Y.; Barquinero, J.; Martí, R. Prospective therapeutic approaches in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Expert Opin. Orphan Drugs 2015, 3, 1167–1182. [Google Scholar] [CrossRef]
- Desgranges, C.; Razaka, G.; Rabaud, M.; Bricaud, H. Catabolism of thymidine in human blood platelets purification and properties of thymidine phosphorylase. Biochim. Biophys. Acta (BBA) Nucleic Acids Protein Synth. 1981, 654, 211–218. [Google Scholar] [CrossRef]
- Yoshimura, A.; Kuwazuru, Y.; Furukawa, T.; Yoshida, H.; Yamada, K.; Akiyama, S.-I. Purification and tissue distribution of human thymidine phosphorylase; high expression in lymphocytes, reticulocytes and tumors. Biochim. Biophys. Acta (BBA) Gen. Subj. 1990, 1034, 107–113. [Google Scholar] [CrossRef]
- Cass, C.E.; Young, J.D.; Baldwin, S.A.; Cabrita, M.A.; Graham, K.A.; Griffiths, M.; Jennings, L.L.; Mackey, J.R.; Ng, A.M.L.; Ritzel, M.W.L.; et al. Nucleoside Transporters of Mammalian Cells. Membr. Transp. Drug Targets 1999, 12, 313–352. [Google Scholar] [CrossRef]
- De Vocht, C.; Ranquin, A.; van Ginderachter, J.; Vanhaecke, T.; Rogiers, V.; van Gelder, P.; Versées, W.; Steyaert, J. Polymeric nanoreactors for enzyme replacement therapy of MNGIE. J. Control. Release 2010, 148, e19–e20. [Google Scholar] [CrossRef]
- De Vocht, C.; Ranquin, A.; Willaert, R.; van Ginderachter, J.A.; Vanhaecke, T.; Rogiers, V.; Versées, W.; van Gelder, P.; Steyaert, J. Assessment of stability, toxicity and immunogenicity of new polymeric nanoreactors for use in enzyme replacement therapy of MNGIE. J. Control. Release 2009, 137, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Moran, N.F.; Bain, M.D.; Muqit, M.; Bax, B.E. Carrier erythrocyte entrapped thymidine phosphorylase therapy for MNGIE. Neurology 2008, 71, 686–688. [Google Scholar] [CrossRef]
- Levene, M.; Coleman, D.G.; Kilpatrick, H.C.; Fairbanks, L.D.; Gangadharan, B.; Gasson, C.; Bax, B.E. Pre-clinical toxicity evaluation of erythrocyte-encapsulated thymidine phosphorylase in BALB/c mice and beagle dogs: An enzyme-replacement therapy for mitochondrial neurogastrointestinal encephalomyopathy. Toxicol. Sci. 2013, 131, 311–324. [Google Scholar] [CrossRef]
- Bax, B.E.; Bain, M.D.; Scarpelli, M.; Filosto, M.; Tonin, P.; Moran, N. Clinical and biochemical improvements in a patient with MNGIE following enzyme replacement. Neurology 2013, 81, 1269–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levene, M.; Bain, M.D.; Moran, N.F.; Nirmalananthan, N.; Poulton, J.; Scarpelli, M.; Filosto, M.; Mandel, H.; MacKinnon, A.D.; Fairbanks, L.; et al. Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in Mitochondrial Neurogastrointestinal Encephalomyopathy. J. Clin. Med. 2019, 8, 457. [Google Scholar] [CrossRef] [Green Version]
- Bax, B.E.; Levene, M.; Bain, M.D.; Fairbanks, L.D.; Filosto, M.; Uçar, S.K.; Klopstock, T.; Kornblum, C.; Mandel, H.; Rahman, S.; et al. Erythrocyte Encapsulated Thymidine Phosphorylase for the Treatment of Patients with Mitochondrial Neurogastrointestinal Encephalomyopathy: Study Protocol for a Multi-Centre, Multiple Dose, Open Label Trial. J. Clin. Med. 2019, 8, 1096. [Google Scholar] [CrossRef] [Green Version]
- Lara, M.C.; Weiss, B.; Illa, I.; Madoz, P.; Massuet, L.; Andreu, A.L.; Valentino, M.L.; Anikster, Y.; Hirano, M.; Marti, R. Infusion of platelets transiently reduces nucleoside overload in MNGIE. Neurology 2006, 67, 1461–1463. [Google Scholar] [CrossRef]
- Hussein, E. Non-myeloablative bone marrow transplant and platelet infusion can transiently improve the clinical outcome of mitochondrial neurogastrointestinal encephalopathy: A case report. Transfus. Apher. Sci. 2013, 49, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Marti, R.; Casali, C.; Tadesse, S.; Uldrick, T.; Fine, B.; Escolar, D.M.; Valentino, M.L.; Nishino, I.; Hesdorffer, C.; et al. Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology 2006, 67, 1458–1460. [Google Scholar] [CrossRef]
- Halter, J.; Schüpbach, W.M.M.; Casali, C.; Elhasid, R.; Fay, K.; Hammans, S.; Illa, I.; Kappeler, L.; Krähenbühl, S.; Lehmann, T.; et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): A consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2010, 46, 330–337. [Google Scholar] [CrossRef]
- Halter, J.P.; Michael, W.; Schupbach, M.; Mandel, H.; Casali, C.; Orchard, K.; Collin, M.; Valcarcel, D.; Rovelli, A.; Filosto, M.; et al. Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain 2015, 138, 2847–2858. [Google Scholar] [CrossRef] [PubMed]
- Filosto, M.; Scarpelli, M.; Tonin, P.; Lucchini, G.; Pavan, F.; Santus, F.; Parini, R.; Donati, M.A.; Cotelli, M.S.; Vielmi, V.; et al. Course and management of allogeneic stem cell transplantation in patients with mitochondrial neurogastrointestinal encephalomyopathy. J. Neurol. 2012, 259, 2699–2706. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Karaa, A.; Goldstein, A.; Ng, Y.S.; Gorman, G.; Feigenbaum, A.; Christodoulou, J.; Haas, R.; Tarnopolsky, M.; Cohen, B.K.; et al. Solid organ transplantation in primary mitochondrial disease: Proceed with caution. Mol. Genet. Metab. 2016, 118, 178–184. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Wang, J.; Dai, H.; Almannai, M.; Scaglia, F.; Craigen, W.J.; Wong, L.J.C. MPV17-related mitochondrial DNA maintenance defect. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Shimura, M.; Kuranobu, N.; Ogawa-Tominaga, M.; Akiyama, N.; Sugiyama, Y.; Ebihara, T.; Fushimi, T.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; et al. Clinical and molecular basis of hepatocerebral mitochondrial DNA depletion syndrome in Japan: Evaluation of outcomes after liver transplantation. Orphanet J. Rare Dis. 2020, 15, 169. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef]
- Kelly, D.A. Liver transplantation: To do or not to do? Pediatr. Transplant. 2000, 4, 170–172. [Google Scholar] [CrossRef]
- Tzoulis, C.; Engelsen, B.A.; Telstad, W.; Aasly, J.; Zeviani, M.; Winterthun, S.; Ferrari, G.; Aarseth, J.H.; Bindoff, L.A. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: A study of 26 cases. Brain 2006, 129, 1685–1692. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.-J.; Naviaux, R.K.; Brunetti-Pierri, N.; Zhang, Q.; Schmitt, E.S.; Truong, C.; Milone, M.; Cohen, B.H.; Wical, B.; Ganesh, J.; et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum. Mutat. 2008, 29, E150–E172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boschetti, E.; D’Alessandro, R.; Bianco, F.; Carelli, V.; Cenacchi, G.; Pinna, A.D.; Del Gaudio, M.; Rinaldi, R.; Stanghellini, V.; Pironi, L.; et al. Liver as a Source for Thymidine Phosphorylase Replacement in Mitochondrial Neurogastrointestinal Encephalomyopathy. PLoS ONE 2014, 9, e96692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, R.; Boschetti, E.; Amore, G.; Costa, R.; Pugliese, A.; Caporali, L.; Gramegna, L.L.; Papa, V.; Vizioli, L.; Capristo, M.; et al. Liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Clinical long-term follow-up and pathogenic implications. J. Neurol. 2020, 267, 3702–3710. [Google Scholar] [CrossRef]
- D’Angelo, R.; Rinaldi, R.; Pironi, L.; Dotti, M.T.; Pinna, A.D.; Boschetti, E.; Capristo, M.; Mohamed, S.; Contin, M.; Caporali, L.; et al. Liver transplant reverses biochemical imbalance in mitochondrial neurogastrointestinal encephalomyopathy. Mitochondrion 2017, 34, 101–102. [Google Scholar] [CrossRef]
- De Giorgio, R.; Pironi, L.; Rinaldi, R.; Boschetti, E.; Caporali, L.; Capristo, M.; Casali, C.; Cenacchi, G.; Contin, M.; D’Angelo, R.; et al. Liver transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Ann. Neurol. 2016, 80, 448–455. [Google Scholar] [CrossRef]
- Kripps, K.; Nakayuenyongsuk, W.; Shayota, B.J.; Berquist, W.; Gomez-Ospina, N.; Esquivel, C.O.; Concepcion, W.; Sampson, J.B.; Cristin, D.J.; Jackson, W.E.; et al. Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Mol. Genet. Metab. 2020, 130, 58–64. [Google Scholar] [CrossRef]
- Taanman, J.-W.; Muddle, J.R.; Muntau, A.C. Mitochondrial DNA depletion can be prevented by dGMP and dAMP supplementation in a resting culture of deoxyguanosine kinase-deficient fibroblasts. Hum. Mol. Genet. 2003, 12, 1839–1845. [Google Scholar] [CrossRef] [Green Version]
- Akman, H.O.; Dorado, B.; Lopez, L.C.; García-Cazorla, Á.; Vilà, M.R.; Tanabe, L.M.; Dauer, W.T.; Bonilla, E.; Tanji, K.; Hirano, M. Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum. Mol. Genet. 2008, 17, 2433–2440. [Google Scholar] [CrossRef] [Green Version]
- Domínguez-González, C.; Hernández-Laín, A.; Rivas, E.; Hernández-Voth, A.; Catalán, J.S.; Fernandez-Torron, R.; Fuiza-Luces, C.; García, J.G.; Morís, G.; Olivé, M.; et al. Late-onset thymidine kinase 2 deficiency: A review of 18 cases. Orphanet J. Rare Dis. 2019, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Camara, Y.; Carreño-Gago, L.; Martín, M.A.; Melia, M.J.; Blázquez, A.; Delmiro, A.; Garrabou, G.; Morén, C.; Díaz-Manera, J.; Gallardo, E.; et al. Severe TK2 enzyme activity deficiency in patients with mild forms of myopathy. Neurology 2015, 84, 2286–2288. [Google Scholar] [CrossRef]
- Garone, C.; Garcia-Diaz, B.; Emmanuele, V.; Lopez, L.C.; Tadesse, S.; Akman, H.O.; Tanji, K.; Quinzii, C.M.; Hirano, M. Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol. Med. 2014, 6, 1016–1027. [Google Scholar] [CrossRef]
- Lopez-Gomez, C.; Levy, R.J.; Sanchez-Quintero, M.J.; Juanola-Falgarona, M.; Barca, E.; Garcia-Diaz, B.; Tadesse, S.; Garone, C.; Hirano, M. Deoxycytidine and Deoxythymidine Treatment for Thymidine Kinase 2 Deficiency. Ann. Neurol. 2017, 81, 641–652. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Solaroli, N.; Bjerke, M.; Stewart, J.B.; Rozell, B.; Johansson, M.; Karlsson, A. Progressive loss of mitochondrial DNA in thymidine kinase 2-deficient mice. Hum. Mol. Genet. 2008, 17, 2329–2335. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gomez, C.; Hewan, H.; Sierra, C.; Akman, H.O.; Sanchez-Quintero, M.J.; Juanola-Falgarona, M.; Tadesse, S.; Tanji, K.; Konofagou, E.E.; Hirano, M. Bioavailability and cytosolic kinases modulate response to deoxynucleoside therapy in TK2 deficiency. EBioMedicine 2019, 46, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Domínguez-González, C.; Madruga-Garrido, M.; Mavillard, F.; Garone, C.; Aguirre-Rodríguez, F.J.; Donati, M.A.; Kleinsteuber, K.; Martí, I.; Martín-Hernández, E.; Morealejo-Aycinena, J.P.; et al. Deoxynucleoside Therapy for Thymidine Kinase 2–Deficient Myopathy. Ann. Neurol. 2019, 86, 293–303. [Google Scholar] [CrossRef]
- Hernandez-Voth, A.; Catalan, J.S.; Blanco, M.C.; Mendez, A.C.; Martin, M.A.; de la Hoz, C.D.F.F.; Garrido, V.V.; Dominguez-Gonzalez, C. Deoxynucleoside therapy for respiratory involvement in adult patients with thymidine kinase 2-deficient myopathy. BMJ Open Respir. Res. 2020, 7, e000774. [Google Scholar] [CrossRef]
- Dominguez-Gonzalez, C.; Badosa, C.; Madruga-Garrido, M.; Marti, I.; Paradas, C.; Ortez, C.; Diaz-Manera, J.; Berardo, A.; Alonso-Perez, J.; Trifunov, S.; et al. Growth Differentiation Factor 15 is a potential biomarker of therapeutic response for TK2 deficient myopathy. Sci. Rep. 2020, 10, 10111. [Google Scholar] [CrossRef]
- Bulst, S.; Holinski-Feder, E.; Payne, B.; Abicht, A.; Krause, S.; Lochmüller, H.; Chinnery, P.F.; Walter, M.C.; Horvath, R. In vitro supplementation with deoxynucleoside monophosphates rescues mitochondrial DNA depletion. Mol. Genet. Metab. 2012, 107, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Pontarin, G.; Ferraro, P.; Bee, L.; Reichard, P.; Bianchi, V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. USA 2012, 109, 13302–13307. [Google Scholar] [CrossRef] [Green Version]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Silva-Pinheiro, P.; Pardo-Hernández, C.; Reyes, A.; Tilokani, L.; Mishra, A.; Cerutti, R.; Li, S.; Rozsivalova, D.-H.; Valenzuela, S.; Dogan, S.A.; et al. DNA polymerase gamma mutations that impair holoenzyme stability cause catalytic subunit depletion. Nucleic Acids Res. 2021, 49, 5230–5248. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Flierl, A.; Chen, Y.; Coskun, P.E.; Samulski, R.J.; Wallace, D.C. Adeno-associated virus-mediated gene transfer of the heart/muscle adenine nucleotide translocator (ANT) in mouse. Gene Ther. 2005, 12, 570–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viscomi, C.; Spinazzola, A.; Maggioni, M.; Fernandez-Vizarra, E.; Massa, V.; Pagano, C.; Vettor, R.; Mora, M.; Zeviani, M. Early-onset liver mtDNA depletion and late-onset proteinuric nephropathy in Mpv17 knockout mice. Hum. Mol. Genet. 2008, 18, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Bottani, E.; Giordano, C.; Civiletto, G.; di Meo, I.; Auricchio, A.; Ciusani, E.; Marchet, S.; Lamperti, C.; D’Amati, G.; Viscomi, C.; et al. AAV-mediated Liver-specific MPV17 Expression Restores mtDNA Levels and Prevents Diet-induced Liver Failure. Mol. Ther. 2014, 22, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Torres-Torronteras, J.; Gomez, A.; Eixarch, H.; Palenzuela, L.; Pizzorno, G.; Hirano, M.; Andreu, A.L.; Barquinero, J.; Marti, R. Hematopoietic gene therapy restores thymidine phosphorylase activity in a cell culture and a murine model of MNGIE. Gene Ther. 2011, 18, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Torres-Torronteras, J.; Cabrera-Perez, R.; Barba, I.; Costa, C.; de Luna, N.; Andreu, A.L.; Barquinero, J.; Hirano, M.; Camara, Y.; Marti, R. Long-Term Restoration of Thymidine Phosphorylase Function and Nucleoside Homeostasis Using Hematopoietic Gene Therapy in a Murine Model of Mitochondrial Neurogastrointestinal Encephalomyopathy. Hum. Gene Ther. 2016, 27, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Yadak, R.; Cabrera-Pérez, R.; Torres-Torronteras, J.; Bugiani, M.; Haeck, J.C.; Huston, M.W.; Bogaerts, E.; Goffart, S.; Jacobs, E.H.; Stok, M.; et al. Preclinical Efficacy and Safety Evaluation of Hematopoietic Stem Cell Gene Therapy in a Mouse Model of MNGIE. Mol. Ther. Methods Clin. Dev. 2018, 8, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Torres-Torronteras, J.; Cabrera-Perez, R.; Vila-Julia, F.; Viscomi, C.; Camara, Y.; Hirano, M.; Zeviani, M.; Marti, R. Long-Term Sustained Effect of Liver-Targeted Adeno-Associated Virus Gene Therapy for Mitochondrial Neurogastrointestinal Encephalomyopathy. Hum. Gene Ther. 2018, 29, 708–718. [Google Scholar] [CrossRef]
- Torres-Torronteras, J.; Viscomi, C.; Cabrera-Pérez, R.; Cámara, Y.; di Meo, I.; Barquinero, J.; Auricchio, A.; Pizzorno, G.; Hirano, M.; Zeviani, M.; et al. Gene Therapy Using a Liver-targeted AAV Vector Restores Nucleoside and Nucleotide Homeostasis in a Murine Model of MNGIE. Mol. Ther. 2014, 22, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Perez, R.; Vila-Julia, F.; Hirano, M.; Mingozzi, F.; Torres-Torronteras, J.; Marti, R. The alpha-1-antitrypsin promoter improves the efficacy of an AAV vector for the treatment of MNGIE. Hum. Gene Ther. 2019, 30, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Vila-Julià, F.; Cabrera-Pérez, R.; Cámara, Y.; Molina-Berenguer, M.; Lope-Piedrafita, S.; Hirano, M.; Mingozzi, F.; Torres-Torronteras, J.; Martí, R. Efficacy of adeno-associated virus gene therapy in a MNGIE murine model enhanced by chronic exposure to nucleosides. EBioMedicine 2020, 62, 103133. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Couto, L.B.; Patarroyo-White, S.; Liu, T.; Nagy, D.; Vargas, J.A.; Zhou, S.; Scallan, C.D.; Sommer, J.; Vijay, S.; et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood 2006, 108, 3321–3328. [Google Scholar] [CrossRef] [Green Version]
- Nathwani, A.C.; Gray, J.T.; Ng, C.Y.C.; Zhou, J.; Spence, Y.; Waddington, S.N.; Tuddenham, E.G.D.; Kemball-Cook, G.; McIntosh, J.; Boon-Spijker, M.; et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 2006, 107, 2653–2661. [Google Scholar] [CrossRef]
- Vercauteren, K.; Hoffman, B.E.; Zolotukhin, I.; Keeler, G.D.; Xiao, J.W.; Basner-Tschakarjan, E.; High, K.; Ertl, H.C.; Rice, C.M.; Srivastava, A.; et al. Superior In Vivo Transduction of Human Hepatocytes Using Engineered AAV3 Capsid. Mol. Ther. 2016, 24, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 Overexpression Ameliorates the Phenotype of Two Mitochondrial Disease Mouse Models. Cell Metab. 2015, 21, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Korwitz, A.; Merkwirth, C.; Richter-Dennerlein, R.; Troeder, S.E.; Sprenger, H.-G.; Quiros, P.M.; López-Otín, C.; Rugarli, E.; Langer, T. Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J. Cell Biol. 2016, 212, 157–166. [Google Scholar] [CrossRef]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Weaver, W.D.; Cohen, B.H. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology 2018, 90, e1212–e1221. [Google Scholar] [CrossRef]
- Szeto, H.H.; Birk, A.V. Serendipity and the Discovery of Novel Compounds That Restore Mitochondrial Plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Pitayu, L.; Baruffini, E.; Rodier, C.; Rotig, A.; Lodi, T.; Delahodde, A. Combined use of Saccharomyces cerevisiae, Caenorhabditis elegans and patient fibroblasts leads to the identification of clofilium tosylate as a potential therapeutic chemical against POLG-related diseases. Hum. Mol. Genet. 2016, 25, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Facchinello, N.; Laquatra, C.; Locatello, L.; Beffagna, G.; Branas Casas, R.; Fornetto, C.; Dinarello, A.; Martorano, L.; Vettori, A.; Risato, G.; et al. Efficient clofilium tosylate-mediated rescue of POLG-related disease phenotypes in zebrafish. Cell Death Dis. 2021, 12, 100. [Google Scholar] [CrossRef]
- Munro, B.; Horvath, R.; Muller, J.S. Nucleoside supplementation modulates mitochondrial DNA copy number in the dguok −/− zebrafish. Hum. Mol. Genet. 2019, 28, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Ruzzenente, B.; Rötig, A.; Metodiev, M.D. Mouse models for mitochondrial diseases. Hum. Mol. Genet. 2016, 25, R115–R122. [Google Scholar] [CrossRef] [Green Version]
- Tyynismaa, H.; Suomalainen, A. Mouse models of mtDNA replication diseases. Methods 2010, 51, 405–410. [Google Scholar] [CrossRef]
- Rahn, J.J.; Bestman, J.E.; Stackley, K.D.; Chan, S.S. Zebrafish lacking functional DNA polymerase gamma survive to juvenile stage, despite rapid and sustained mitochondrial DNA depletion, altered energetics and growth. Nucleic Acids Res. 2015, 43, 10338–10352. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Takeda, S.; Sagiya, Y.; Gotoh, M.; Nakamura, Y.; Arakawa, H. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat. Genet. 2003, 34, 440–445. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.N.; Paetau, A.; Suomalainen-Wartiovaara, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Curbo, S.; Zhao, Q.; Krishnan, S.; Kuiper, R.; Karlsson, A. Severe mtDNA depletion and dependency on catabolic lipid metabolism in DGUOK knockout mice. Hum. Mol. Genet. 2019, 28, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, B.; Garone, C.; Barca, E.; Mojahed, H.; Gutierrez, P.; Pizzorno, G.; Tanji, K.; Arias-Mendoza, F.; Quinzii, C.M.; Hirano, M. Deoxynucleoside stress exacerbates the phenotype of a mouse model of mitochondrial neurogastrointestinal encephalopathy. Brain 2014, 137, 1337–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darin, N.; Oldfors, A.; Moslemi, A.R.; Holme, E.; Tulinius, M. The incidence of mitochondrial encephalomyopathies in childhood: Clinical features and morphological, biochemical, and DNA abnormalities. Ann. Neurol. 2001, 49, 377–383. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, R.; Rinaldi, R.; Carelli, V.; Boschetti, E.; Caporali, L.; Capristo, M.; Casali, C.; Cenacchi, G.; Gramegna, L.L.; Lodi, R.; et al. ITA-MNGIE: An Italian regional and national survey for mitochondrial neuro-gastro-intestinal encephalomyopathy. Neurol. Sci. 2016, 37, 1149–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Category | Gene | Protein | Protein Function/Pathway | Clinical Features | Type of Inheritance | Type of mtDNA Aberration | OMIM # (Gene) | Reference * (Year) | |
|---|---|---|---|---|---|---|---|---|---|
| mtDNA replication machinery | POLG | DNA polymerase gamma | Polymerase | Alpers–Huttenlocher syndrome/ataxia/PEO | AR/AD | D/MD/PM | 174763 | [16] (2001) | |
| POLG2 | DNA polymerase subunit gamma-2 | Polymerase (ancillary) | PEO/hepatic failure | AD/AR | MD/D | 604983 | [17] (2006) | ||
| TWNK | Twinkle | Helicase | Perrault syndrome/PEO/ataxia/encephalopathy/IOSCA | AD/AR | D/MD/PM | 606075 | [18] (2001) | ||
| MGME1 | Mitochondrial genome maintenance exonuclease 1 | Exonuclease | PEO/emaciation | AR | D/MD | 615076 | [19] (2013) | ||
| DNA2 | DNA replication ATP-dependent helicase/nuclease DNA2 | Helicase/nuclease | PEO/myopathy/Seckel syndrome | AD | MD | 601810 | [20] (2013) | ||
| RNASEH1 | Ribonuclease H1 | Ribonuclease | PEO/muscle weakness/dysphagia/spinocerebellar signs | AR | D/MD | 604123 | [21] (2015) | ||
| TFAM | Mitochondrial transcription factor A | Transcription factor | Neonatal liver failure | AR | D | 600438 | [22] (2016) | ||
| TOP3A | DNA topoisomerase 3 alpha | Topoisomerase | PEO/Bloom syndrome-like disorder | AR | MD/D | 601243 | [23] (2018) | ||
| SSBP1 | Mitochondrial single strand binding protein | ssDNA stabilization | Optic atrophy/liver failure/neurological syndrome /retinopathy | AD/AR | D | 600439 | [24] (2019) | ||
| LIG3 | Ligase III | Mitochondrial DNA ligase | MNGIE-like | AR | D | 600940 | [25] (2021) | ||
| dNTP metabolism | TYMP | Thymidine phosphorylase | Nucleoside catabolism | MNGIE | AR | D/MD/PM | 603041 | [26] (1999) | |
| TK2 | Thymidine kinase 2 | dNTP anabolism | Myopathy/PEO | AR | D/MD | 188250 | [27] (2001) | ||
| DGUOK | Deoxyguanosine kinase | dNTP anabolism | Neurohepatopathy/myopathy/PEO | AR | D/MD | 601465 | [28] (2001) | ||
| RRM2B | p53-subunit of ribonucleotide reductase | dNTP anabolism | Encephalomyopathy/PEO /MNGIE/KSS/neuropathy/deafness/tubulopathy | AR / AD | D/MD | 604712 | [29] (2007) | ||
| Mitochondrial dynamics | OPA1 | Dynamin-like 120 kDa protein, mitochondrial | GTPase/mitochondrial fusion | Optic atrophy/Behr syndrome | AD | MD | 605290 | [30] (2008) | |
| MFN2 | Mitofusin-2 | GTPase/mitochondrial fusion | Optic atrophy/myopathy/axonal neuropathy/Charcot-Marie-Tooth | AR / AD | D/MD | 608507 | [31] (2012) | ||
| SPG7 | Paraplegin | Subunit of m-AAA protease | PEO/spastic paraplegia | AR | MD | 602783 | [32] (2014) | ||
| AFG3L2 | AFG3-like protein 2 | Subunit of m-AAA protease | PEO/ataxia | AD | MD | 604581 | [33] (2015) | ||
| MSTO1 | Protein misato homolog 1 | Mitochondrial fusion | Muscular dystrophy with cerebellar involvement/myopathy/ataxia | AR | D | 617619 | [34] (2017) | ||
| MICOS13 | MICOS complex subunit MIC13 | Maintenance of cristae structure | Hepato-encephalopathy | AR | D | 616658 | [35] (2019) | ||
| Unknown pathomechanism | Membrane channels | SLC25A4 | Adenine nucleotide translocator | ADP/ATP carrier | PEO/cardiomyopathy/myopathy | AD / AR | MD | 103220 | [36] (2000) |
| MPV17 | Protein mpv17 | Membrane channel/unknown | Neurohepatopathy/neuropathy/leukoencephalopathy/Charcot–Marie–Tooth | AR | D/MD | 137960 | [37] (2006) | ||
| SLC25A21 | Mitochondrial 2-oxodicarboxylate carrier | Transmembrane transporter | Spinal muscular atrophy-like | AR | D | 607571 | [38] (2018) | ||
| SLC25A10 | Mitochondrial dicarboxylate carrier | Transmembrane transporter | Epileptic encephalopathy | AR | D | 606794 | [39] (2018) | ||
| Other function / unknown function | SUCLA2 | β-subunit, Succinate-CoA ligase | Krebs cycle | Encephalomyopathy | AR | D | 603921 | [40] (2005) | |
| SUCLG1 | α-subunit, Succinate-CoA ligase | Krebs cycle | Encephalomyopathy | AR | D | 611224 | [41] (2007) | ||
| AGK | Acylglycerol kinase | Lipid metabolism | Congenital cataract/hypertrophic cardiomyopathy/skeletal myopathy and lactic acidosis/Sengers syndrome | AD | D | 610345 | [42] (2012) | ||
| GFER | Growth factor, augmenter of liver regeneration | Growth factor | Progressive myopathy/congenital cataract/sensorineural hearing loss/developmental delay | AR | MD | 600924 | [43] (2009) | ||
| ABAT | 4-aminobutyrate aminotransferase | Aminotransferase | Encephalomyopathy | AR | D | 137150 | [44] (2015) | ||
| FBXL4 | F-box/LRR-repeat protein 4 | Protein homeostasis | Encephalomyopathy | AR | D | 605654 | [45] (2013) | ||
| MRM2 | rRNA methyltransferase 2, mitochondrial | Mito rRNA maturation | MELAS-like | AR | D | 606906 | [46] (2017) | ||
| C1QBP | Complement component 1 Q subcomponent-binding protein, mitochondrial | Inflammation/nuclear transcription/mitoribosome biogenesis/apoptosis | Cardiopathy-multisystemic/PEO-myopathy | AR | MD | 601269 | [47] (2017) | ||
| NCT04378075 | A Study to Evaluate Efficacy and Safety of Vatiquinone for Treating Mitochondrial Disease in Participants with Refractory Epilepsy |
| Condition | POLG |
| Study type/phase | Interventional (phase 2 and phase 3) |
| Intervention | Vatiquinone administration |
| Status | Recruiting |
| Estimated study completion | 1 April 2023 |
| Outcomes | Change in the number of observable motor seizures Occurrence or recurrence of epilepsy Participants who require rescue seizure medication |
| Sponsor | PTC therapeutics |
| NCT01370447 | EPI-743 for Mitochondrial Respiratory Chain Diseases |
| Condition | POLG |
| Study type/phase | Interventional (phase 2) |
| Intervention | EPI-743 |
| Status | Active, not recruiting |
| Estimated study completion | 31 December 2021 |
| Outcomes | Change in neuromuscular function Number of subjects experiencing adverse events Change in Newcastle Paediatric Mitochondrial Disease Score Pharmacokinetics of EPI-743 |
| Sponsor | PTC Therapeutics |
| NCT02023866 | Open-Label, Dose-Escalating Study Assessing Safety, Tolerability, Efficacy, of RP103 in Mitochondrial Disease |
| Condition | POLG-TYMP |
| Study type/phase | Interventional (phase 2) |
| Intervention | Cysteamine bitartrate |
| Status | Completed |
| Estimated study completion | October 2016 |
| Outcomes | Change in Newcastle Paediatric Mitochondrial Disease Scale (NPMDS) Score |
| Sponsor | Horizon Pharma USA, Inc. |
| NCT02473445 | A Long-Term Extension of Study RP103-MITO-001 (NCT02023866) to Assess Cysteamine Bitartrate Delayed-Release Capsules (RP103) in Children with Inherited Mitochondrial Disease |
| Condition | POLG-TYMP |
| Study type/phase | Interventional (phase 2) |
| Intervention | Cysteamine bitartrate |
| Status | Completed |
| Estimated study completion | 6 March 2017 |
| Outcomes | Change in Newcastle Paediatric Mitochondrial Disease Scale (NPMDS) Score |
| Sponsor | Horizon Pharma USA, Inc. |
| NCT03701568 | A RETROspective Study of Patients with TK2d |
| Condition | TK2 |
| Study type/phase | Observational |
| Intervention | dCtd/dThd |
| Status | Completed |
| Estimated study completion | 31 May 2019 |
| Outcomes | Clinical course Motor function and ambulatory assessments |
| Sponsor | Modis Therapeutics, Inc. |
| NCT03845712 | An Open-Label Study of Continuation Treatment with Combination Pyrimidine Nucleosides in Patients With TK2 |
| Condition | TK2 |
| Study type/phase | Interventional (phase 2) |
| Intervention | MT1621 |
| Status | Active, not recruiting |
| Estimated study completion | 31 January 2022 |
| Outcomes | Safety Motor function assessments Respiratory status Growth/nutrition Pharmacokinetics Quality of life through patient questionnaire |
| Sponsor | Modis Therapeutics, Inc. |
| NCT04581733 | A Study of the Efficacy and Safety of MT1621 in Thymidine Kinase 2 (TK2) Deficiency |
| Condition | TK2 |
| Study type/phase | Interventional (phase 3) |
| Intervention | MT1621 |
| Status | Not yet recruiting |
| Estimated study completion | March 2025 |
| Outcomes | Time to loss/acquisition of any motor milestone Overall survival |
| Sponsor | Modis Therapeutics, Inc. |
| NCT03639701 | Treatment of TK2 Deficiency with Thymidine and Deoxycytidine |
| Condition | TK2 |
| Study type/phase | Interventional (phase 1 and phase 2) |
| Intervention | dThd |
| Status | Enrolling by invitation |
| Estimated study completion | 1 April 2024 |
| Outcomes | Safety Efficacy measured by different ways |
| Sponsor | Columbia University |
| NCT03866954 | Trial of Erythrocyte Encapsulated Thymidine Phosphorylase in Mitochondrial Neurogastrointestinal Encephalomyopathy |
| Condition | TYMP |
| Study type/phase | Interventional (phase 2) |
| Intervention | EETP |
| Status | Not yet recruiting |
| Estimated study completion | September 2022 |
| Outcomes | Safety of procedure Pharmacodynamic effects Efficacy of EETP Changes in clinical assessments |
| Sponsor | St George’s, University of London |
| NCT02427178 | MNGIE Allogeneic Hematopoietic Stem Cell Transplant Safety Study |
| Condition | TYMP |
| Study type/phase | Interventional (phase 1) |
| Intervention | Hematopoietic allogenic stem cells |
| Status | Recruiting |
| Estimated study completion | June 2023 |
| Outcomes | Engraftment success Survival Blood levels of dThd and dUrd |
| Sponsor | Columbia University |
| NCT00804102 | Transcorneal Electrical Stimulation Therapy for Retinal Disease |
| Condition | OPA1 |
| Study type/phase | Interventional (not phase applicable) |
| Intervention | Transcorneal electrical stimulation |
| Status | Completed |
| Estimated study completion | April 2011 |
| Outcomes | Enhanced field of vision Enhanced visual acuity Lower threshold for electrical evoked phosphenes |
| Sponsor | Okuvision GmbH |
| NCT03011541 | Stem Cell Ophthalmology Treatment Study II |
| Condition | OPA1 |
| Study type/phase | Interventional (not phase applicable) |
| Intervention | Administration of autologous bone marrow derived stem cells |
| Status | Recruiting |
| Estimated study completion | January 2022 |
| Outcomes | Visual acuity Visual fields Optical coherence tomography |
| Sponsor | MD Stem Cells |
| NCT01648634 | Nebivolol for the Prevention of Left Ventricular Systolic Dysfunction in Patients with Duchenne Muscular Dystrophy |
| Condition | SUCLA2 |
| Study type/phase | Interventional (phase 3) |
| Intervention | Nevibolol |
| Status | Active, not recruiting |
| Estimated study completion | June 2021 |
| Outcomes | Left ventricular systolic dysfunction Right ventricular ejection fraction Hospitalizations Mortality |
| Sponsor | Assistance Publique-Hôpitaux de Paris |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramón, J.; Vila-Julià, F.; Molina-Granada, D.; Molina-Berenguer, M.; Melià, M.J.; García-Arumí, E.; Torres-Torronteras, J.; Cámara, Y.; Martí, R. Therapy Prospects for Mitochondrial DNA Maintenance Disorders. Int. J. Mol. Sci. 2021, 22, 6447. https://doi.org/10.3390/ijms22126447
Ramón J, Vila-Julià F, Molina-Granada D, Molina-Berenguer M, Melià MJ, García-Arumí E, Torres-Torronteras J, Cámara Y, Martí R. Therapy Prospects for Mitochondrial DNA Maintenance Disorders. International Journal of Molecular Sciences. 2021; 22(12):6447. https://doi.org/10.3390/ijms22126447
Chicago/Turabian StyleRamón, Javier, Ferran Vila-Julià, David Molina-Granada, Miguel Molina-Berenguer, Maria Jesús Melià, Elena García-Arumí, Javier Torres-Torronteras, Yolanda Cámara, and Ramon Martí. 2021. "Therapy Prospects for Mitochondrial DNA Maintenance Disorders" International Journal of Molecular Sciences 22, no. 12: 6447. https://doi.org/10.3390/ijms22126447