Cyclophilin Inhibition Protects Against Experimental Acute Kidney Injury and Renal Interstitial Fibrosis

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
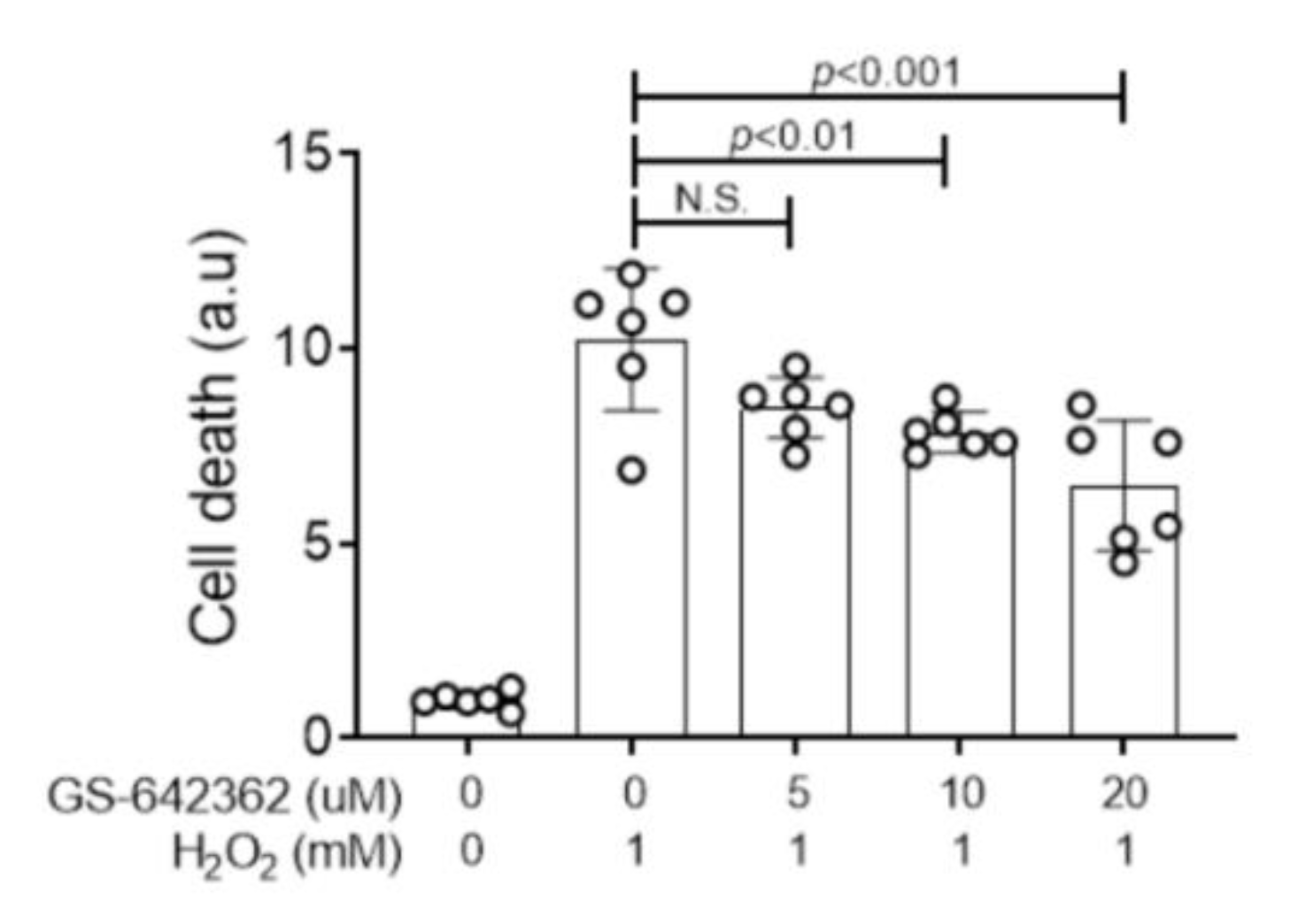
2.1. GS-642362 Prevents Oxygen Radical Induced Tubular Cell Death
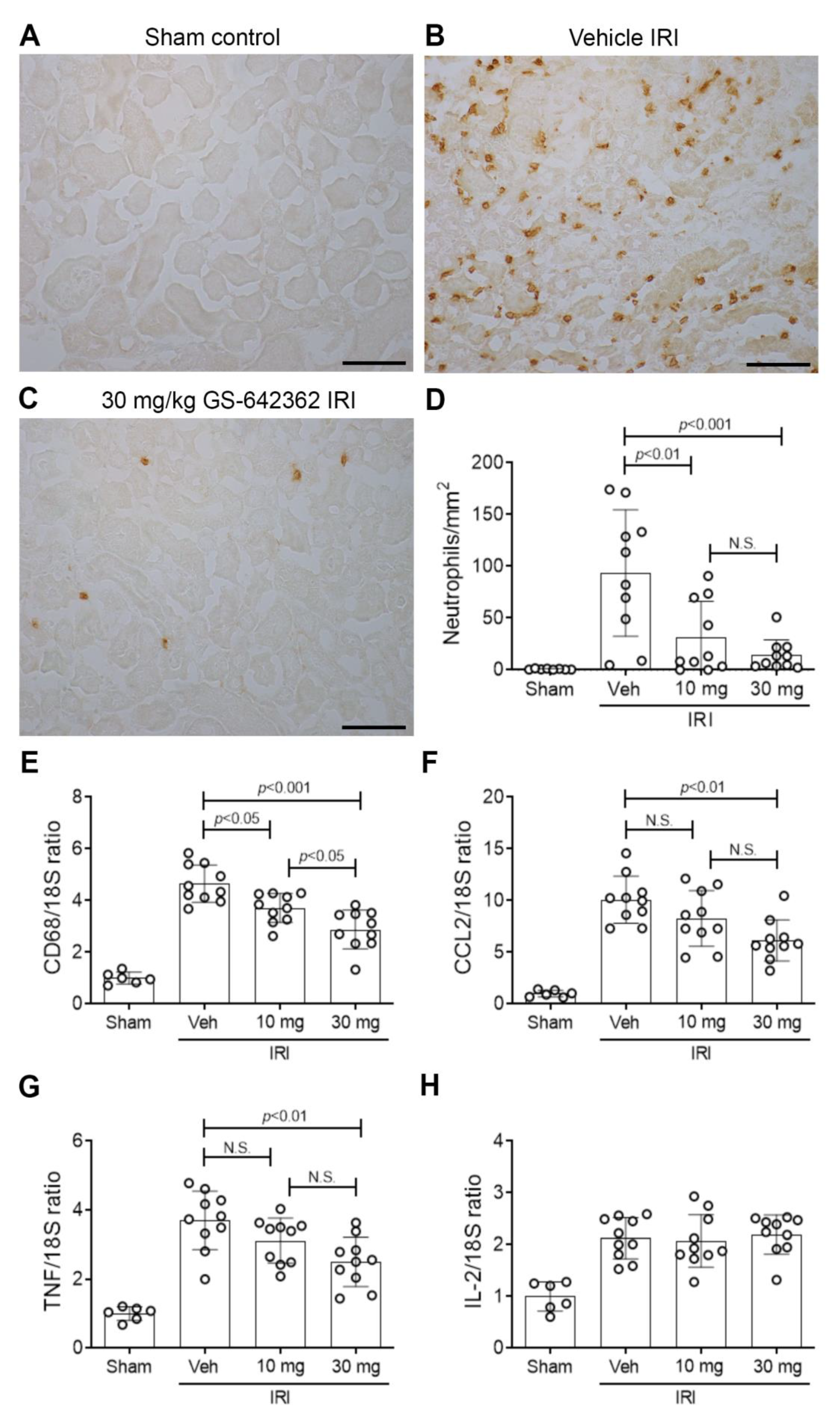
2.2. GS-642362 Protects Against Renal Ischemia/Reperfusion Injury (IRI)
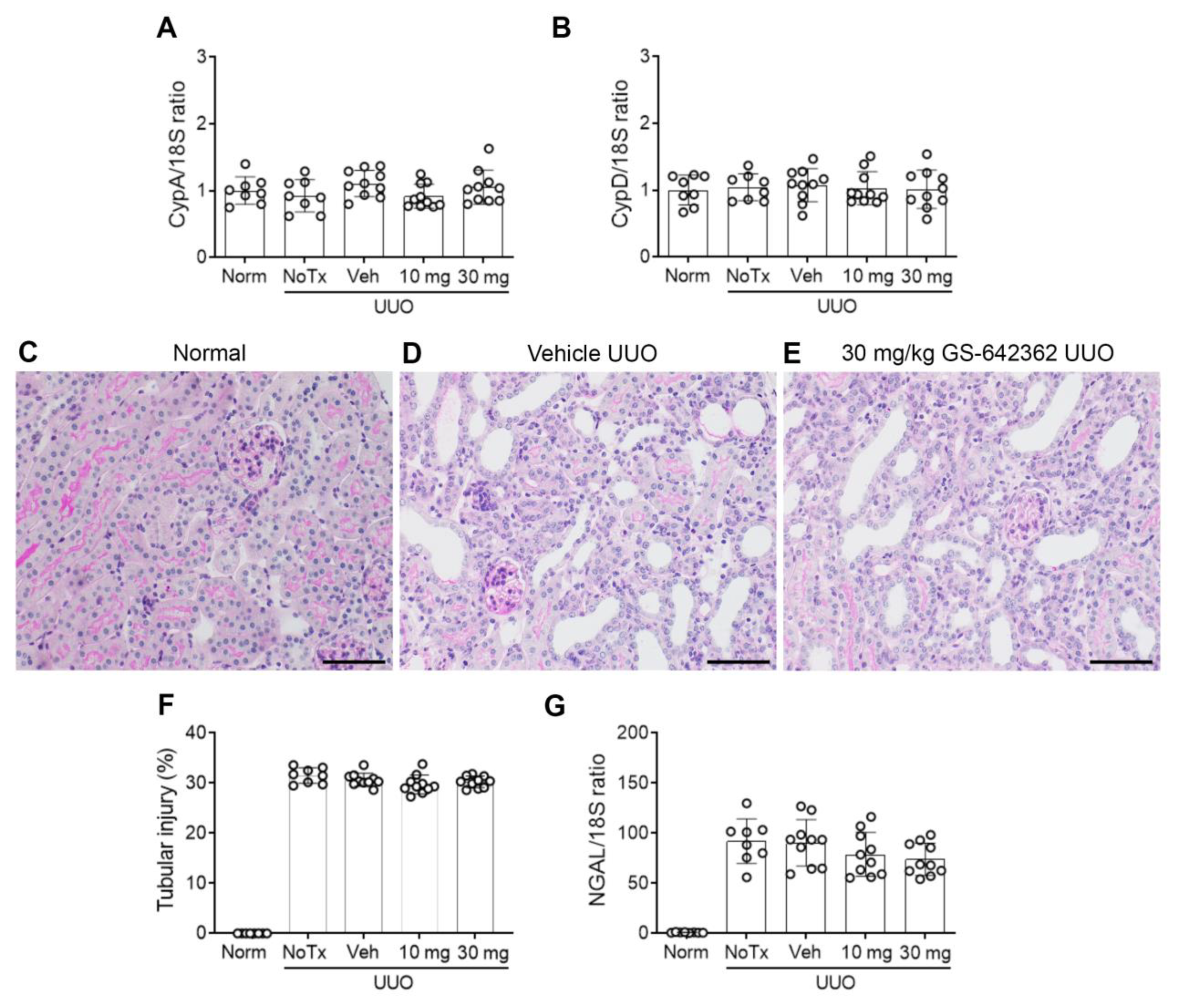
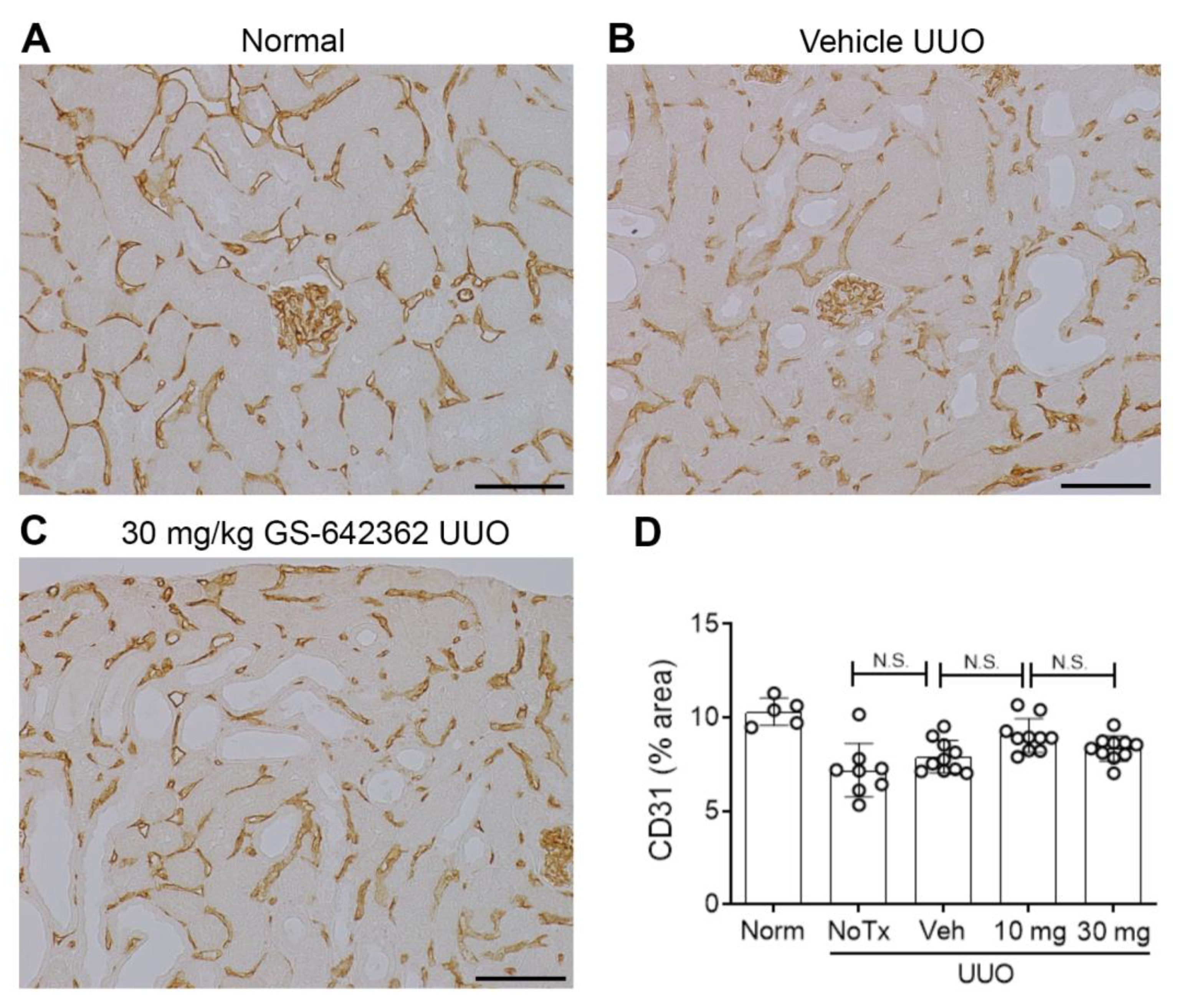
2.3. GS-642362 Reduces Fibrosis in the Unilateral Ureteric Obstruction (UUO) Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Renal Ischaemia/Reperfusion Injury (IRI)
4.4. Unilateral Ureteric Obstruction (UUO)
4.5. Measurement of Peak Plasma Levels of GS-642362
4.6. Immunohistochemistry
4.7. Tubular Cell Culture
4.8. Real Time Polymerase Chain Reaction (RT-PCR)
4.9. Western Blotting
4.10. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Cyp | Cyclophilin |
| IRI | Ischemia/reperfusion injury |
| JNK | Jun amino terminal kinase |
| MAPK | Mitogen-activated protein kinase |
| PDGF-B | Platelet-derived growth factor-B |
| SMA | Smooth muscle actin |
| TUNEL | Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling |
| UUO | Unilateral ureteric obstruction |
References
- Qu, X.; Wang, C.; Zhang, J.; Qie, G.; Zhou, J. The Roles of CD147 and/or Cyclophilin A in Kidney Diseases. Mediat. Inflamm. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukrinsky, M. Extracellular cyclophilins in health and disease. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 2087–2095. [Google Scholar] [CrossRef]
- Wang, P.; Heitman, J. The cyclophilins. Genome Biol. 2005, 6, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dear, J.W.; Simpson, K.J.; Nicolai, M.P.J.; Catterson, J.H.; Street, J.; Huizinga, T.; Craig, D.G.; Dhaliwal, K.; Webb, S.; Bateman, D.N.; et al. Cyclophilin A is a Damage-Associated Molecular Pattern Molecule That Mediates Acetamino-phen-Induced Liver Injury. J. Immunol. 2011, 187, 3347. [Google Scholar] [CrossRef] [Green Version]
- Seizer, P.; Gawaz, M.; May, A.E. Cyclophilin A and EMMPRIN (CD147) in cardiovascular diseases. Cardiovasc. Res. 2014, 102, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Ge, H.; He, B. Pro-inflammatory activities induced by CyPA-EMMPRIN interaction in monocytes. Atherosclerosis 2010, 213, 415–421. [Google Scholar] [CrossRef]
- Argaud, L.; Gateau-Roesch, O.; Muntean, D.; Chalabreysse, L.; Loufouat, J.; Robert, D.; Ovize, M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J. Mol. Cell. Cardiol. 2005, 38, 367–374. [Google Scholar] [CrossRef]
- Heinzmann, D.; Bangert, A.; Muller, A.M.; von Ungern-Sternberg, S.N.; Emschermann, F.; Schonberger, T.; Chatterjee, M.; Mack, A.F.; Klingel, K.; Kandolf, R.; et al. The Novel Extracellular Cyclophilin A (CyPA)—Inhibitor MM284 Reduces Myocardial Inflammation and Re-modeling in a Mouse Model of Troponin I—Induced Myocarditis. PLoS ONE 2015, 10, e0124606. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Kuznetsov, A.V. Mitochondrial Permeability Transition and Cell Death: The Role of Cyclophilin, D. Front. Physiol. 2013, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Rieusset, J.; Fauconnier, J.; Paillard, M.; Belaidi, E.; Tubbs, E.; Chauvin, M.-A.; Durand, A.; Bravard, A.; Teixeira, G.; Bartosch, B.; et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 2015, 59, 614–623. [Google Scholar] [CrossRef] [Green Version]
- Matouschek, A.; Rospert, S.; Schmid, K.; Glick, B.S.; Schatz, G. Cyclophilin catalyzes protein folding in yeast mitochondria. Proc. Natl. Acad. Sci. USA 1995, 92, 6319–6323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability tran-sition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cy-clophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Devalaraja-Narashimha, K.; Diener, A.M.; Padanilam, B.J. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Physiol. 2009, 297, F749–F759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, M.W.; Handschumacher, R.E. Cyclophilin, a Primary Molecular Target for Cyclosporine. Transplant 1988, 46, 29S–35S. [Google Scholar] [CrossRef]
- Fliri, H.; Baumann, G.; Enz, A.; Kallen, J.; Luyten, M.; Mikol, V.; Movva, R.; Quesniaux, V.; Schreier, M.; Walkinshaw, M.; et al. Cyclosporins. Structure-activity relationships. Ann. N. Y. Acad. Sci. 1993, 696, 47–53. [Google Scholar] [CrossRef]
- Borel, J.F.; Feurer, C.; Magnée, C.; Stähelin, H. Effects of the new anti-lymphocytic peptide cyclosporin A in animals. Immunology 1977, 32, 1017–1025. [Google Scholar]
- Bennett, W.M.; Norman, D.J. Action and toxicity of cyclosporine. Ann. Rev. Med. 1986, 37, 215–224. [Google Scholar] [CrossRef]
- Yang, H.; Li, R.; Zhang, L.; Zhang, S.; Dong, W.; Chen, Y.; Wang, W.; Li, C.; Ye, Z.; Zhao, X.; et al. p53-cyclophilin D mediates renal tubular cell apoptosis in ischemia-reperfusion-induced acute kidney in-jury. Am. J. Physiol. Renal. Physiol. 2019, 317, F1311–F1317. [Google Scholar] [CrossRef]
- Ar’Rajab, A.; Dawidson, I.J.; Harris, R.B.; Mileski, W.J.; Sentementes, J.T. Deleterious effect of cyclosporins on the ischemic kidney in the rat and the protection by the calcium antagonist verapamil. J. Am. Soc. Nephrol. 1994, 5, 93–101. [Google Scholar] [PubMed]
- Johnson, D.W.; Saunders, H.J.; Johnson, F.J.; Huq, S.O.; Field, M.J.; Pollock, C.A. Fibrogenic Effects of Cyclosporin A on the Tubulointerstitium: Role of Cytokines and Growth Factors. Nephron Exp. Nephrol. 1999, 7, 470–478. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, J.B.; Shaw, A.D.; Iv, F.T.B. Acute kidney injury following cardiac surgery: Current understanding and future directions. Crit. Care 2016, 20, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Parr, S.K.; Matheny, M.E.; Abdel-Kader, K.; Greevy, R.A.; Bian, A.; Fly, J.; Chen, G.; Speroff, T.; Hung, A.M.; Ikizler, T.A.; et al. Acute kidney injury is a risk factor for subsequent proteinuria. Kidney Int. 2018, 93, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Triverio, P.-A.; Martin, P.-Y.; Romand, J.; Pugin, J.; Perneger, T.; Saudan, P. Long-term prognosis after acute kidney injury requiring renal replacement therapy. Nephrol. Dial. Transplant. 2009, 24, 2186–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackman, R.L.; Steadman, V.A.; Dean, D.K.; Jansa, P.; Poullennec, K.G.; Appleby, T.; Austin, C.; Blakemore, C.A.; Cai, R.; Cannizzaro, C.; et al. Discovery of a Potent and Orally Bioavailable Cyclophilin Inhibitor Derived from the Sanglifehrin Macrocycle. J. Med. Chem. 2018, 61, 9473–9499. [Google Scholar] [CrossRef]
- Hou, W.; Leong, K.G.; Ozols, E.; Tesch, G.H.; Nikolic-Paterson, D.J.; Ma, F.Y. Cyclophilin D promotes tubular cell damage and the development of interstitial fibrosis in the obstructed kidney. Clin. Exp. Pharmacol. Physiol. 2018, 45, 250–260. [Google Scholar] [CrossRef]
- Linkermann, A.; Brasen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate is-chemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Pasupulati, R.; Feldkamp, T.; Roeser, N.F.; Weinberg, J.M. Cyclophilin D and the mitochondrial permeability transition in kidney proximal tubules after hypoxic and ischemic injury. Am. J. Physiol. Physiol. 2011, 301, F134–F150. [Google Scholar] [CrossRef] [Green Version]
- Klausner, J.M.; Paterson, I.S.; Goldman, G.; Kobzik, L.; Rodzen, C.; Lawrence, R.; Valeri, C.R.; Shepro, D.; Hechtman, H.B. Postischemic renal injury is mediated by neutrophils and leukotrienes. Am. J. Physiol. Content 1989, 256, 794–802. [Google Scholar] [CrossRef]
- Ryan, J.; Kanellis, J.; Blease, K.; Ma, F.Y.; Nikolic-Paterson, D.J. Spleen Tyrosine Kinase Signaling Promotes Myeloid Cell Recruitment and Kidney Damage after Renal Ischemia/Reperfusion Injury. Am. J. Pathol. 2016, 186, 2032–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singbartl, K.; Ley, K. Protection from ischemia-reperfusion induced severe acute renal failure by blocking E-selectin. Crit. Care Med. 2000, 28, 2507–2514. [Google Scholar] [CrossRef]
- Perrucci, G.; Gowran, A.; Zanobini, M.; Capogrossi, M.C.; Pompilio, G.; Nigro, P. Peptidyl-prolyl isomerases: A full cast of critical actors in cardiovascular diseases. Cardiovasc. Res. 2015, 106, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofferson, D.E.; Yuan, J. Cyclophilin A release as a biomarker of necrotic cell death. Cell Death Differ. 2010, 17, 1942–1943. [Google Scholar] [CrossRef] [PubMed]
- Constant, S.L.; Heine, S.J.; Olive, D.; Murphy, P.M.; Bukrinsky, M.I.; Gao, J.-L. Cyclophilin A cooperates with MIP-2 to augment neutrophil migration. J. Inflamm. Res. 2011, 4, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, K.G.; Ozols, E.; Kanellis, J.; Nikolic-Paterson, D.J.; Ma, F.Y. Cyclophilin A Promotes Inflammation in Acute Kidney Injury but Not in Renal Fibrosis. Int. J. Mol. Sci. 2020, 21, 3667. [Google Scholar] [CrossRef]
- Ma, F.Y.; Han, Y.; Ozols, E.; Chew, P.; Vesey, D.A.; Gobe, G.C.; Morais, C.; Lohman, R.J.; Suen, J.Y.; Johnson, D.W.; et al. Protease-activated receptor 2 does not contribute to renal inflammation or fibrosis in the ob-structed kidney. Nephrology 2019, 24, 983–991. [Google Scholar]
- Ma, F.Y.; Tesch, G.H.; Nikolic-Paterson, D.J. ASK1/p38 signaling in renal tubular epithelial cells promotes renal fibrosis in the mouse obstructed kidney. Am. J. Physiol. Physiol. 2014, 307, F1263–F1273. [Google Scholar] [CrossRef]
- Ma, F.Y.; Flanc, R.S.; Tesch, G.H.; Han, Y.; Atkins, R.C.; Bennett, B.L.; Friedman, G.C.; Fan, J.-H.; Nikolic-Paterson, D.J. A Pathogenic Role for c-Jun Amino-Terminal Kinase Signaling in Renal Fibrosis and Tubular Cell Apoptosis. J. Am. Soc. Nephrol. 2007, 18, 472–484. [Google Scholar] [CrossRef]
- Stambe, C.; Atkins, R.C.; Tesch, G.H.; Masaki, T.; Schreiner, G.F.; Nikolic-Paterson, D.J. The role of p38alpha mito-gen-activated protein kinase activation in renal fibrosis. J. Am. Soc. Nephrol. 2004, 15, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Sarró, E.; Durán, M.; Rico, A.; Bou-Teen, D.; Fernández-Majada, V.; Croatt, A.J.; Nath, K.A.; Salcedo, M.T.; Gundelach, J.H.; Batlle, D.; et al. Cyclophilins A and B oppositely regulate renal tubular epithelial cell phenotype. J. Mol. Cell Biol. 2020, 12, 499–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seizer, P.; Klingel, K.; Sauter, M.; Westermann, D.; Ochmann, C.; Schönberger, T.; Schleicher, R.; Stellos, K.; Schmidt, E.-M.; Borst, O.; et al. Cyclophilin A affects inflammation, virus elimination and myocardial fibrosis in coxsackievirus B3-induced myocarditis. J. Mol. Cell. Cardiol. 2012, 53, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.; Bobardt, M.; Chatterji, U.; Mayo, P.R.; Trepanier, D.J.; Foster, R.T.; Gallay, P.A.; Ure, D.R. A Pan-Cyclophilin Inhibitor, CRV431, Decreases Fibrosis and Tumor Development in Chronic Liver Disease Models. J. Pharmacol. Exp. Ther. 2019, 371, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindblom, R.S.; Higgins, G.C.; Nguyen, T.-V.; Arnstein, M.; Henstridge, D.C.; Granata, C.; Snelson, M.; Thallas-Bonke, V.; Cooper, M.E.; Forbes, J.M.; et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin. Sci. 2020, 134, 239–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, H.Y.; Mu, W.; Nikolic-Paterson, D.J.; Atkins, R.C. A novel, simple, reliable, and sensitive method for multiple immu-noenzyme staining: Use of microwave oven heating to block antibody crossreactivity and retrieve antigens. J. Histochem. Cyto-chem. 1995, 43, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.Y.; Tesch, G.H.; Ozols, E.; Xie, M.; Schneider, M.D.; Nikolic-Paterson, D.J. TGF-beta1-activated kinase-1 regulates inflammation and fibrosis in the obstructed kidney. Am. J. Physiol. Renal. Physiol. 2011, 300, F1410–F1421. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leong, K.G.; Ozols, E.; Kanellis, J.; Badal, S.S.; Liles, J.T.; Nikolic-Paterson, D.J.; Ma, F.Y. Cyclophilin Inhibition Protects Against Experimental Acute Kidney Injury and Renal Interstitial Fibrosis. Int. J. Mol. Sci. 2021, 22, 271. https://doi.org/10.3390/ijms22010271
Leong KG, Ozols E, Kanellis J, Badal SS, Liles JT, Nikolic-Paterson DJ, Ma FY. Cyclophilin Inhibition Protects Against Experimental Acute Kidney Injury and Renal Interstitial Fibrosis. International Journal of Molecular Sciences. 2021; 22(1):271. https://doi.org/10.3390/ijms22010271
Chicago/Turabian StyleLeong, Khai Gene, Elyce Ozols, John Kanellis, Shawn S. Badal, John T. Liles, David J. Nikolic-Paterson, and Frank Y. Ma. 2021. "Cyclophilin Inhibition Protects Against Experimental Acute Kidney Injury and Renal Interstitial Fibrosis" International Journal of Molecular Sciences 22, no. 1: 271. https://doi.org/10.3390/ijms22010271