
Analysis of Circulating miRNA Profile in Plasma Samples of Glioblastoma Patients


 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
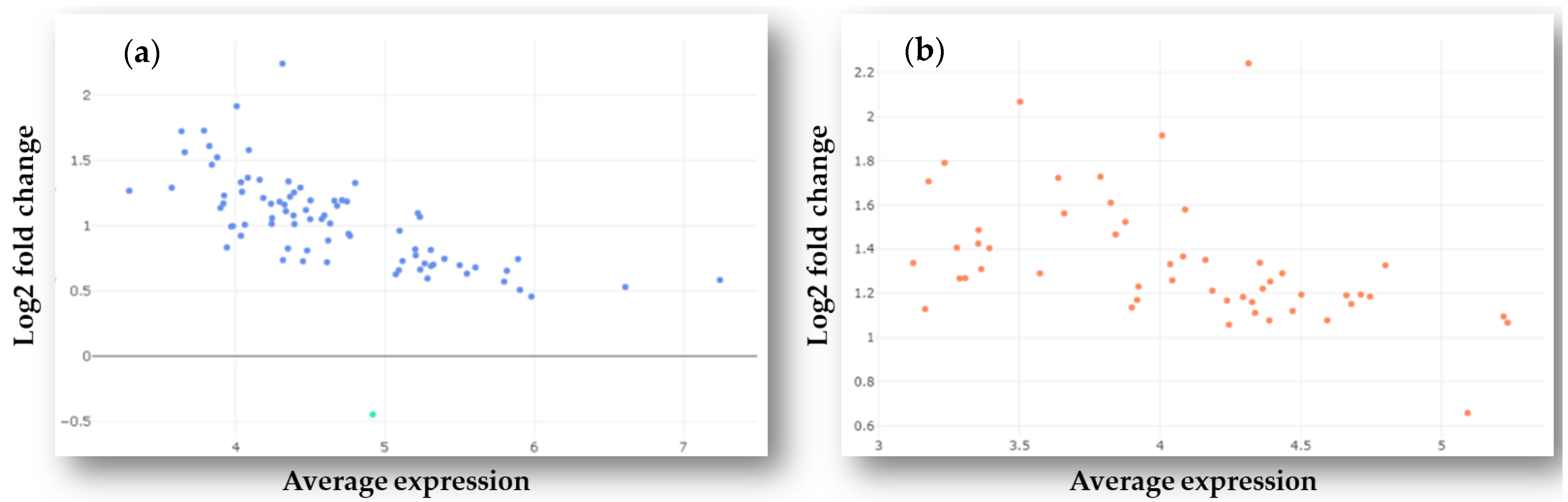
2.1. Identification of Differently Expressed MicroRNAs in Plasma Samples of GBM Patients and Healthy Controls
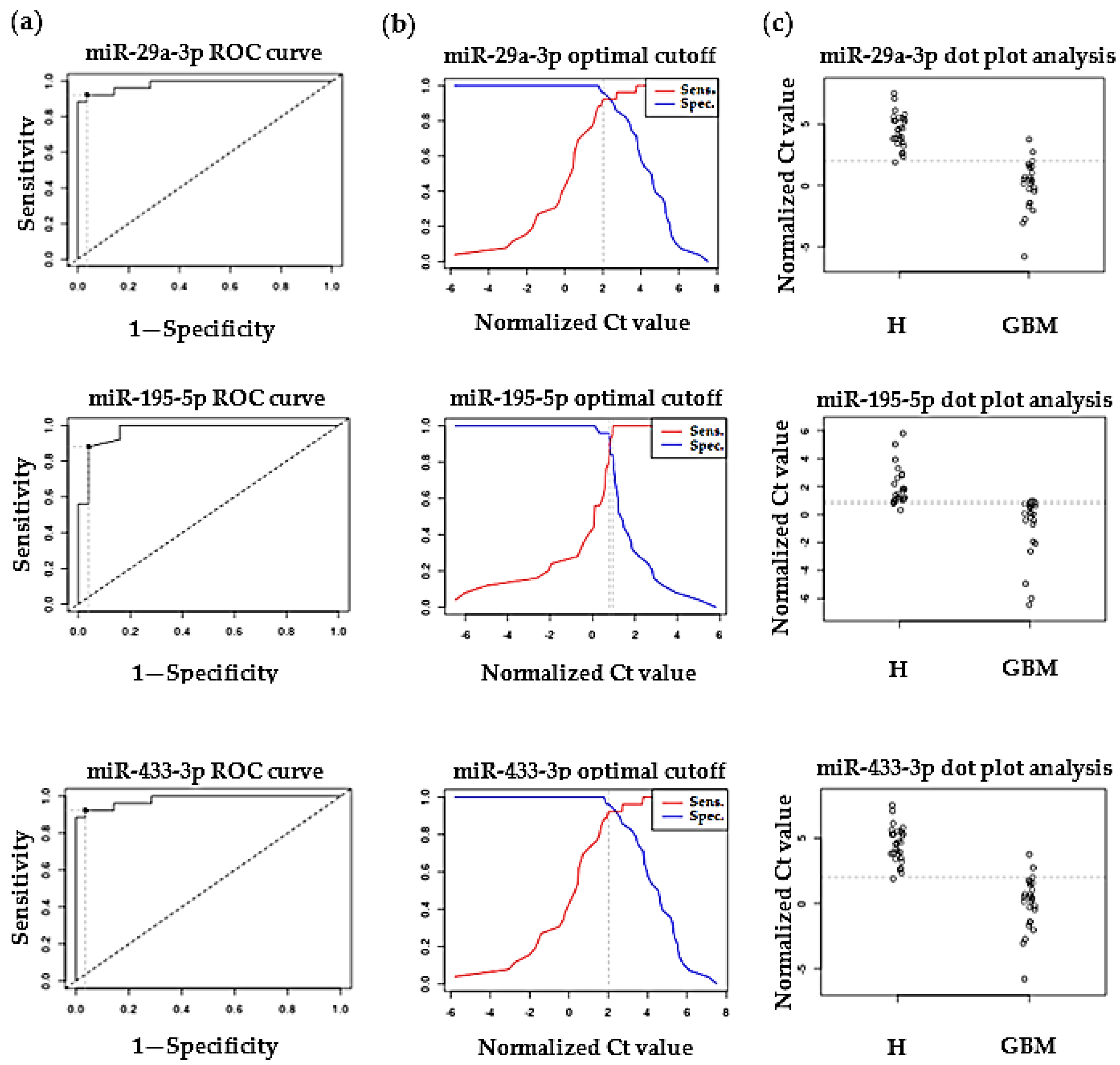
2.2. Validation of Differentially Expressed MiRNAs by RT-qPCR
2.3. MiRNA Ranking, Target Gene Prediction, and Analysis
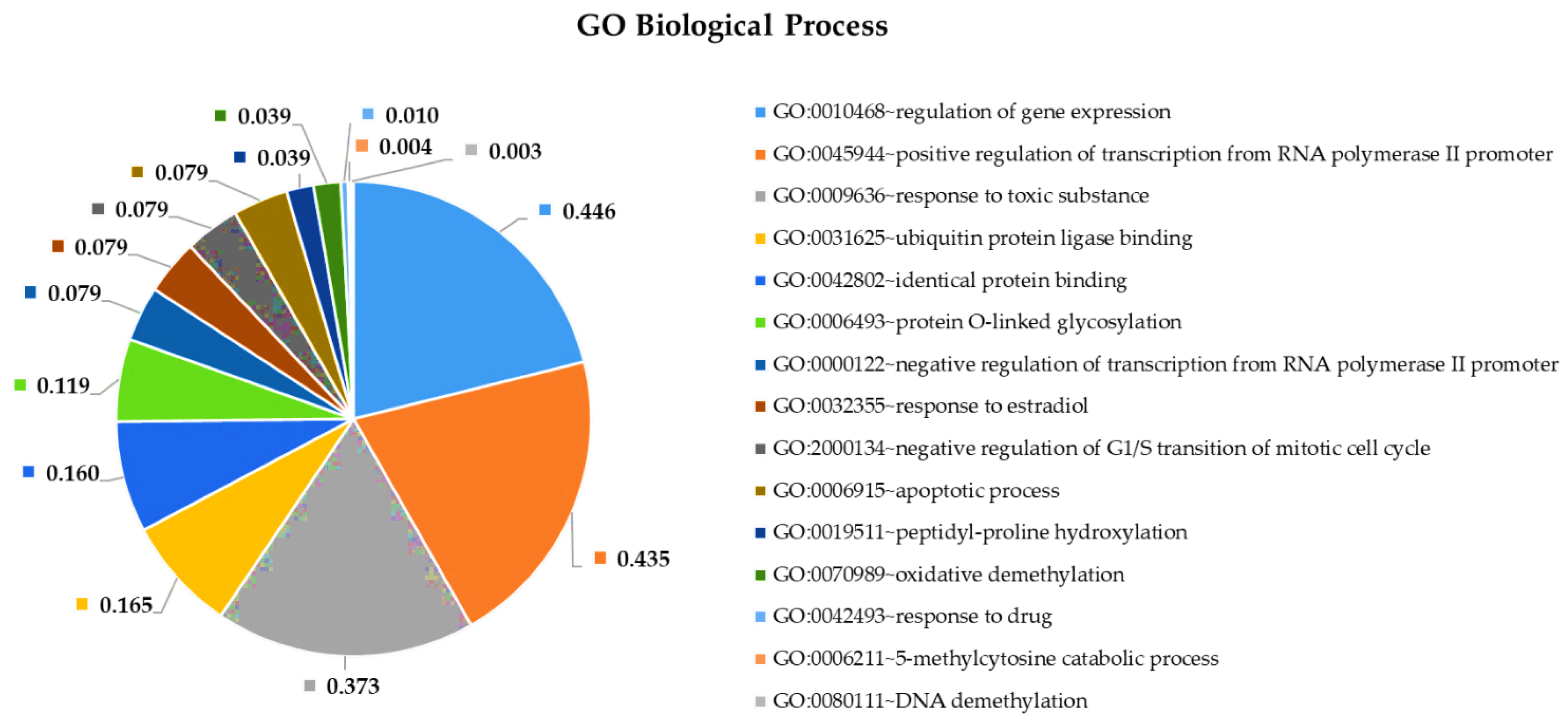
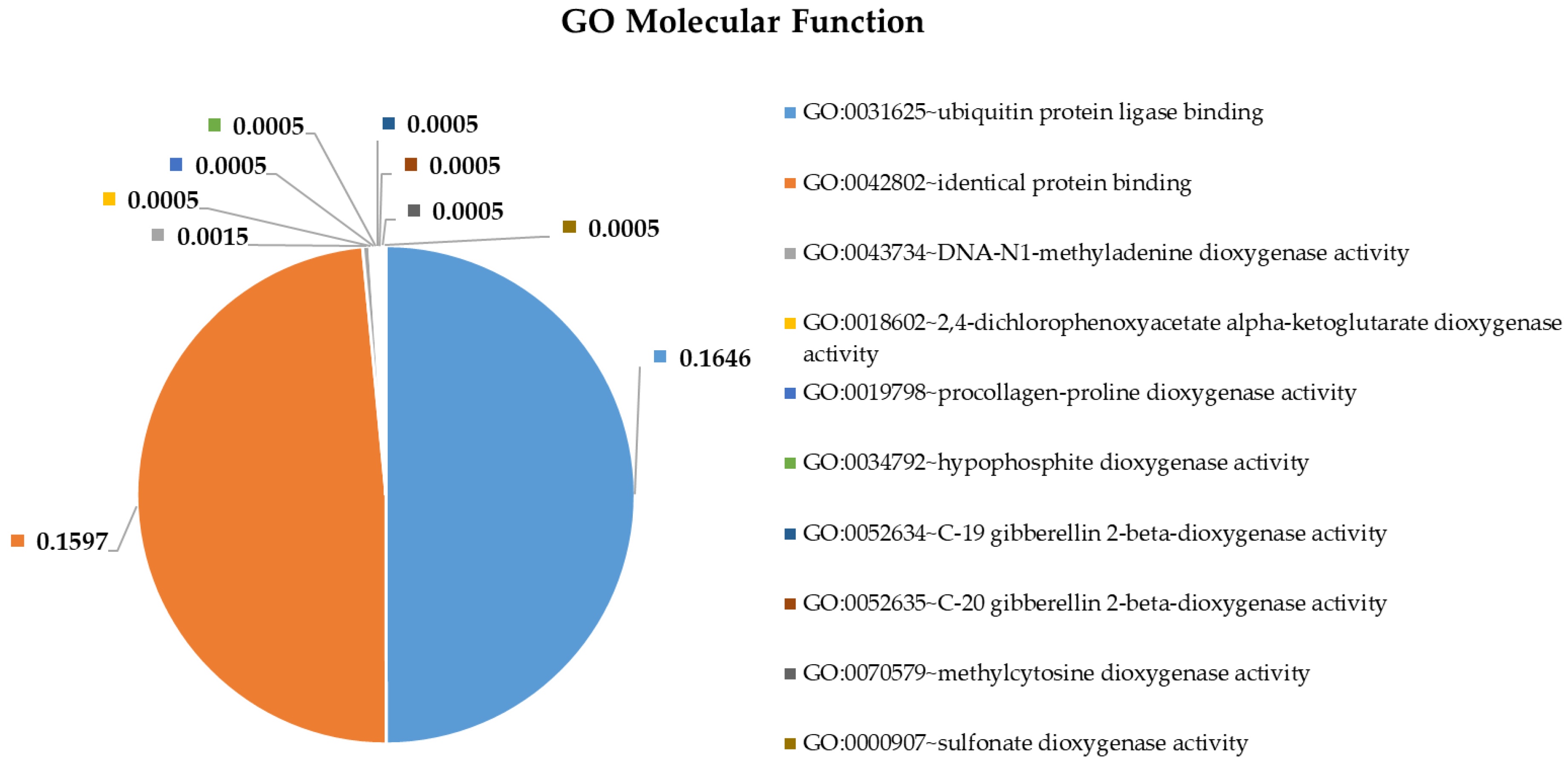
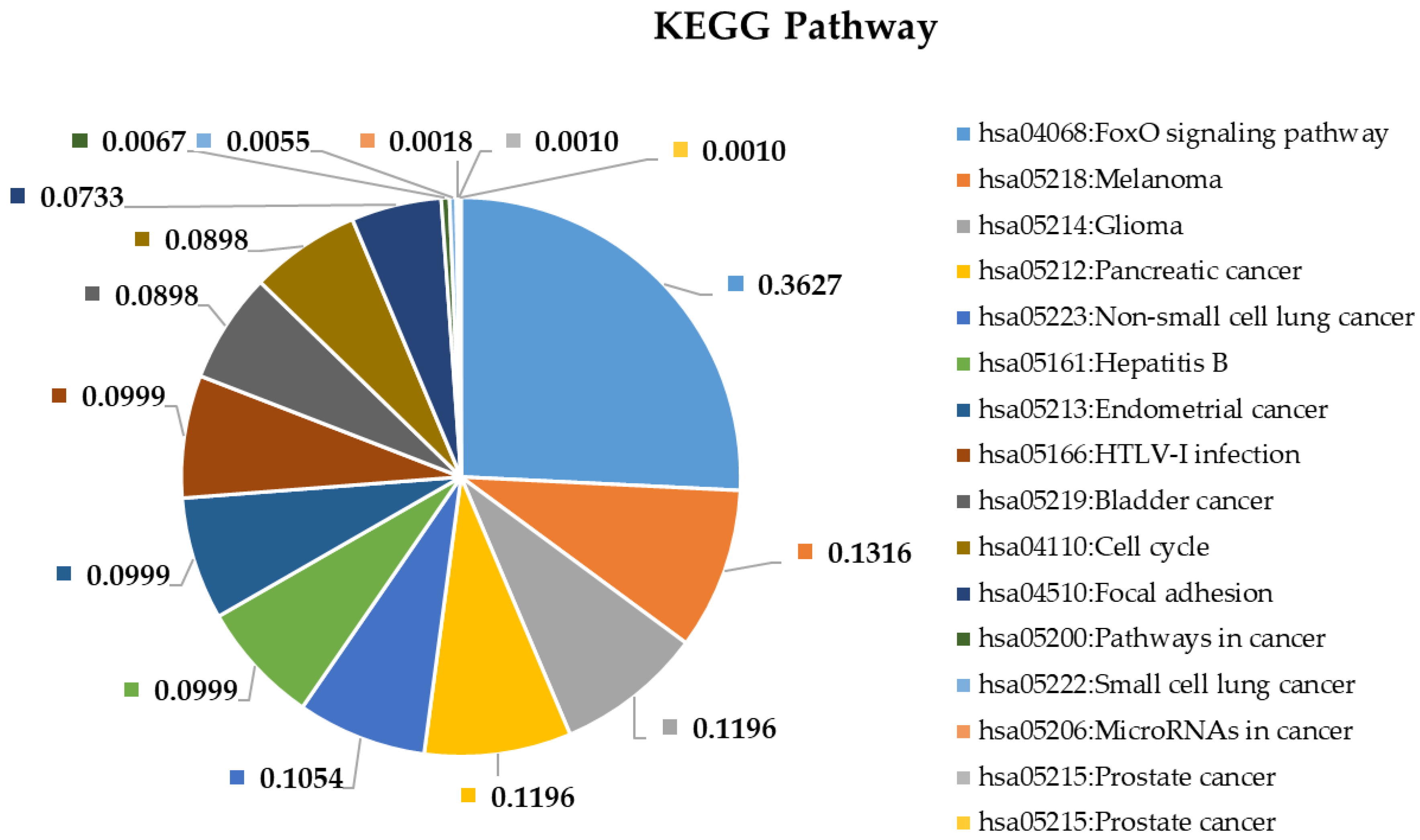
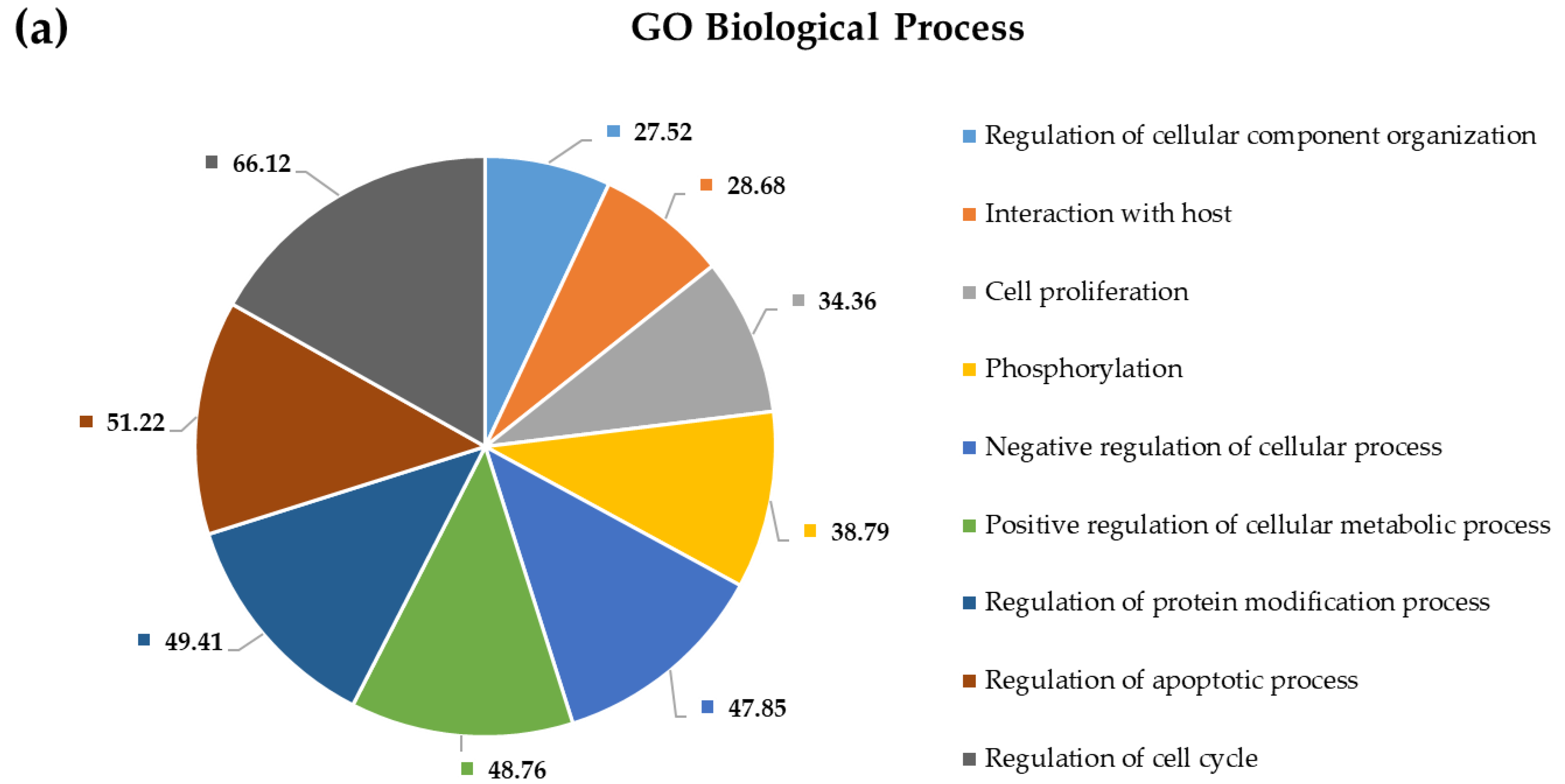
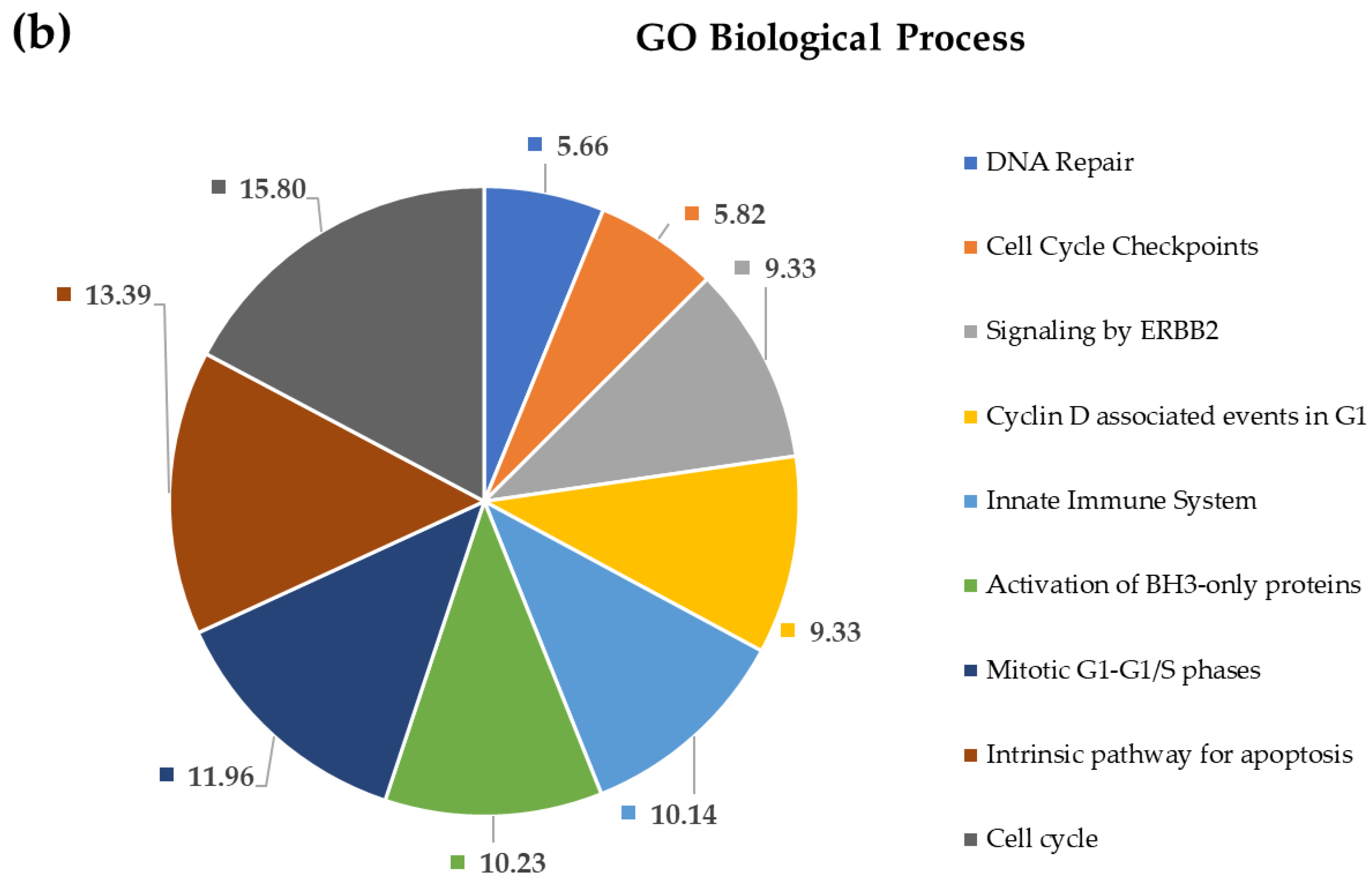
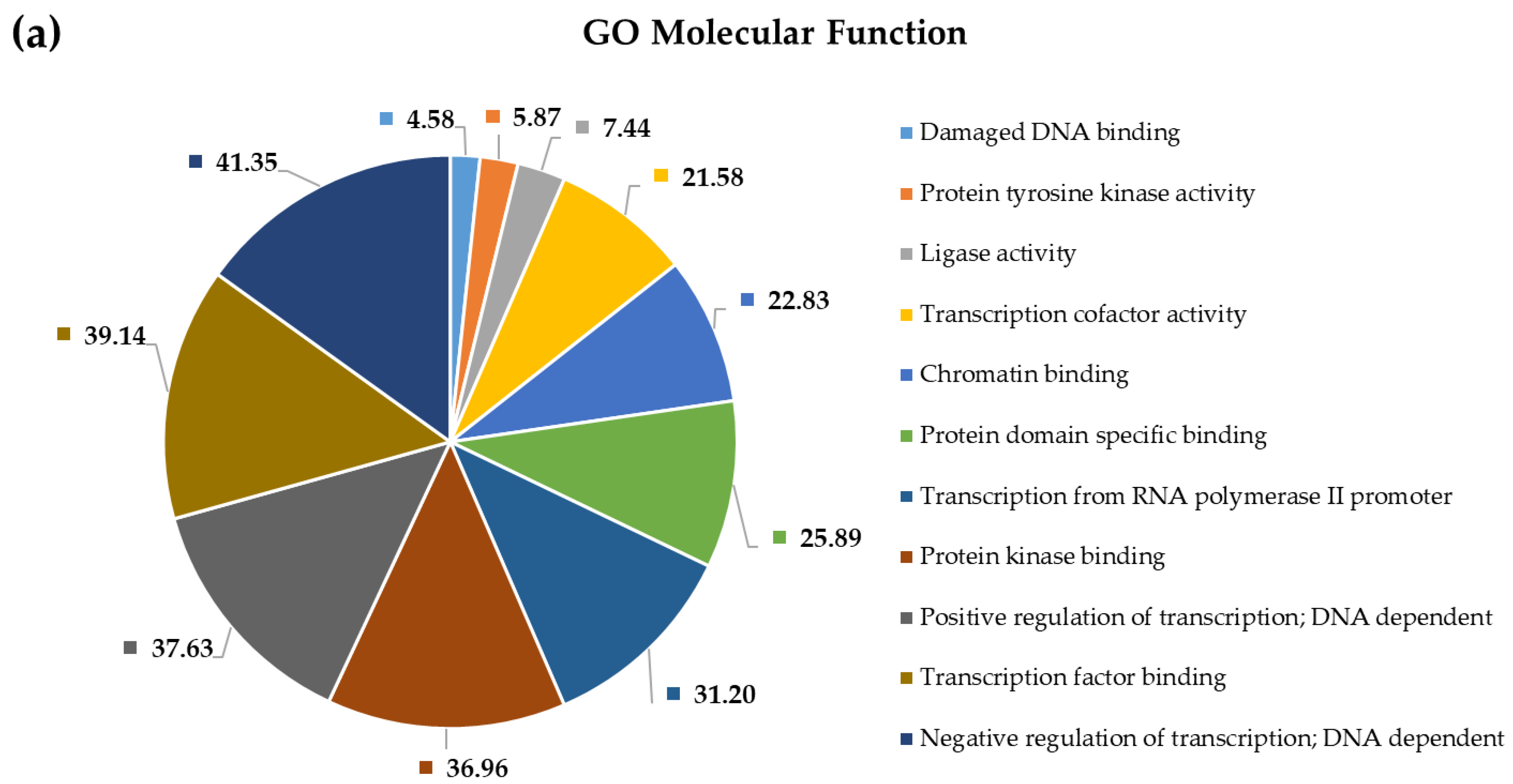
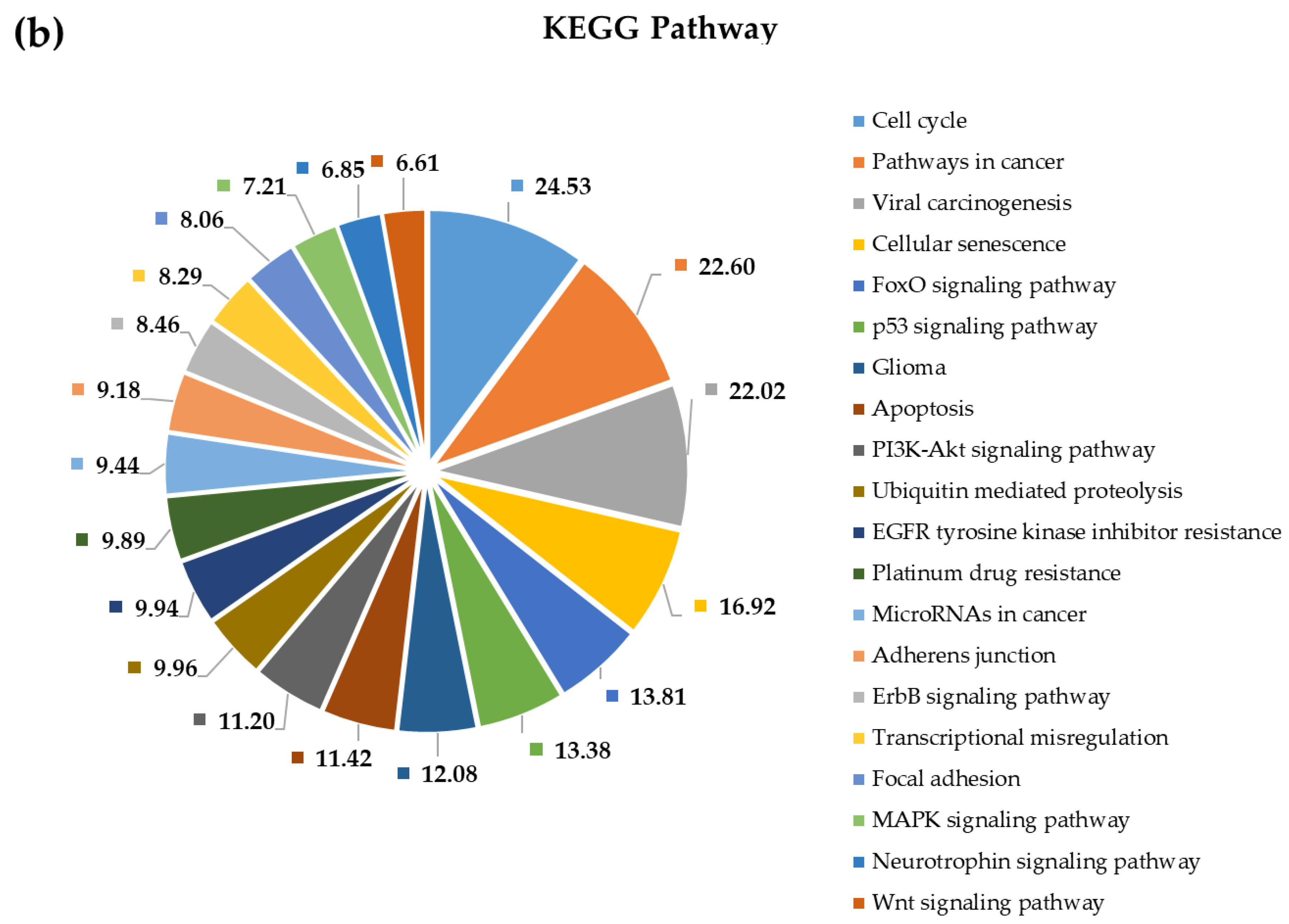
2.4. Pathway and Gene Ontology Enrichment Analysis of MiRNA Targets
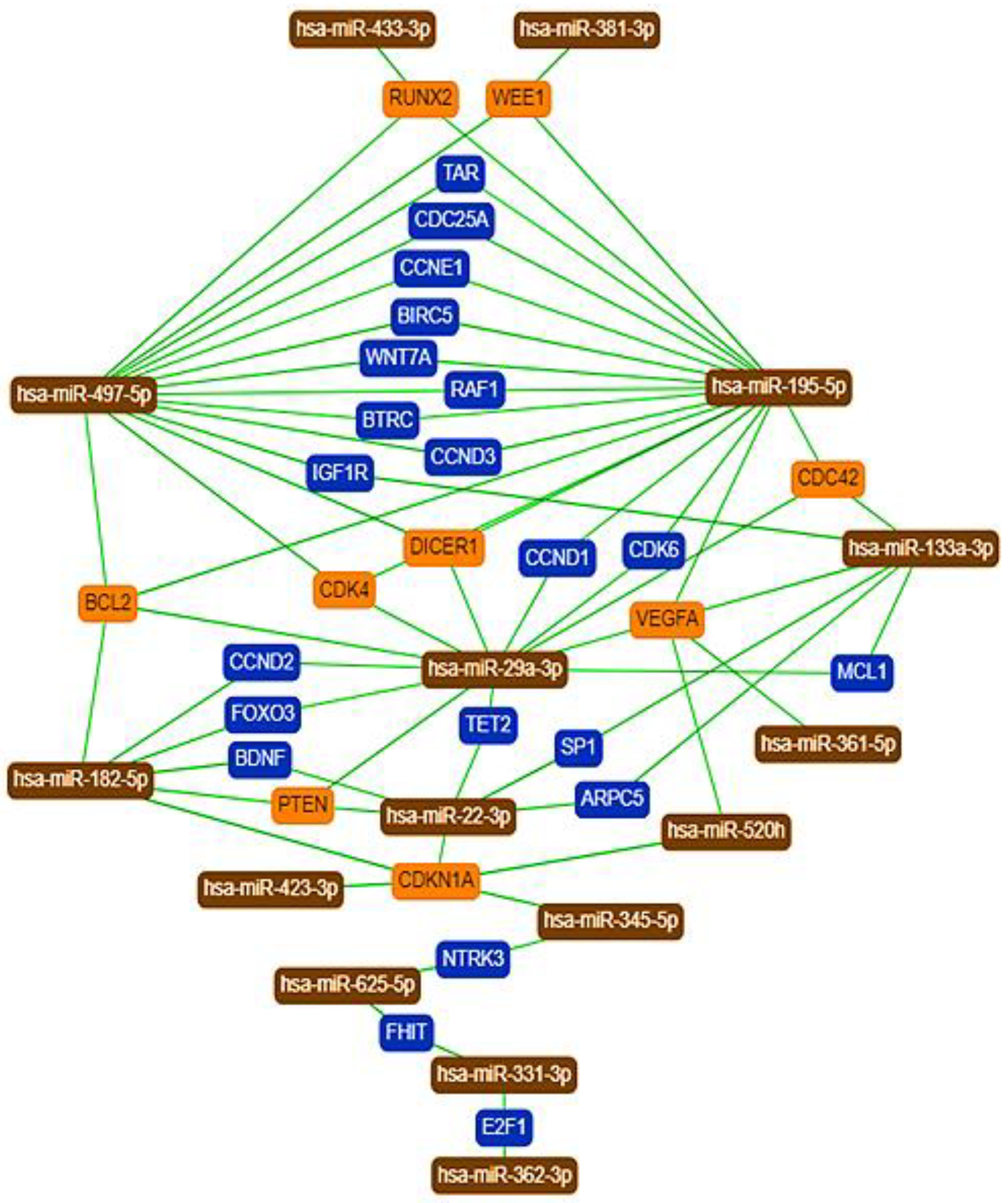
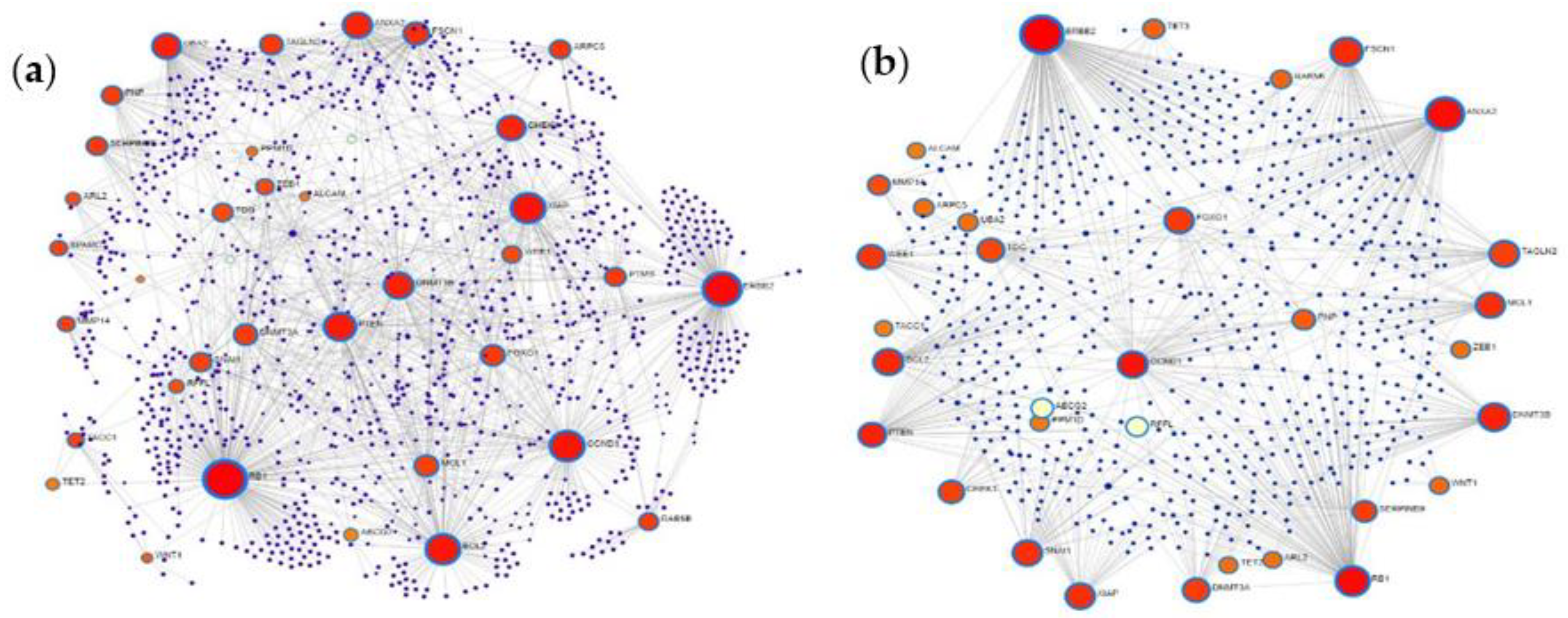
2.5. Protein–Protein Interaction Network Analysis of MiRNA Targets
3. Discussion
3.1. Identification of Differently Expressed MiRNAs in Plasma Samples of GBM Patients and Healthy Controls
3.1.1. NanoString Analysis and Identification of Differentially Expressed MiRNAs
3.1.2. Differently Expressed MiRNAs Specific for GBM
3.1.3. The Role of Hsa-miR-433-3p in Cancer Development
3.2. Validation of Differentially Expressed MiRNAs by RT-qPCR
3.3. MiRNA Ranking, Target Gene Prediction, and Analysis
3.4. Pathway and Gene Ontology Enrichment Analysis of MiRNA Targets
3.5. Protein–Protein Interaction Network Analysis of MiRNA Targets
4. Materials and Methods
4.1. Patients and Samples
4.2. RNA Isolation and Purification for NanoString Measurement
4.3. NanoString Analysis and Identification of Differentially Expressed MiRNAs
4.4. Prediction of Targets of Differentially Expressed MiRNA, Functional Annotation, and Pathway Enrichment Analysis
4.5. Validation of Hsa-miRNA Expression by Quantitative Real-Time PCR (RT-qPCR)
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.-K.; Wen, P.Y.; Olson, J.J. Exciting New Advances in Neuro-Oncology: The Avenue to a Cure for Malignant Glioma. CA A Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef] [PubMed]
- Birkó, Z.; Nagy, B.; Klekner, Á.; Virga, J. Novel Molecular Markers in Glioblastoma—Benefits of Liquid Biopsy. Int. J. Mol. Sci. 2020, 21, 7522. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Zhao, C.; Tian, F.; Yu, Y.; Liu, G.; Zan, L.; Updike, M.S.; Song, J. miRNA-dysregulation associated with tenderness variation induced by acute stress in Angus cattle. J. Anim. Sci. Biotechnol. 2012, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Wang, J.; Ding, N.; Hu, W.; Zhang, X.; Wang, B.; Hua, J.; Wei, W.; Zhu, Q. Exosome-mediated microRNA transfer plays a role in radiation-induced bystander effect. RNA Biol. 2015, 12, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular Vesicles in Cancer: Exosomes, Microvesicles and the Emerging Role of Large Oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of MicroRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wang, C.; Chen, X.; Chen, S.; Zhang, Y.; Zhi, F.; Wang, J.; Li, L.; Zhou, X.; Li, N.; et al. Identification of seven serum microRNAs from a genome-wide serum microRNA expression profile as potential noninvasive biomarkers for malignant astrocytomas. Int. J. Cancer 2012, 132, 116–127. [Google Scholar] [CrossRef]
- Qu, S.; Guan, J.; Liu, Y. Identification of microRNAs as novel biomarkers for glioma detection: A meta-analysis based on 11 articles. J. Neurol. Sci. 2015, 348, 181–187. [Google Scholar] [CrossRef]
- Roth, P.; Wischhusen, J.; Happold, C.; Chandran, P.A.; Hofer, S.; Eisele, G.; Weller, M.; Keller, A. A specific miRNA signature in the peripheral blood of glioblastoma patients. J. Neurochem. 2011, 118, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Nguyen, H.P.T.; Luwor, R.B.; Stylli, S.S.; Gogos, A.; Paradiso, L.; Kaye, A.H.; Morokoff, A.P. A comprehensive meta-analysis of circulation miRNAs in glioma as potential diagnostic biomarker. PLoS ONE 2018, 13, e0189452. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okura, H.; Golbourn, B.J.; Shahzad, U.; Agnihotri, S.; Sabha, N.; Krieger, J.R.; Figueiredo, C.A.; Chalil, A.; Landon-Brace, N.; Riemenschneider, A.; et al. A role for activated Cdc42 in glioblastoma multiforme invasion. Oncotarget 2016, 7, 56958–56975. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, W.; Kim, T.M.; Jung, Y.; Menon, L.G.; Xing, H.; Li, H.; Carroll, R.S.; Park, P.J.; Yang, H.W.; et al. MicroRNA-29a activates a multi-component growth and invasion program in glioblasto-ma. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Xu, S.; Zhong, Y.; Tu, T.; Xu, Y.; Li, X.; Wang, B.; Yang, F. High gene expression levels of VEGFA and CXCL8 in the peritumoral brain zone are associated with the recurrence of glioblastoma: A bioinformatics analysis. Oncol. Lett. 2019, 18, 6171–6179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, F.E.; Ironside, J.W.; Gregor, A.; Wyatt, B.; Stewart, M.; Rye, R.; Adams, J.; Potts, H.W. The prognostic influence of bcl-2 in malignant glioma. Br. J. Cancer 2002, 86, 1899–1904. [Google Scholar] [CrossRef] [Green Version]
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Mazurek, M.; Grochowski, C.; Litak, J.; Osuchowska, I.; Maciejewski, R.; Kamieniak, P. Recent Trends of microRNA Significance in Pediatric Population Glioblastoma and Current Knowledge of Micro RNA Function in Glioblastoma Multiforme. Int. J. Mol. Sci. 2020, 21, 3046. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Ye, X.; Piantadosi, S.; Desideri, S.; Nabors, L.B.; Rosenfeld, M.; Fisher, J. NABTT CNS Consortium Survival of Patients with Newly Diagnosed Glioblastoma Treated with Radiation and Temozolomide in Research Studies in the United States. Clin. Cancer Res. 2010, 16, 2443–2449. [Google Scholar] [CrossRef] [Green Version]
- Klekner, Á.; Szivos, L.; Virga, J.; Árkosy, P.; Bognár, L.; Birkó, Z.; Nagy, B. Significance of liquid biopsy in glioblastoma–A review. J. Biotechnol. 2019, 298, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2009, 220, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.-D.; Lin, F.-M.; Wu, W.-Y.; Liang, C.; Huang, W.-C.; Chan, W.-L.; Tsai, W.-T.; Chen, G.-Z.; Lee, C.-J.; Chiu, C.-M.; et al. miRTarBase: A database curates experimentally validated microRNA–target interactions. Nucleic Acids Res. 2010, 39, D163–D169. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Ding, Q.; Han, H.; Wu, D. miRCancer: A microRNA-cancer association database constructed by text mining on literature. Bioinformatics 2013, 29, 638–644. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Jiang, Y.; Liu, L.; Zuo, Z.; Zeng, C. Circulating MicroRNAs as Promising Diagnostic Biomarkers for Patients With Glioma: A Meta-Analysis. Front. Neurol. 2021, 11. [Google Scholar] [CrossRef]
- Rodrigues-Junior, D.M.; Pelarin, M.F.A.; Nader, H.B.; Vettore, A.L.; Pinhal, M.A.S. MicroRNA-1252-5p Associated with Extracellular Vesicles Enhances Bortezomib Sensitivity in Multiple Myeloma Cells by Targeting Heparanase. OncoTargets Ther. 2021, ume 14, 455–467. [Google Scholar] [CrossRef]
- Huh, J.H.; Kim, T.H.; Kim, K.; Song, J.-A.; Jung, Y.J.; Jeong, J.-Y.; Lee, M.J.; Kim, Y.K.; Lee, D.H.; An, H.J. Dysregulation of miR-106a and miR-591 confers paclitaxel resistance to ovarian cancer. Br. J. Cancer 2013, 109, 452–461. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Vartanian, A.; Burrell, K.; Zadeh, G. A microRNA Link to Glioblastoma Heterogeneity. Cancers 2012, 4, 846–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Zhang, H.; Zhang, Z.; Zhang, X.; Ning, B.; Guo, J.; Nie, N.; Liu, B.; Wu, X. Down-regulated miR-9 and miR-433 in human gastric carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 82. [Google Scholar] [CrossRef] [Green Version]
- Estep, M.; Armistead, D.; Hossain, N.; Elarainy, H.; Goodman, Z.; Baranova, A.; Chandhoke, V.; Younossi, Z.M. Differential expression of miRNAs in the visceral adipose tissue of patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2010, 32, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, L.J.; Tan, Y.-X.; Ren, H.; Qi, Z.-T. Identification of deregulated miRNAs and their targets in hepatitis B virus-associated hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 5442–5453. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, X.; Xu, X.; Di, H.; Du, J.; Xu, B.; Wang, Q.; Wang, J. MiR-433-3p suppresses cell growth and enhances chemosensitivity by targeting CREB in human glioma. Oncotarget 2016, 8, 5057–5068. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.; Mo, F.; Ding, D.; Li, L.; Han, X.; Zhao, N.; Foltz, G.; Lin, B.; Lan, Q.; Huang, Q. A Catalogue of Glioblastoma and Brain MicroRNAs Identified by Deep Sequencing. OMICS 2012, 16, 690–699. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, F.; Wang, L.; Jiao, Z.; Fang, J.; Li, J. MicroRNA-433 regulates apoptosis by targeting PDCD4 in human osteosarcoma cells. Oncol. Lett. 2017, 14, 2353–2358. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-Y.; Lu, W.-C. Reduced Expression of hsa-miR-338-3p Contributes to the Development of Glioma Cells by Targeting Mitochondrial 3-Oxoacyl-ACP Synthase (OXSM) in Glioblastoma (GBM). OncoTargets Ther. 2020, 13, 9513–9523. [Google Scholar] [CrossRef]
- Kannan, R.; Lyon, I.; Baker, N. Dietary control of lipogenesis in vivo in host tissues and tumors of mice bearing Ehrlich ascites carcinoma. Cancer Res. 1980, 40, 4606–4611. [Google Scholar]
- Jia, Y.; Tian, Y.; An, S.; Yang, D. Effects of microRNA-195 on the Prognosis of Glioma Patients and the Proliferation and Apoptosis of Human Glioma Cells. Pathol. Oncol. Res. 2020, 26, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Cheng, Z.; Zhang, J.; Li, R.; Zhao, P.; Fu, Z.; You, Y. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008, 1236, 185–193. [Google Scholar] [CrossRef]
- Yamada, D.; Fujikawa, K.; Kawabe, K.; Furuta, T.; Nakada, M.; Takarada, T. RUNX2 Promotes Malignant Progression in Glioma. Neurochem. Res. 2018, 43, 2047–2054. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, S.; Gao, F.; Li, L.; Zheng, S.; Yung, W.K.A.; Koul, D. Activation of WEE1 confers resistance to PI3K inhibition in glioblastoma. Neuro Oncol. 2017, 20, 78–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Xiao, A.; Floyd, D.; Olmez, I.; Lee, J.; Godlewski, J.; Bronisz, A.; Bhat, K.P.; Sulman, E.P.; Nakano, I.; et al. CDK4/6 inhibition is more active against the glioblastoma proneural subtype. Oncotarget 2017, 8, 55319–55331. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.; Brännström, T.; Bergenheim, A.T.; Henriksson, R. Spatial expression of VEGF-A in human glioma. J. Neuro Oncol. 2002, 59, 1–6. [Google Scholar] [CrossRef]
- Benitez, J.A.; Ma, J.; D’Antonio, M.; Boyer, A.; Camargo, M.F.; Zanca, C.; Kelly, S.; Khodadadi-Jamayran, A.; Jameson, N.M.; Andersen, M.; et al. PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3. Nat. Commun. 2017, 8, 15223. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 524. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Géczi, D.; Nagy, B.; Szilágyi, M.; Penyige, A.; Klekner, Á.; Jenei, A.; Virga, J.; Birkó, Z. Analysis of Circulating miRNA Profile in Plasma Samples of Glioblastoma Patients. Int. J. Mol. Sci. 2021, 22, 5058. https://doi.org/10.3390/ijms22105058
Géczi D, Nagy B, Szilágyi M, Penyige A, Klekner Á, Jenei A, Virga J, Birkó Z. Analysis of Circulating miRNA Profile in Plasma Samples of Glioblastoma Patients. International Journal of Molecular Sciences. 2021; 22(10):5058. https://doi.org/10.3390/ijms22105058
Chicago/Turabian StyleGéczi, Dóra, Bálint Nagy, Melinda Szilágyi, András Penyige, Álmos Klekner, Adrienn Jenei, József Virga, and Zsuzsanna Birkó. 2021. "Analysis of Circulating miRNA Profile in Plasma Samples of Glioblastoma Patients" International Journal of Molecular Sciences 22, no. 10: 5058. https://doi.org/10.3390/ijms22105058