Knockout of ACTB and ACTG1 with CRISPR/Cas9(D10A) Technique Shows that Non-Muscle β and γ Actin Are Not Equal in Relation to Human Melanoma Cells’ Motility and Focal Adhesion Formation

 , ,
, ,
Abstract
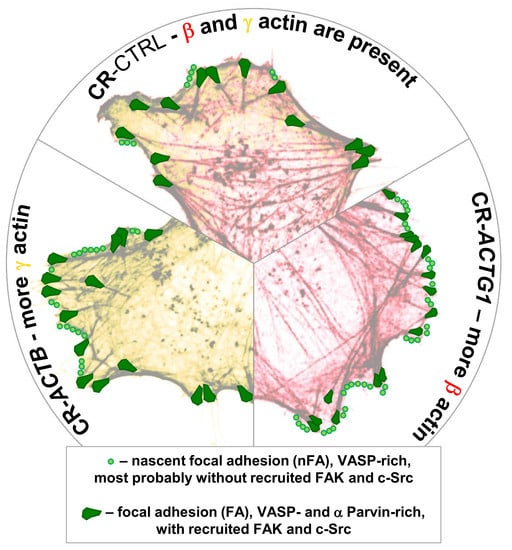
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Both Non-Muscle Actins Are Highly Abundant in A375 Cells but Occupy Different Areas of the Cell
2.2. Successful Knockout of Genes Coding for β and γ Actin Leads to Altered Non-Muscle Isoactin Expression and Affects Cells’ Migration
2.3. Migration and Invasion Are Affected in the CR-ACTB Clones and Greatly Affected in the CR-ACTG1 Clones
2.4. Bundling of Stress Fibers Is Altered in Clones that Lack Either β or γ Actin
2.5. Lamellipodial Dynamics Are Altered in Cells Devoid of Either β or γ Actin
2.6. There Are Differences in the Distribution and Levels of Actin Polymerization Nucleators within the Leading Edge
2.7. Focal Adhesion (FA) Formation Is Changed in the Clones Devoid of Either β or γ Actin
2.8. Signaling Connected to FAs Is Altered in the Clones Devoid of Either β or γ Actin
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines and Cell Culture Conditions
4.3. CRISPR/Cas9(D10A) Inactivation of ACTB and ACTG1 Genes
4.4. Invasion Assay
4.5. Cell Migration Assays
4.6. Immunocytochemistry and Confocal Microscopy
4.7. Live Imaging and Kymograph Analysis
4.8. Western Blot Analysis
4.9. Quantitative Polymerase Chain Reaction (qPCR)
4.10. 2-D PAGE
4.11. Rho-GTPases Assays
4.12. F:G Actin Ratio Assay
4.13. Focal Adhesion and Cell Area Analysis
4.14. Lamellipodium Width and Thickness Analysis
4.15. Stress Fibers’ Formation Analysis
4.16. Gelatin Digestion Assay
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABPs | Actin-binding proteins |
| ACTB | gene coding for β actin |
| ACTG1 | gene coding for γ actin |
| CR-CTRL | control cells |
| CR-ACTB | cells with knocked out ACTB |
| CR-ACTG1 | cells with knocked out ACTG1 |
| F-actin | filamentous, polymerized actin |
| G-actin | globular, monomeric actin |
| FA | focal adhesion |
| LPA | lysophosphatidic acid |
| PMA | phorbol-12-myristate-13-acetate |
| nFA | nascent focal adhesion |
References
- Perrin, B.J.; Ervasti, J.M. The actin gene family: Function follows isoform. Cytoskeleton (Hoboken) 2010, 67, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Hurst, V.; Shimada, K.; Gasser, S.M. Nuclear Actin and Actin-Binding Proteins in DNA Repair. Trends Cell Biol. 2019, 29, 462–476. [Google Scholar] [CrossRef] [Green Version]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winder, S.J.; Ayscough, K.R. Actin-binding proteins. J. Cell Sci. 2005, 118, 651–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, B.; Robinson, R.C. Guardians of the actin monomer. Eur. J. Cell Biol. 2013, 92, 316–332. [Google Scholar] [CrossRef]
- Sibony-Benyamini, H.; Gil-Henn, H. Invadopodia: The leading force. Eur. J. Cell Biol. 2012, 91, 896–901. [Google Scholar] [CrossRef]
- Simiczyjew, A.; Pietraszek-Gremplewicz, K.; Mazur, A.J.; Nowak, D. Are non-muscle actin isoforms functionally equivalent? Histol. Histopathol. 2017, 11896. [Google Scholar]
- Ampe, C.; Van Troys, M. Mammalian Actins: Isoform-Specific Functions and Diseases. Handb. Exp. Pharmacol. 2016, 1–37. [Google Scholar]
- Vandekerckhove, J.; Weber, K. At least six different actins are expressed in a higher mammal: An analysis based on the amino acid sequence of the amino-terminal tryptic peptide. J. Mol. Biol. 1978, 126, 783–802. [Google Scholar] [CrossRef]
- Kashina, A.S. Regulation of actin isoforms in cellular and developmental processes. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Bergeron, S.E.; Zhu, M.; Thiem, S.M.; Friderici, K.H.; Rubenstein, P.A. Ion-dependent Polymerization Differences between Mammalian β- and γ-Nonmuscle Actin Isoforms. J. Biol. Chem. 2010, 285, 16087–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Saha, S.; Shabalina, S.A.; Kashina, A. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science 2010, 329, 1534–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vedula, P.; Kashina, A. The makings of the ‘actin code’: Regulation of actin’s biological function at the amino acid and nucleotide level. J. Cell Sci. 2018, 131, jcs215509. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.A.; Gunning, P. Beta and gamma actin mRNAs are differentially located within myoblasts. J. Cell Biol. 1993, 122, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Vedula, P.; Kurosaka, S.; Leu, N.A.; Wolf, Y.I.; Shabalina, S.A.; Wang, J.; Sterling, S.; Dong, D.W.; Kashina, A. Diverse functions of homologous actin isoforms are defined by their nucleotide, rather than their amino acid sequence. Elife 2017, 6, e31661. [Google Scholar] [CrossRef]
- Verloes, A.; Di Donato, N.; Masliah-Planchon, J.; Jongmans, M.; Abdul-Raman, O.A.; Albrecht, B.; Allanson, J.; Brunner, H.; Bertola, D.; Chassaing, N.; et al. Baraitser–Winter cerebrofrontofacial syndrome: Delineation of the spectrum in 42 cases. Eur. J. Hum. Genet. 2015, 23, 292. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, N.; Rump, A.; Koenig, R.; Der Kaloustian, V.M.; Halal, F.; Sonntag, K.; Krause, C.; Hackmann, K.; Hahn, G.; Schrock, E.; et al. Severe forms of Baraitser-Winter syndrome are caused by ACTB mutations rather than ACTG1 mutations. Eur. J. Hum. Genet. 2014, 22, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Kemper, K.; de Goeje, P.L.; Peeper, D.S.; van Amerongen, R. Phenotype switching: Tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res. 2014, 74, 5937–5941. [Google Scholar] [CrossRef] [Green Version]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Oser, M.; Condeelis, J. The cofilin activity cycle in lamellipodia and invadopodia. J. Cell. Biochem. 2009, 108, 1252–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoumacher, M.; Louvard, D.; Vignjevic, D. Cytoskeleton networks in basement membrane transmigration. Eur. J. Cell Biol. 2011, 90, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Schönichen, A.; Mannherz, H.G.; Behrmann, E.; Mazur, A.J.; Kühn, S.; Silván, U.; Schoenenberger, C.-A.; Fackler, O.T.; Raunser, S.; Dehmelt, L.; et al. FHOD1 is a combined actin filament capping and bundling factor that selectively associates with actin arcs and stress fibers. J. Cell Sci. 2013, 126, 1891–1901. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.L.; Ravid, S.; Spudich, J.A. Control of Nonmuscle Myosins by Phosphorylation. Annu. Rev. Biochem. 1992, 61, 721–759. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Carlier, M.-F.; Shekhar, S. Global treadmilling coordinates actin turnover and controls the size of actin networks. Nat. Rev. Mol. Cell Biol. 2017. [Google Scholar] [CrossRef]
- Harbeck, B.; Hüttelmaier, S.; Schlüter, K.; Jockusch, B.M.; Illenberger, S. Phosphorylation of the vasodilator-stimulated phosphoprotein regulates its interaction witn actin. J. Biol. Chem. 2000, 275, 30817–30825. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors This paper is dedicated to the memory of Prof. Donald F. Steiner (1930–2014)—Advisor, mentor, and discoverer of proinsulin. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Chen, H.; Cheng, C.Y. Focal Adhesion Kinase (FAK). In Encyclopedia of Signaling Molecules; Springer International Publishing: New York, NY, USA, 2018; pp. 1800–1812. [Google Scholar]
- Goidin, D.; Mamessier, A.; Staquet, M.-J.; Schmitt, D.; Berthier-Vergnes, O. Ribosomal 18S RNA Prevails over Glyceraldehyde-3-Phosphate Dehydrogenase and β-Actin Genes as Internal Standard for Quantitative Comparison of mRNA Levels in Invasive and Noninvasive Human Melanoma Cell Subpopulations. Anal. Biochem. 2001, 295, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugina, V.; Zwaenepoel, I.; Gabbiani, G.; Clément, S.; Chaponnier, C. Beta and gamma-cytoplasmic actins display distinct distribution and functional diversity. J. Cell Sci. 2009, 122, 2980–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugina, V.; Khromova, N.; Rybko, V.; Blizniukov, O.; Shagieva, G.; Chaponnier, C.; Kopnin, B.; Kopnin, P. Tumor promotion by γ and suppression by β non-muscle actin isoforms. Oncotarget 2015, 6, 14556–14571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shum, M.S.Y.; Pasquier, E.; Po’uha, S.T.; O’Neill, G.M.; Chaponnier, C.; Gunning, P.W.; Kavallaris, M. γ-Actin regulates cell migration and modulates the ROCK signaling pathway. FASEB J. 2011, 25, 4423–4433. [Google Scholar] [CrossRef] [Green Version]
- Hoock, T.C.; Newcomb, P.M.; Herman, I.M. Beta actin and its mRNA are localized at the plasma membrane and the regions of moving cytoplasm during the cellular response to injury. J. Cell Biol. 1991, 112, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, E.; Tuset, M.-P.; Sinnappan, S.; Carnell, M.; Macmillan, A.; Kavallaris, M. γ-Actin plays a key role in endothelial cell motility and neovessel maintenance. Vasc. Cell 2015, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Khaitlina, S.Y. Functional specificity of actin isoforms. Int. Rev. Cytol. 2001, 202, 35–98. [Google Scholar]
- Latham, S.L.; Ehmke, N.; Reinke, P.Y.A.; Taft, M.H.; Eicke, D.; Reindl, T.; Stenzel, W.; Lyons, M.J.; Friez, M.J.; Lee, J.A.; et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat. Commun. 2018, 9, 4250. [Google Scholar] [CrossRef]
- Moradi, M.; Sivadasan, R.; Saal, L.; Lüningschrör, P.; Dombert, B.; Rathod, R.J.; Dieterich, D.C.; Blum, R.; Sendtner, M. Differential roles of α-, β-, and γ-actin in axon growth and collateral branch formation in motoneurons. J. Cell Biol. 2017, 216, 793–814. [Google Scholar] [CrossRef]
- Bunnell, T.M.; Burbach, B.J.; Shimizu, Y.; Ervasti, J.M. β-Actin specifically controls cell growth, migration, and the G-actin pool. Mol. Biol. Cell 2011, 22, 4047–4058. [Google Scholar] [CrossRef]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peckham, M.; Miller, G.; Wells, C.; Zicha, D.; Dunn, G.A. Specific changes to the mechanism of cell locomotion induced by overexpression of beta-actin. J. Cell Sci. 2001, 114, 1367–1377. [Google Scholar]
- Suarez, C.; Kovar, D.R. Internetwork competition for monomers governs actin cytoskeleton organization. Nat. Rev. Mol. Cell Biol. 2016, 17, 799–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, T.A.; Christensen, J.R.; Barone, E.; Suarez, C.; Sirotkin, V.; Kovar, D.R. Homeostatic actin cytoskeleton networks are regulated by assembly factor competition for monomers. Curr. Biol. 2014, 24, 579–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugesan, S.; Hong, J.; Yi, J.; Li, D.; Beach, J.R.; Shao, L.; Meinhardt, J.; Madison, G.; Wu, X.; Betzig, E.; et al. Formin-generated actomyosin arcs propel t cell receptor microcluster movement at the immune synapse. J. Cell Biol. 2016, 215, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Antkowiak, A.; Guillotin, A.; Boiero Sanders, M.; Colombo, J.; Vincentelli, R.; Michelot, A. Sizes of actin networks sharing a common environment are determined by the relative rates of assembly. PLoS Biol. 2019, 17, e3000317. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.L.; Wiedemann, U.; Stuurman, N.; Vale, R.D. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J. Cell Biol. 2003, 162, 1079–1088. [Google Scholar] [CrossRef]
- Nicholson-Dykstra, S.M.; Higgs, H.N. Arp2 depletion inhibits sheet-like protrusions but not linear protrusions of fibroblasts and lymphocytes. Cell Motil. Cytoskeleton 2008, 65, 904–922. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Asokan, S.B.; Berginski, M.E.; Haynes, E.M.; Sharpless, N.E.; Griffith, J.D.; Gomez, S.M.; Bear, J.E. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 2012, 148, 973–987. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Diensthuber, R.P.; Chizhov, I.; Claus, P.; Heissler, S.M.; Preller, M.; Taft, M.H.; Manstein, D.J. Distinct Functional Interactions between Actin Isoforms and Nonsarcomeric Myosins. PLoS ONE 2013, 8, e70636. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Dang, I.; Gautreau, A. Evidence for a cell cycle checkpoint that senses branched actin in the lamellipodium. BioEssays 2012, 34, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1692, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Schiller, H.B.; Fässler, R. Mechanosensitivity and compositional dynamics of cell–matrix adhesions. EMBO Rep. 2013, 14, 509–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, E.R.; Humphries, J.D.; James, J.; Jones, M.C.; Askari, J.A.; Humphries, M.J. The integrin adhesome network at a glance. J. Cell Sci. 2016, 129, 4159–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, K.; Guilluy, C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 2016, 343, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchsbaum, R.J. Rho activation at a glance. J. Cell Sci. 2007, 120, 1149–1152. [Google Scholar] [CrossRef] [Green Version]
- Mannherz, H.G. The Molecular Basis of Directional Cell Migration. In Cell Signaling and Growth Factors in Development; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2005; pp. 321–345. [Google Scholar]
- Tondeleir, D.; Lambrechts, A.; Muller, M.; Jonckheere, V.; Doll, T.; Vandamme, D.; Bakkali, K.; Waterschoot, D.; Lemaistre, M.; Debeir, O.; et al. Cells Lacking -Actin are Genetically Reprogrammed and Maintain Conditional Migratory Capacity. Mol. Cell. Proteomics 2012, 11, 255–271. [Google Scholar] [CrossRef] [Green Version]
- Dormoy-Raclet, V.; Ménard, I.; Clair, E.; Kurban, G.; Mazroui, R.; Di Marco, S.; von Roretz, C.; Pause, A.; Gallouzi, I.-E. The RNA-binding protein HuR promotes cell migration and cell invasion by stabilizing the beta-actin mRNA in a U-rich-element-dependent manner. Mol. Cell. Biol. 2007, 27, 5365–5380. [Google Scholar] [CrossRef] [Green Version]
- Makowiecka, A.; Malek, N.; Mazurkiewicz, E.; Mrówczyńska, E.; Nowak, D.; Mazur, A.J. Thymosin β4 Regulates Focal Adhesion Formation in Human Melanoma Cells and Affects Their Migration and Invasion. Front. Cell Dev. Biol. 2019, 7, 304. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92. [Google Scholar] [CrossRef] [PubMed]
- Dratkiewicz, E.; Pietraszek-Gremplewicz, K.; Simiczyjew, A.; Mazur, A.J.; Nowak, D. Gefitinib or lapatinib with foretinib synergistically induce a cytotoxic effect in melanoma cell lines. Oncotarget 2018, 9, 18254–18268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simiczyjew, A.; Mazur, A.J.; Popow-Woźniak, A.; Malicka-Błaszkiewicz, M.; Nowak, D. Effect of overexpression of β- and γ-actin isoforms on actin cytoskeleton organization and migration of human colon cancer cells. Histochem. Cell Biol. 2014, 142, 307–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simiczyjew, A.; Mazur, A.J.; Dratkiewicz, E.; Nowak, D. Involvement of β- and γ-actin isoforms in actin cytoskeleton organization and migration abilities of bleb-forming human colon cancer cells. PLoS ONE 2017, 12, e0173709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simiczyjew, A.; Mazur, A.J.; Ampe, C.; Malicka-Błaszkiewicz, M.; van Troys, M.; Nowak, D. Active invadopodia of mesenchymally migrating cancer cells contain both β and γ cytoplasmic actin isoforms. Exp. Cell Res. 2015, 339, 206–219. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malek, N.; Mrówczyńska, E.; Michrowska, A.; Mazurkiewicz, E.; Pavlyk, I.; Mazur, A.J. Knockout of ACTB and ACTG1 with CRISPR/Cas9(D10A) Technique Shows that Non-Muscle β and γ Actin Are Not Equal in Relation to Human Melanoma Cells’ Motility and Focal Adhesion Formation. Int. J. Mol. Sci. 2020, 21, 2746. https://doi.org/10.3390/ijms21082746
Malek N, Mrówczyńska E, Michrowska A, Mazurkiewicz E, Pavlyk I, Mazur AJ. Knockout of ACTB and ACTG1 with CRISPR/Cas9(D10A) Technique Shows that Non-Muscle β and γ Actin Are Not Equal in Relation to Human Melanoma Cells’ Motility and Focal Adhesion Formation. International Journal of Molecular Sciences. 2020; 21(8):2746. https://doi.org/10.3390/ijms21082746
Chicago/Turabian StyleMalek, Natalia, Ewa Mrówczyńska, Aleksandra Michrowska, Ewa Mazurkiewicz, Iuliia Pavlyk, and Antonina Joanna Mazur. 2020. "Knockout of ACTB and ACTG1 with CRISPR/Cas9(D10A) Technique Shows that Non-Muscle β and γ Actin Are Not Equal in Relation to Human Melanoma Cells’ Motility and Focal Adhesion Formation" International Journal of Molecular Sciences 21, no. 8: 2746. https://doi.org/10.3390/ijms21082746