Autocrine Bradykinin Release Promotes Ischemic Preconditioning-Induced Cytoprotection in Bovine Aortic Endothelial Cells

 , , , , , and
, , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
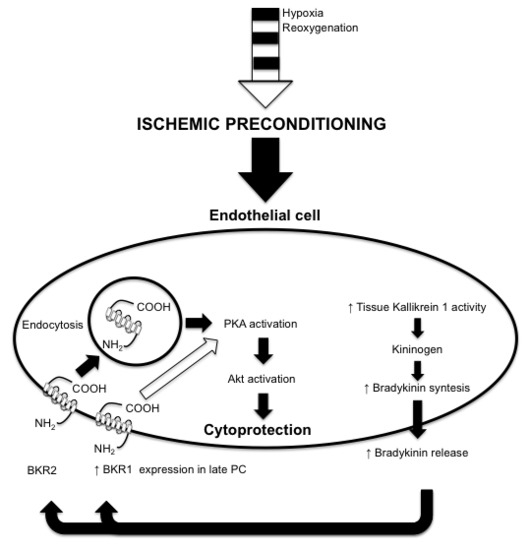
1. Introduction
2. Results
2.1. PC Evokes Bradykinin Release from Endothelial Cells
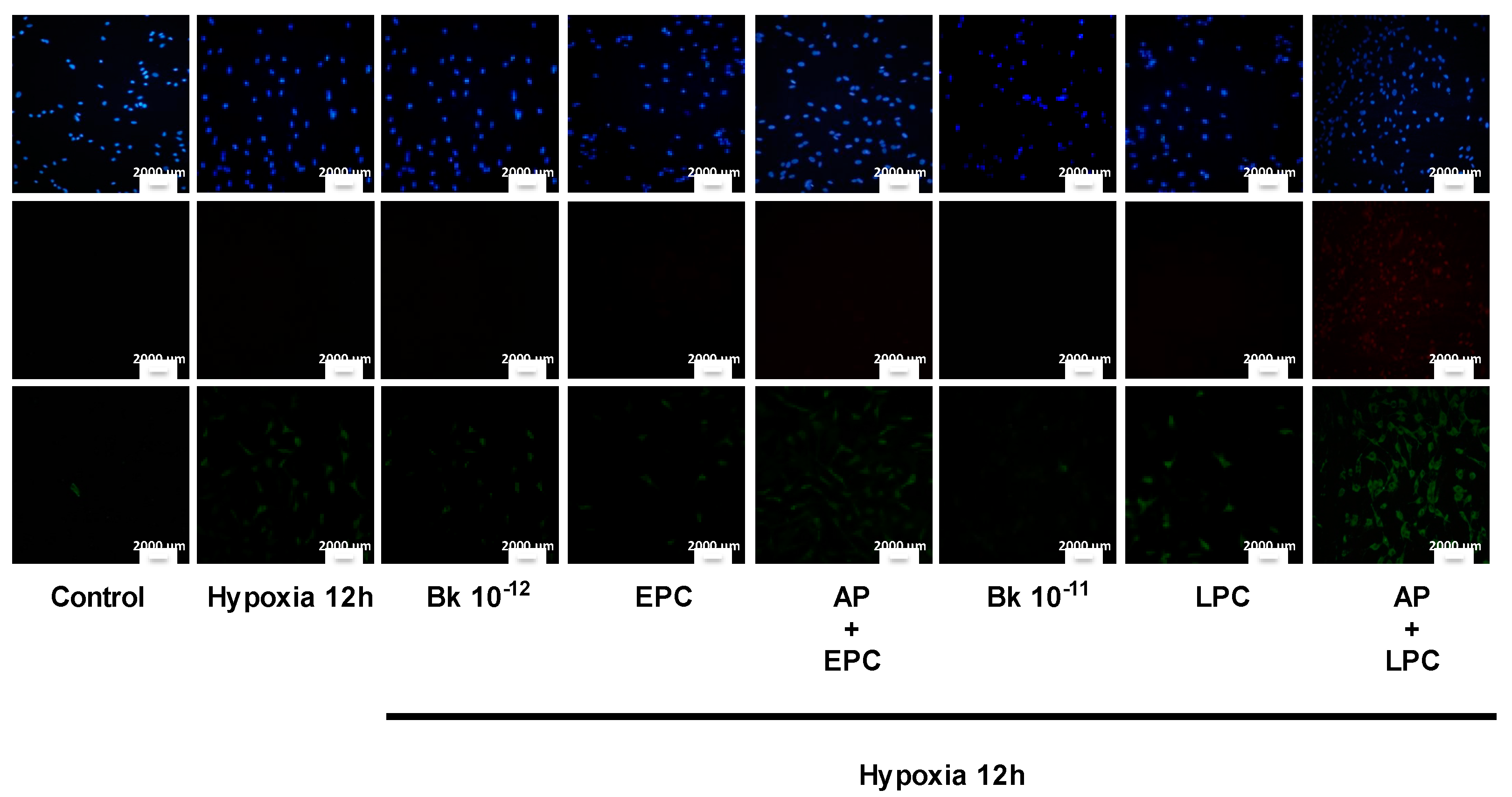
2.2. PC-Induced Bk Synthesis Promotes Cytoprotection against Hypoxia-Induced Apoptosis
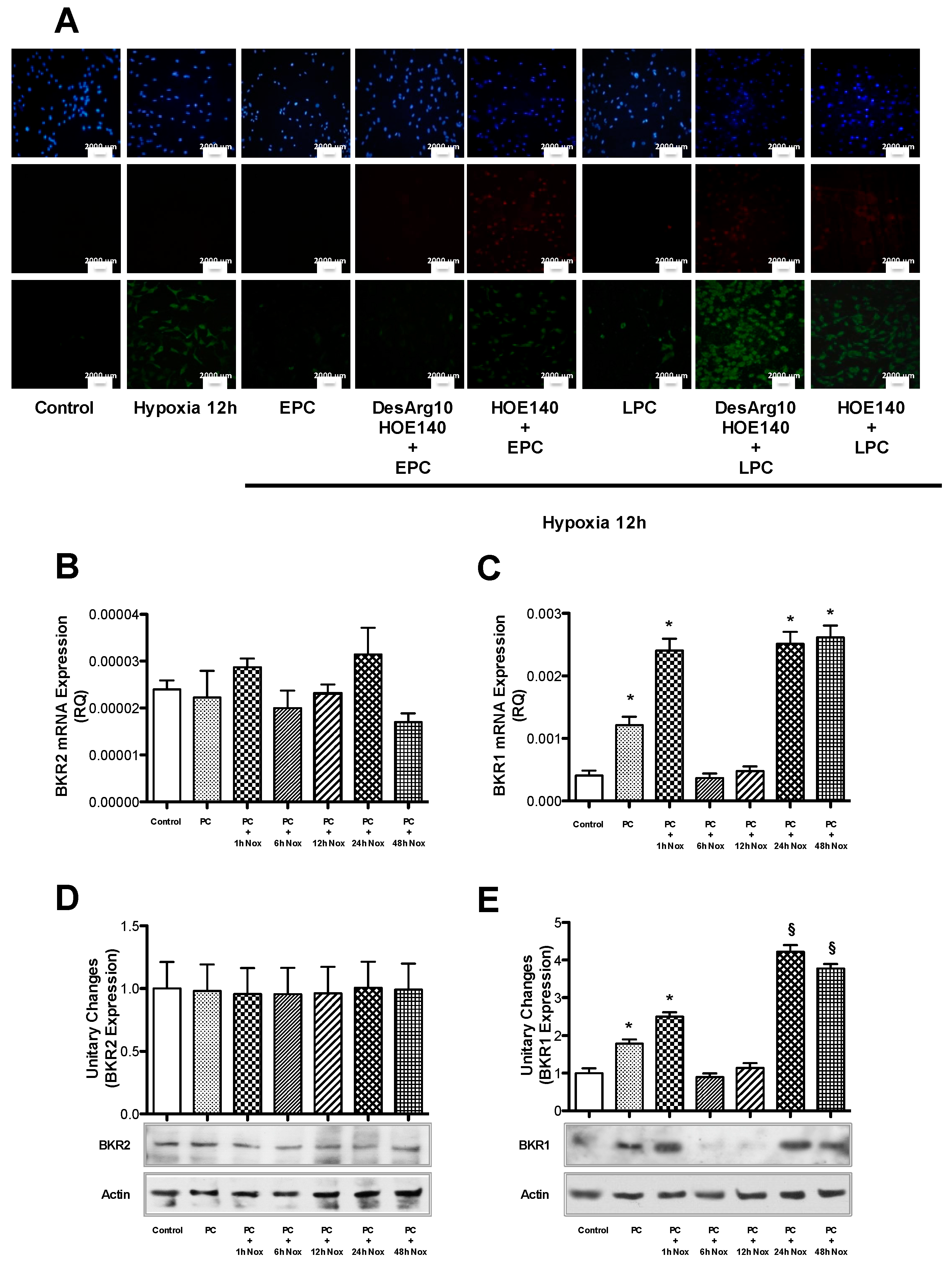
2.3. PC Induces Cytoprotection through BKRs
2.4. BKR2 Endocytosis and Recycling Induce Cytoprotection During PC
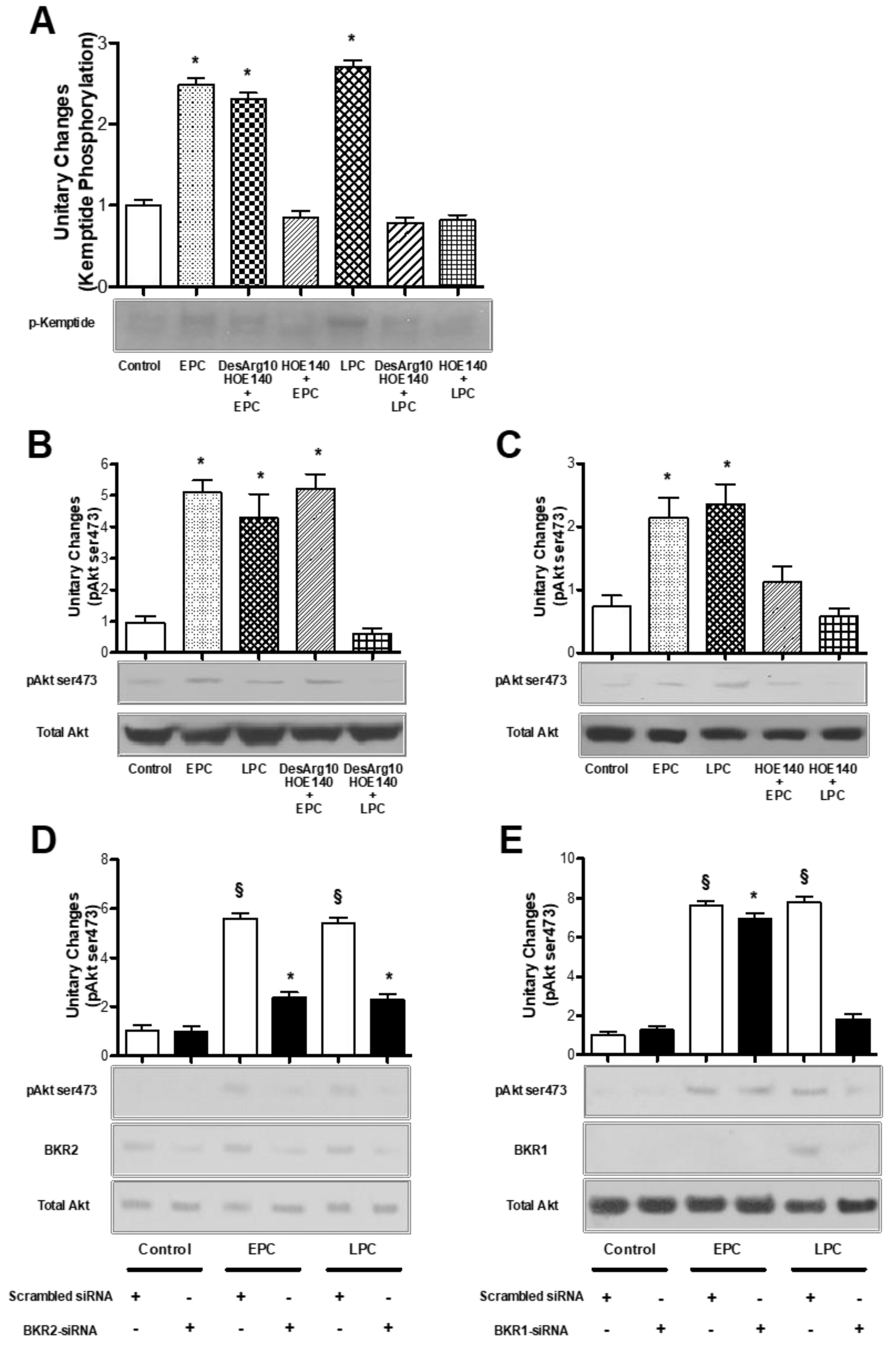
2.5. BKRs Mediate PKA and Akt Activation during PC
2.6. BKRs Induce Crosstalk between PKA and Akt during PC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Hypoxia
4.3. Estimation of Bradykinin (Bk) Production
4.4. Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
4.5. Measurement of Tissue Kallikrein Activity (KLK1) in Endothelial Cells
4.6. Immunoblotting and Immunoprecipitation
4.7. Apoptosis and Necrosis Detection
4.8. Gene Silencing by Transient Transfection
4.9. Preparation of Membrane and Cytosol Extracts
4.10. PKA Activity Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP | Aprotinin |
| bAECs | Bovine aortic endothelial cells |
| Bk | Bradykinin |
| Bradykinin receptor 1 | BKR1 |
| Bradykinin receptor 2 | BKR2 |
| Kemptide | Kp |
| Kininogen | Kn |
| PC | Ischemic preconditioning |
| PI3K | phosphatidil-inositol-3-kinase |
| PKA | Protein Kinase A |
| RT-PCR | Reverse Transcriptase Polymerase Chain Reaction |
| Tissue kallikrein | KLK1 |
References
- Jalowy, A.; Schulz, R.; Dorge, H.; Behrends, M.; Heusch, G. Infarct size reduction by AT1-receptor blockade through a signal cascade of AT2-receptor activation, bradykinin and prostaglandins in pigs. J. Am. Coll. Cardiol. 1998, 32, 1787–1796. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Mancardi, D.; Tullio, F.; Pagliaro, P. Postconditioning and intermittent bradykinin induced cardioprotection require cyclooxygenase activation and prostacyclin release during reperfusion. Basic Res. Cardiol. 2008, 103, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Post, H.; Vahlhaus, C.; Heusch, G. Ischemic preconditioning in pigs: A graded phenomenon: Its relation to adenosine and bradykinin. Circulation 1998, 98, 1022–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Randhawa, P.K.; Singh, N.; Jaggi, A.S. Bradykinin in ischemic conditioning-induced tissue protection: Evidences and possible mechanisms. Eur. J. Pharmacol. 2015, 768, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Tang, X.L.; Park, S.W.; Sun, J.Z.; Kalya, A.; Bolli, R. The early and late phases of ischemic preconditioning: A comparative analysis of their effects on infarct size, myocardial stunning, and arrhythmias in conscious pigs undergoing a 40-min coronary occlusion. Circ. Res. 1997, 80, 730–742. [Google Scholar] [CrossRef]
- Kokkonen, J.O.; Kuoppala, A.; Saarinen, J.; Lindstedt, K.A.; Kovanen, P.T. Kallidin- and bradykinin-degrading pathways in human heart: Degradation of kallidin by aminopeptidase M-like activity and bradykinin by neutral endopeptidase. Circulation 1999, 99, 1984–1990. [Google Scholar] [CrossRef] [Green Version]
- Kokkonen, J.O.; Lindstedt, K.A.; Kuoppala, A.; Kovanen, P.T. Kinin-degrading pathways in the human heart. Trends Cardiovasc. Med. 2000, 10, 42–45. [Google Scholar] [CrossRef]
- Goto, M.; Liu, Y.; Yang, X.M.; Ardell, J.L.; Cohen, M.V.; Downey, J.M. Role of bradykinin in protection of ischemic preconditioning in rabbit hearts. Circ. Res. 1995, 77, 611–621. [Google Scholar] [CrossRef]
- Linz, W.; Wiemer, G.; Scholkens, B.A. Beneficial effects of bradykinin on myocardial energy metabolism and infarct size. Am. J. Cardiol. 1997, 80, 118A–123A. [Google Scholar] [CrossRef]
- Schoemaker, R.G.; van Heijningen, C.L. Bradykinin mediates cardiac preconditioning at a distance. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1571–H1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellis, A.; Castaldo, D.; Trimarco, V.; Monti, M.G.; Chivasso, P.; Sadoshima, J.; Trimarco, B.; Morisco, C. Cross-talk between PKA and Akt protects endothelial cells from apoptosis in the late ischemic preconditioning. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nakamura, H.; Yodoi, J.; Bloom, E.T. Redox regulation of the signaling pathways leading to eNOS phosphorylation. Free Radic. Biol. Med. 2005, 38, 1231–1242. [Google Scholar] [CrossRef]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [Green Version]
- Graf, K.; Grafe, M.; Auch-Schwelk, W.; Baumgarten, C.R.; Scheffer, H.; Hildebrandt, A.; Fleck, E. Tissue kallikrein activity and kinin release in human endothelial cells. Eur. J. Clin. Chem. Clin. Biochem. J. Forum Eur. Clin. Chem. Soc. 1994, 32, 495–500. [Google Scholar] [CrossRef] [Green Version]
- Heitsch, H.; Brovkovych, S.; Malinski, T.; Wiemer, G. Angiotensin-(1-7)-Stimulated Nitric Oxide and Superoxide Release From Endothelial Cells. Hypertension 2001, 37, 72–76. [Google Scholar] [CrossRef]
- Condorelli, G.; Roncarati, R.; Ross, J., Jr.; Pisani, A.; Stassi, G.; Todaro, M.; Trocha, S.; Drusco, A.; Gu, Y.; Russo, M.A.; et al. Heart-targeted overexpression of caspase3 in mice increases infarct size and depresses cardiac function. Proc. Natl. Acad. Sci. USA 2001, 98, 9977–9982. [Google Scholar] [CrossRef] [Green Version]
- Blaukat, A.; Alla, S.A.; Lohse, M.J.; Muller-Esterl, W. Ligand-induced phosphorylation/dephosphorylation of the endogenous bradykinin B2 receptor from human fibroblasts. J. Biol. Chem. 1996, 271, 32366–32374. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Brovkovych, V.; Zhang, Y.; Tan, F.; Skidgel, R.A. Downregulation of kinin B1 receptor function by B2 receptor heterodimerization and signaling. Cell. Signal. 2015, 27, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef]
- Bawolak, M.T.; Roy, C.; Gera, L.; Marceau, F. Prolonged signalling and trafficking of the bradykinin B2 receptor stimulated with the amphibian peptide maximakinin: Insight into the endosomal inactivation of kinins. Pharmacol. Res. 2012, 65, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, H.; Takagi, H.; Oh, H.; Suzuma, K.; Suzuma, I.; Miyamoto, N.; Uemura, A.; Watanabe, D.; Murakami, T.; Sugaya, T.; et al. Phosphatidylinositol 3-kinase/Akt regulates angiotensin II-induced inhibition of apoptosis in microvascular endothelial cells by governing survivin expression and suppression of caspase-3 activity. Circ. Res. 2004, 94, 785–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.; Gelpi, R.J. Adenosine and cardioprotection during reperfusion--an overview. Mol. Cell. Biochem. 2003, 251, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Headrick, J.P.; Hack, B.; Ashton, K.J. Acute adenosinergic cardioprotection in ischemic-reperfused hearts. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1797–1818. [Google Scholar] [CrossRef] [Green Version]
- Puhl, S.L.; Kazakov, A.; Muller, A.; Fries, P.; Wagner, D.R.; Bohm, M.; Maack, C.; Devaux, Y. Adenosine A1 receptor activation attenuates cardiac hypertrophy and fibrosis in response to alpha1 -adrenoceptor stimulation in vivo. Br. J. Pharmacol. 2016, 173, 88–102. [Google Scholar] [CrossRef] [Green Version]
- Pugsley, M.K. The diverse molecular mechanisms responsible for the actions of opioids on the cardiovascular system. Pharmacol. Ther. 2002, 93, 51–75. [Google Scholar] [CrossRef]
- Peart, J.N.; Gross, E.R.; Gross, G.J. Opioid-induced preconditioning: Recent advances and future perspectives. Vasc. Pharmacol. 2005, 42, 211–218. [Google Scholar] [CrossRef]
- Schultz, J.E.; Rose, E.; Yao, Z.; Gross, G.J. Evidence for involvement of opioid receptors in ischemic preconditioning in rat hearts. Am. J. Physiol. 1995, 268, H2157–H2161. [Google Scholar] [CrossRef]
- Shimizu, M.; Tropak, M.; Diaz, R.J.; Suto, F.; Surendra, H.; Kuzmin, E.; Li, J.; Gross, G.; Wilson, G.J.; Callahan, J.; et al. Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: Evidence suggesting cross-species protection. Clin. Sci. 2009, 117, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.E.; Hsu, A.K.; Gross, G.J. Ischemic preconditioning in the intact rat heart is mediated by delta1- but not mu- or kappa-opioid receptors. Circulation 1998, 97, 1282–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatta, A.J.; Kin, H.; Yoshishige, D.; Jiang, R.; Wang, N.; Reeves, J.G.; Mykytenko, J.; Guyton, R.A.; Zhao, Z.Q.; Caffrey, J.L.; et al. Evidence that cardioprotection by postconditioning involves preservation of myocardial opioid content and selective opioid receptor activation. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1444–H1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, G.T.; Li, R.; Jiang, L.L.; Irwin, M.G. Remifentanil post-conditioning attenuates cardiac ischemia-reperfusion injury via kappa or delta opioid receptor activation. Acta Anaesthesiol. Scand. 2010, 54, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Boengler, K.; Schulz, R. Cardioprotection: Nitric oxide, protein kinases, and mitochondria. Circulation 2008, 118, 1915–1919. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Bianchi, C.; Sandmeyer, J.L.; Sellke, F.W. Bradykinin preconditioning improves the profile of cell survival proteins and limits apoptosis after cardioplegic arrest. Circulation 2005, 112, I190–I195. [Google Scholar] [CrossRef]
- Oeseburg, H.; Iusuf, D.; van der Harst, P.; van Gilst, W.H.; Henning, R.H.; Roks, A.J. Bradykinin protects against oxidative stress-induced endothelial cell senescence. Hypertension 2009, 53, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Griol-Charhbili, V.; Messadi-Laribi, E.; Bascands, J.L.; Heudes, D.; Meneton, P.; Giudicelli, J.F.; Alhenc-Gelas, F.; Richer, C. Role of tissue kallikrein in the cardioprotective effects of ischemic and pharmacological preconditioning in myocardial ischemia. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1172–1174. [Google Scholar] [CrossRef]
- Yoshida, H.; Zhang, J.J.; Chao, L.; Chao, J. Kallikrein gene delivery attenuates myocardial infarction and apoptosis after myocardial ischemia and reperfusion. Hypertension 2000, 35, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.P.; Liu, Y.H.; Scicli, G.M.; Webb, C.R.; Carretero, O.A. Role of kinins in the cardioprotective effect of preconditioning: Study of myocardial ischemia/reperfusion injury in B2 kinin receptor knockout mice and kininogen-deficient rats. Hypertension 1997, 30, 735–740. [Google Scholar] [CrossRef]
- Kositprapa, C.; Ockaili, R.A.; Kukreja, R.C. Bradykinin B2 receptor is involved in the late phase of preconditioning in rabbit heart. J. Mol. Cell. Cardiol. 2001, 33, 1355–1362. [Google Scholar] [CrossRef]
- Duka, A.; Kintsurashvili, E.; Duka, I.; Ona, D.; Hopkins, T.A.; Bader, M.; Gavras, I.; Gavras, H. Angiotensin-converting enzyme inhibition after experimental myocardial infarct: Role of the kinin B1 and B2 receptors. Hypertension 2008, 51, 1352–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieg, T.; Qin, Q.; Philipp, S.; Alexeyev, M.F.; Cohen, M.V.; Downey, J.M. Acetylcholine and bradykinin trigger preconditioning in the heart through a pathway that includes Akt and NOS. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2606–H2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.W.; Kim, H.S.; Cha, Y.N.; Park, Y.S.; Jo, S.A.; Jo, I. Rapid increase in endothelial nitric oxide production by bradykinin is mediated by protein kinase A signaling pathway. Biochem. Biophys. Res. Commun. 2003, 306, 981–987. [Google Scholar] [CrossRef]
- Shah, A.M. Paracrine modulation of heart cell function by endothelial cells. Cardiovasc. Res. 1996, 31, 847–867. [Google Scholar] [CrossRef] [Green Version]
- Savarese, G.; Costanzo, P.; Cleland, J.G.; Vassallo, E.; Ruggiero, D.; Rosano, G.; Perrone-Filardi, P. A meta-analysis reporting effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in patients without heart failure. J. Am. Coll. Cardiol. 2013, 61, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Torrado, J.; Cain, C.; Mauro, A.G.; Romeo, F.; Ockaili, R.; Chau, V.Q.; Nestler, J.A.; Devarakonda, T.; Ghosh, S.; Das, A.; et al. Sacubitril/Valsartan Averts Adverse Post-Infarction Ventricular Remodeling and Preserves Systolic Function in Rabbits. J. Am. Coll. Cardiol. 2018, 72, 2342–2356. [Google Scholar] [CrossRef]
- Eitel, I.; Stiermaier, T.; Rommel, K.P.; Fuernau, G.; Sandri, M.; Mangner, N.; Linke, A.; Erbs, S.; Lurz, P.; Boudriot, E.; et al. Cardioprotection by combined intrahospital remote ischaemic perconditioning and postconditioning in ST-elevation myocardial infarction: The randomized LIPSIA CONDITIONING trial. Eur. Heart J. 2015, 36, 3049–3057. [Google Scholar] [CrossRef]
- Morisco, C.; Marrone, C.; Trimarco, V.; Crispo, S.; Monti, M.G.; Sadoshima, J.; Trimarco, B. Insulin resistance affects the cytoprotective effect of insulin in cardiomyocytes through an impairment of MAPK phosphatase-1 expression. Cardiovasc. Res. 2007, 76, 453–464. [Google Scholar] [CrossRef]
- Portela, P.; Howell, S.; Moreno, S.; Rossi, S. In vivo and in vitro phosphorylation of two isoforms of yeast pyruvate kinase by protein kinase A. J. Biol. Chem. 2002, 277, 30477–30487. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellis, A.; Sorriento, D.; Fiordelisi, A.; Izzo, R.; Sadoshima, J.; Mauro, C.; Cerasuolo, F.; Mancusi, C.; Barbato, E.; Pilato, E.; et al. Autocrine Bradykinin Release Promotes Ischemic Preconditioning-Induced Cytoprotection in Bovine Aortic Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 2965. https://doi.org/10.3390/ijms21082965
Bellis A, Sorriento D, Fiordelisi A, Izzo R, Sadoshima J, Mauro C, Cerasuolo F, Mancusi C, Barbato E, Pilato E, et al. Autocrine Bradykinin Release Promotes Ischemic Preconditioning-Induced Cytoprotection in Bovine Aortic Endothelial Cells. International Journal of Molecular Sciences. 2020; 21(8):2965. https://doi.org/10.3390/ijms21082965
Chicago/Turabian StyleBellis, Alessandro, Daniela Sorriento, Antonella Fiordelisi, Raffaele Izzo, Junichi Sadoshima, Ciro Mauro, Federica Cerasuolo, Costantino Mancusi, Emanuele Barbato, Emanuele Pilato, and et al. 2020. "Autocrine Bradykinin Release Promotes Ischemic Preconditioning-Induced Cytoprotection in Bovine Aortic Endothelial Cells" International Journal of Molecular Sciences 21, no. 8: 2965. https://doi.org/10.3390/ijms21082965