Tumor Milieu Controlled by RB Tumor Suppressor

1
Department of Cell Biology, Cancer Institute, Japanese Foundation for Cancer Research, Tokyo 135-8550, Japan
2
Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA
3
Division of Oncology and Molecular Biology, Cancer Research Institute, Kanazawa University, Ishikawa 920-1192, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(7), 2450; https://doi.org/10.3390/ijms21072450
Submission received: 18 March 2020
/
Accepted: 31 March 2020
/
Published: 1 April 2020
(This article belongs to the Special Issue Tumor Microenvironment 3.0)
Abstract
:The RB gene is one of the most frequently mutated genes in human cancers. Canonically, RB exerts its tumor suppressive activity through the regulation of the G1/S transition during cell cycle progression by modulating the activity of E2F transcription factors. However, aberration of the RB gene is most commonly detected in tumors when they gain more aggressive phenotypes, including metastatic activity or drug resistance, rather than accelerated proliferation. This implicates RB controls’ malignant progression to a considerable extent in a cell cycle-independent manner. In this review, we highlight the multifaceted functions of the RB protein in controlling tumor lineage plasticity, metabolism, and the tumor microenvironment (TME), with a focus on the mechanism whereby RB controls the TME. In brief, RB inactivation in several types of cancer cells enhances production of pro-inflammatory cytokines, including CCL2, through upregulation of mitochondrial reactive oxygen species (ROS) production. These factors not only accelerate the growth of cancer cells in a cell-autonomous manner, but also stimulate non-malignant cells in the TME to generate a pro-tumorigenic niche in a non-cell-autonomous manner. Here, we discuss the biological and pathological significance of the non-cell-autonomous functions of RB and attempt to predict their potential clinical relevance to cancer immunotherapy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The retinoblastoma (RB) tumor suppressor protein plays a pivotal role in the control of cell cycle, terminal differentiation, and various other biological events. RB is genetically or functionally inactivated in many human cancers, including retinoblastoma, small cell lung cancer (SCLC), prostate cancer, and breast cancer [1,2,3,4]. The canonical pathway whereby RB exerts its tumor suppressive activity entails the formation of a transcriptional repression complex with E2F transcription factors and various chromatin modifiers, such as histone deacetylases (HDACs). This complex orchestrates the G1/S transition during cell cycle progression primarily by controlling E2F target genes [1,2,3,4]. Most mitogenic signals commonly merge on the transcriptional upregulation of D-type cyclins and then stimulate cyclin-dependent kinases, including CDK4/6. D-type cyclin-CDK4/6 complexes have been proposed to promote mono-phosphorylation on RB, which allow it to exert early G1 functions by starting the release of E2Fs [1,5,6,7]. E2F target genes, including cyclin E and A, in cooperation with CDK2 or CDK1, are responsible for full phosphorylation of RB at 13 remaining sites. This allows cells to enter the S and M phases [1,5,6,7]. Since uncontrolled cell proliferation is a hallmark of cancer cells, it has been postulated that genes inhibiting RB function, including CCND1 and CDK4, act as oncogenes. Conversely, genes activating RB function, including cyclin-dependent kinase inhibitors (e.g., CDKN1A, CDKN1B, and CDKN2A), are well-known to act as tumor suppressor genes. The fact that genetic and/or epigenetic aberrations of the components in the RB pathway tend to be mutually exclusive in the patients [8,9] might implicate a linearity of the RB pathway (Figure 1).
Over several decades, many researchers have attempted to pharmacologically target the aberrant status of the RB pathway in cancer cells. Most compounds developed early on did not achieve clinical benefits due to the lack of selectivity and potency. However, current highly-specific and potent CDK4/6 inhibitors, such as palbociclib (PD0332991), ribociclib (LEE011), and abemaciclib (LY2835219), have shown promising efficacy in patients, especially with RB-intact estrogen receptor (ER)-positive and HER2-negative breast cancer [10,11]. This led US Food and Drug Administration (FDA) to endow accelerated approval to these drugs as breakthrough therapies in 2015. Guidelines recommend using these in combination with an aromatase inhibitor or estrogen antagonist. The therapeutic efficacy of these inhibitors depends mostly on the long-term maintenance of the unphosphorylated status of the RB protein. Therefore, loss-of-function RB mutations would result in a gain of resistance to the treatment with CDK4/6 inhibitors. Even in the presence of intact RB, many events (e.g., FAT1 loss, Cyclin E1 or CDK6 overexpression, PI3KCA mutation) were reported to cause resistance to these compounds in breast cancers [12]. The emergence of novel CDK4/6 inhibitors could be praised as one of the triumphs achieved by RB research. However, understanding the methods for maximizing the utility of these compounds and resolving the resistance to them remains necessary.
In addition to the canonical RB pathway driven by its interaction with E2Fs and HDACs, the RB protein also has functions independent of E2Fs, acting as transcriptional activators. For instance, chromatin immunoprecipitation and sequencing (ChIP-seq) revealed that the RB protein can also bind to intronic and intergenic regions as well as in promoters with the E2Fs-binding sites [13,14,15]. One of the most well-established, non-canonical functions of RB is to maintain genome stability during DNA replication and mitosis [16]. For example, the RB-E2F complex recruits condensin II to secure chromosomal condensation and subsequent DNA segregation [17]. RB inactivation, therefore, induces aneuploidy. In fact, according to a comprehensive genomic analysis of the human tumor genome, genomic instability tends to be higher in tumors with mutations in the RB pathway [17,18]. Recent studies have shown that inhibition of kinases related to cell cycle check points, including checkpoint kinase 1 (CHK1), polo-like kinase 1 (PLK1), or aurora kinase A or B, exhibits synthetic lethality in combination with RB deficiency in triple-negative breast cancer (TNBC) or SCLC [19,20]. This indicates that genomic instability in RB-deficient cancer cells may be a pharmacologically vulnerable target. In addition to its role in genome stability, it is becoming increasingly clear that RB possesses multifaceted functions in controlling cell death, differentiation, metabolism, stemness, and innate immune signaling [3,16,21,22,23,24,25,26]. To date, more than 300 proteins have been reported to bind with the RB protein. The variability in these binding partners could explain the multifunctional aspect of the RB protein. In this article, among such a variety of RB functions, we focused on those known to regulate lineage plasticity, cancer metabolism, and inflammatory signaling. We extended the discussion toward understanding how these functions allow RB to orchestrate the tumor microenvironment (TME) through the regulation of inflammatory signaling.
2. Beyond Cell Cycle Regulation
2.1. Increased Lineage Plasticity Induced by RB Inactivation
Although RB is primarily implicated in the regulation of the cell cycle, RB mutation is frequently observed in late-stage cancer or at metastatic sites in which uncontrolled cell proliferation is likely to be established prior to RB inactivation [16]. It has been reported that RB promotes differentiation that is independent of the cell cycle regulation and RB-inactivated cells, therefore, exhibit defective terminal differentiation [1,2,3]. Interestingly, aberration of the RB gene often correlates with appearance of phenotypes associated with dedifferentiation or transdifferentiation in lung cancer, prostate cancer, and breast cancer [27,28,29,30,31]. The lineage plasticity induced by RB inactivation would promote the resistance to therapies by epidermal growth factor receptor (EGFR) inhibitors, estrogen and androgen receptor antagonists, and androgen deprivation (castration) because these treatments generally target cell lineage-specific characteristics of tumors [28,29,32,33,34]. Several groups have reported that simultaneous inactivation of multiple RB family members (e.g., RB, RB2/p130, and RB3/p107) induces not only cell cycle re-entry but also increases lineage plasticity in post-mitotic cells. For example, mouse embryonic fibroblasts (MEFs), in which all RB family proteins are inactivated, show a resistance to G1 cell cycle arrest and acquire characteristics similar to those of stem cells, as depicted by elevated sphere-forming activity and expression of pluripotent genes [35]. RB depletion in an ARF-null genetic background induces cell cycle re-entry and dedifferentiation in post-mitotic muscle cells [36]. Moreover, the RB-E2F1 complex suppresses the expression of pluripotent factors, such as SOX2 and POU5F1, by directly binding to their regulatory regions [14]. Consistent with these reports, RB inactivation increases the generation efficacy of inducible pluripotent stem (iPS) cells from human fibroblasts [14]. In prostate cancer cells, RB inactivation increases cellular plasticity, which increases resistance to anti-androgen therapy and promotes metastasis via induction of SOX2, especially in a TP53-deficient background [29,30]. Another paper revealed that RB depletion, together with p53 inactivation, induces transdifferentiation from adenocarcinoma to neuroendocrine-type cancers in prostate and lung cancer cells [37]. Mice heterozygous for Rb generate thyroid medullary cancer (MTC) from calcitonin-producing neuroendocrine cells as a consequence of biallelic loss of Rb. We reported that thyroid medullary cancer derived from calcitonin-producing cells as a consequence of biallelic loss of Rb; these cells exhibit dedifferentiated characteristics, depicted by a lower expression of calcitonin in a Trp53-null but not in genetic backgrounds lacking other p53 pathway genes, such as Arf and Cdkn1a [38]. Many papers have described that loss of p53 function synergizes with RB inactivation towards upregulation of cancer lineage plasticity. In prostate cancer cells, it has been demonstrated that RB and p53 cooperate to suppress the transcription of epigenetic reprogramming factors, such as SOX2 and EZH2 [29,30]. The p53 pathway is activated following RB inactivation depending on E2F-1 and their target ARF, which may provide another mechanism underlying the synergism between RB and p53 inactivation [39]. p53 appears to strongly counterbalance the effects of RB loss on the cell cycle, survival, and presumably many unidentified cellular behaviors; thus, p53 inactivation seems to promote a malignant phenotype induced by RB inactivation. Indeed, in some fraction of cancers, unique phenotypes are observed only with double inactivation of both RB and p53 [27,40,41,42,43].
2.2. Metabolic Reprogramming Induced by RB Inactivation
Since cell cycle progression requires the production of large amounts of energy and biomass for cell division, it is not surprising that RB regulates metabolic pathway involving glycolysis, the TCA cycle, and oxidative phosphorylation (OXPHOS) as well as cell cycle progression. In general, rapidly proliferating cells, including cancer cells, require a shift to anabolic metabolism in order to supply nucleotides, amino acids, and fatty acids for the synthesis of DNA/RNA, protein, and lipid membranes to produce daughter cells [44,45]. Of note, many oncogene and tumor suppressor proteins, including RAS, PTEN, and p53, directly regulate multiple metabolic pathways [46,47,48] and glutaminolysis is especially a well-studied pathway regulated by RB-E2Fs. For example, RB depletion increased glutamine consumption through E2F-dependent upregulation of a glutamine transporter encoded by the ASCT2/SLC15A gene [49]. In addition, Rb depletion in the fly promotes glutamine metabolism [50]. Moreover, the expression of pyruvate dehydrogenase kinases (PDKs), which inhibit the conversion of pyruvate to acetyl-CoA, is negatively regulated by E2F-1 in cooperation with histone demethylase KDM4A [51]. Hence, RB depletion causes cells to be more reliant on glutaminolysis rather than glycolysis for supplementation of carbon sources to the TCA cycle and OXPHOS [51]. The biological significance of upregulated glutaminolysis in RB-depleted cells is under debate; cells may have an increased necessity to synthesize glutathione, a representative intracellular antioxidant, from glutamine to quench reactive oxygen species (ROS) that are excessively produced as a consequence of accelerated cell proliferation induced by the RB depletion.
3. Enhanced Cytokines Secretion by Oxidative Stress Induced by RB
3.1. Increased Cellular Stress Induced by RB Inactivation Promotes Cytokine and Chemokine Secretion
In general, accumulation of cellular stresses caused by rapid proliferation and high metabolic demand stimulates production of a variety of growth factors, cytokines, and chemokines, which act on cell homeostasis. The variance among these factors depends on the cellular context. The accumulation of multiple kinds of cellular stress (e.g., DNA damage, replicative stress, oxidative stress, ER stress), likely to be induced by aberration of the RB pathway, might affect the production of growth factors, cytokines, and chemokines from cancer cells. We demonstrated that RB inactivation forces the mitochondria to produce higher levels of ROS, leading to the accumulation of oxidative stress and subsequent increased secretion of cytokines [52]. Cancer cells commonly depend on glycolysis rather than OXPHOS for energy production, which is known as the Warburg effect [53]. Warburg speculated that cancer cells might depend less on mitochondrial activity for survival. However, mitochondrial metabolism is indispensable for the growth of cancer cells [54]. OXPHOS is typically upregulated in cancer cells compared to that in normal cells, which is considered to enable cells to generate enough ATP to drive a variety of ATP-demanding anabolic pathways. Treatment with inhibitors targeting OXPHOS therefore suppresses the growth of cancer cells [55]. Higher ATP production contributes not only to rapid cell proliferation but also to enhanced cell motility and dissemination [25]. Of note, RB/E2Fs directly regulate mitochondrial protein translation and OXPHOS. E2F-1 directly binds to the promoter and induces the expression of mitochondrial protein-coding genes, such as MRPL37 and TOMM4 [56]. RB depletion in breast cancer cells increased the expression of mitochondrial proteins and stimulated cytokine secretion depending on the elevated mitochondrial ROS production [52]. RB depletion in breast cancer cells increased production of multiple cytokines and chemokines, including IL-6 and CCL2. The involvement of mitochondrial ROS in this phenomenon was demonstrated by a rescue experiment using antioxidants, including N-acetyl-L-cysteine (NAC) and mitoubiquinone (MitoQ) [52].
It has been reported that RB is required for the induction of autophagy that occurs under a hypoxic condition [57,58]. RB-inactivated cells therefore show the accumulation of damaged mitochondrial due to the failure in proper scavenging of dysfunctional mitochondria by mitophagy, which may lead to increased ROS production [59]. In general, accumulation of damaged or aged mitochondria is supposed to cause higher mitochondrial ROS production due to an insufficiency to quench the superoxide produced by OXPHOS [60]. In fact, comprehensive proteomic analysis of RB-inactivated cells identified functional abnormalities in the mitochondria [61]. The expression of pro-inflammatory cytokines is also directly regulated by the RB-E2F complexes as well. For example, activation of E2Fs following RB inactivation directly induces PTGS2, prostaglandin-endoperoxide synthase 2, in basal-like breast cancer cells [62]. Furthermore, RB depletion induces IL-6 secretion in Cdkn1a-deficient epidermis [63]. E2F binding sites were found to reside in the regulatory region of genes of a series of cytokines and their receptors; the RB-E2F complex suppresses the transactivation of genes for cytokine and chemokine, including CXCL1, CXCL2 and IL-8 [15]. To date, several reports have revealed that RB inactivation is associated with enhanced cytokine expression in several types of cancer cells. This may explain why RB inactivation enhances hormone-independent growth and metastasis of tumor cells.
3.2. RB Is Involved in the Mechanism to Keep Redox Balance at a Nontoxic Level
It is well-established that pro-inflammatory cytokines, such as IL-6 and IL-8, play crucial roles in the generation and maintenance of the stem cell-like fraction in tumor cells through stimulating NF-κB and STAT3 signaling, which promotes therapy resistance, tumor recurrence, and metastasis [59,64]. For example, administration of IL-6 or IL-8 is sufficient to induce mammosphere-forming activity in luminal-type breast cancer cells [65,66]. Similarly, several studies demonstrated that activation of the IL-6-STAT3 pathway induces expansion of the stem cell-like population and acquisition of drug resistance, including hormone-independent growth in breast cancer cells [67,68,69,70,71]. The IL-6-STAT3 pathway is reported to contribute to acquisition of drug resistance in lung and liver cancer cells as well [72,73,74].
A number of cytokines that are induced by RB inactivation seem to share an antioxidative function, which is required to overcome the excessive ROS accumulation. The treatment of RB-deficient breast cancer cells with an antibody that is capable of neutralizing IL-6 activity accelerated the accumulation of mitochondrial superoxide and subsequent cell death [52]. In contrast, the treatment of breast cancer cells with recombinant IL-6 diminished mitochondrial superoxide production, which was associated with increased mammosphere-forming activity and hormone-independent growth [52]. These findings indicate that RB controls mitochondrial superoxide production through bivalent pathways: one involves mitochondrial ROS and the other involves IL-6, which counterbalances the former to keep the redox balance at a nontoxic level. Consistent with antioxidative function of IL-6, the promoter region of the IL-6 gene contains an antioxidant responsive element (ARE), which is a binding sequence for NRF2, a master regulator of the transcriptional response to oxidative stress [75]. On the other hand, NRF2 can interfere with the induction of pro-inflammatory cytokines from macrophages [76]. The role of NRF2 in RB-mediated control of cytokine secretion and the involvement of oxidative stress in it requires further debate.
Investigation of the mechanism whereby RB controls IL-6 production indicated that RB inactivation stimulates fatty acid oxidation (FAO) and increases ROS production from mitochondria, which then activates the Jun kinase (JNK) pathway. Mechanistically, RB inactivation increased AMP-activated protein kinase (AMPK) phosphorylation and subsequent acetyl-CoA carboxylase (ACC) phosphorylation, resulting in the downregulation of malonyl-CoA synthesis. Decrease in malonyl-CoA allows CPT1 to transport long-chain fatty acids into the mitochondria for FAO. This pathway is demonstrated by the antagonistic effect of JNK inhibitors, FAO inhibitors, and antioxidants on IL-6 upregulation induced by RB inactivation [52]. In addition, downregulation of microRNA-140 (mir-140) following Rb inactivation in mouse sarcoma cells resulted in the upregulated expression of multiple pro-inflammatory cytokines, including Il-6, Vegfα, and other growth factors that have a mir-140 binding sequence in their 3’ UTR. Although the molecular mechanism that underlies the regulation of mir-140 by RB is still unclear, we speculate that RB functions to destabilize the mRNA of these factors through micro RNAs [77]. Collectively, we proposed a novel role for RB in controlling the production of pro-inflammatory cytokines: this pathway controls metabolic reprogramming leading to FAO alteration, mitochondrial ROS, JNK pathway, and antioxidative pathways to maintain cellular redox balance at a nontoxic level in a cell-autonomous manner.
4. RB Impacts the Tumor Microenvironment via Chemokine
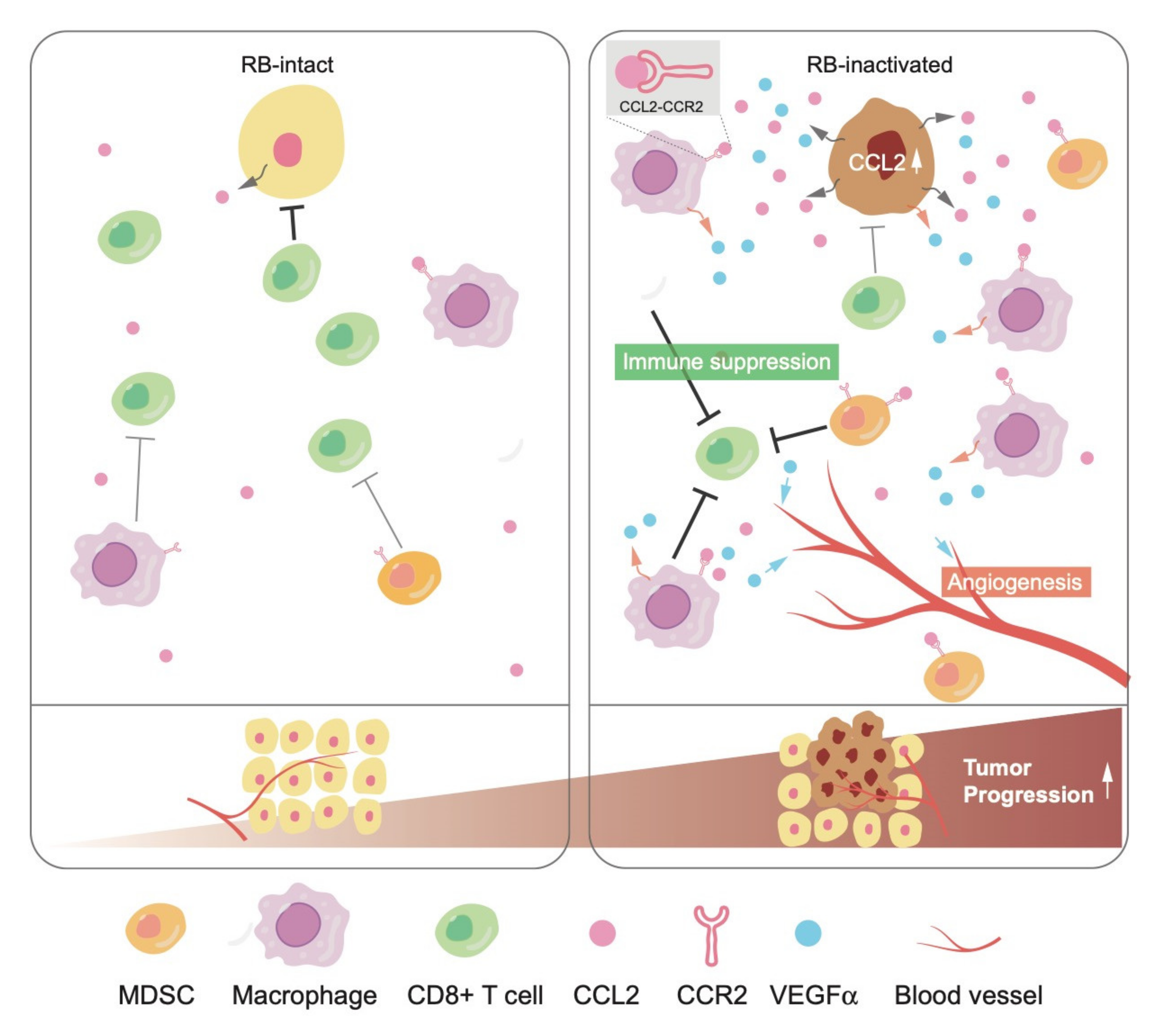
In tumor tissues, elevated cytokine secretion following RB inactivation may stimulate not only cancer cells themselves but also the surrounding non-malignant cells, called TME, which include immune cells, fibroblasts, and vascular networks. CCL2 is a well-characterized chemokine that stimulates the infiltration of its receptor CCR2-positive cells, including monocytes and/or macrophages, into the TME. The recruitment of these immune cells further stimulates angiogenesis to nourish cancer cells mostly in a vascular endothelial growth factor (VEGF)-dependent manner [78]. Recent studies indicated that CCL2 plays a critical role in triggering the infiltration of immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs) and regulatory T-cells (T-regs), causing a shift to pro-tumorigenic TME [79,80]. Accordingly, many groups are currently attempting to control malignancies by the pharmacological blockade of CCL2-CCR2.
We once established a ‘soft tissue sarcoma system’, derived from a well-differentiated leiomyosarcoma developed in a Trp53-null mouse, in which additional Rb inactivation induces a phenotypic change to undifferentiated type. We identified significant upregulation of CCL2 and a pro-angiogenic cytokine Vegfα following Rb depletion in this system [81]. Interestingly, mouse soft tissue sarcoma cells did not express CCR2. Consistent with higher secretion of CCL2 and Vegfα, upon orthotopic tumor engraftment, the TME of Rb-depleted cells showed higher angiogenesis and infiltration of CCR2-positive tumor-associated macrophages (TAMs) and MDSCs. These phenotypes were significantly suppressed when Rb-depleted cells were xenografted to Ccr2-null mice. Interestingly, infiltration of T-regs into the TME was upregulated as well following RB depletion in a CCL2-CCR2-independent manner. This indicated a possibility that RB controls chemokines other than CCL2. Taken together, these observations indicate that RB exerts non-cell autonomous functions through the regulation of cytokine secretion (Figure 2).
CCL2 is upregulated following RB inactivation in human breast cancer cells, not only in mouse sarcoma cells. We observed elevated CCL2 expression in the mammary glands of these mice regardless of the Ccr2 genotype. We detected hyperplastic growth and prominent infiltration of macrophages in the mammary gland of Ccr2+/+; MMTV-Cre; Rbflox/flox mice. However, these phenotypes were less significant in Ccr2-/-; MMTV-Cre; Rbflox/flox mice, suggesting the importance of CCL2-CCR2 in carcinogenesis caused by RB deficiency. In addition to CCL2, we identified several other cytokines and chemokines as possible targets of RB, which may contribute to the foundation of pro-tumorigenic TME as well. As is in case of IL-6, the mechanism whereby RB controls CCL2 seems to entail mitochondrial ROS production and the JNK pathway (Figure 3). Further studies are required to uncover the molecular mechanisms underlying the regulation of cytokines and chemokines by RB.
5. Therapeutic Strategy Targeting Innate Immune Signaling in RB-Inactive Cancer
Cancer immunotherapy with immune checkpoint blockade (ICB) using anti-PD-1/PD-L1 or anti-CTLA4 antibodies enhances antitumor activity of the immune components of the TME, including cytotoxic CD8 positive T cells and natural killer cells [82]. ICB exhibits significant therapeutic efficacy for many types of cancer patients, including non-small cell lung cancer (NSCLC), melanoma, and renal cell carcinoma. However, the benefits of ICB are limited for the majority of patients and predictive markers of sensitivity to ICB are still under investigation [83,84,85].
Given that RB inactivation in cancer cells promotes the infiltration of immunosuppressive cells, such as MDSCs and TAMs, into the TME, at least in part through the CCL2-CCR2. RB inactivation due to genetic mutation or transcriptional suppression via DNA hypermethylation in the RB gene promoter could be one of potential markers of a so-called ‘cold tumor’, which exhibits poor immunogenicity leading to the resistance to ICB. If so, treatment inhibiting CCL2-CCR2 using agents, such as neutralizing antibody to CCL2, might cooperate with ICB. Since cancers prevalently carry inactivation of the RB pathway, a significant fraction of cancers might respond to such therapy. Interestingly, it has been recently reported that RB mutation is correlated with the resistance to anti-PD-1 therapy in advanced NSCLC patients using nivolumab and pembrolizumab [86]. Meanwhile, several papers reported that RB-inactive cells show less responsiveness to the stimuli activating the anti-viral response driven by viral double-stranded RNA or interferon (IFN) gamma [87,88]. Inversely, the treatment with CDK4/6 inhibitor, which reactivates RB through keeping it in an unphosphorylated status, promotes cytokines secretion related to the anti-viral response and reduces the infiltration of immunosuppressive immune cells, such as MDSCs and T-regs [89,90,91]. Since the anti-viral response has recently emerged as a key pathway of antitumor immunity [92,93,94], downregulation of the anti-viral response by RB inactivation might contribute to the resistance to ICB via immune evasion as well, although the molecular mechanisms of how RB regulates anti-viral pathways are not yet completely elucidated.
In general, cellular senescence is strongly dependent on the RB pathway. Senescent cells show a marked increase in the secretion of multiple cytokines, so-called the senescence-associated secretory phenotype (SASP) [95]. Interestingly, cytokines induced during SASP largely overlap with those that are upregulated following the treatment of CDK4/6 inhibitors. However, it is not yet certain whether they constitute a part of the senescence program [96]. A recent study has shown that the RB protein, phosphorylated by CDK4/6-cyclin D complex, specifically suppresses NF-κB p65 activity through direct binding and thereby inhibits the expression of NF-κB targets, including PD-L1 [97], which suggests another mechanism that promotes immune evasion in RB-deficient cancer cells.
It has recently become clear that the accumulation of DNA damage in tumors resulting from disruption of the DNA replication/repair pathways, DNA-damaging chemotherapy, or radiation therapy promotes an antitumor immune response [98]. In general, aberrant cytoplasmic DNA accumulation or micronuclei formation following DNA damaging events are detected by cyclic GMP-AMP synthase (cGAS), which produces the second messenger cyclic GMP-AMP (cGAMP) that directly activates its downstream stimulator of interferon genes (STING) [99]. Activation of STING subsequently induces upregulation of its downstream cytokines, including type I interferon and CXCL10, which promotes T-cell-mediated therapeutic antitumor immunity via enhancing neoantigen presentation and T-cell recruitment into the TME [100]. Not only DNA damage accumulation but also excessive ER stress activate the STING pathway [101]. Considering these mechanisms, it is not surprising that the increased genomic instability and metabolic stress that accumulates in RB-inactive cancer cells potentiate the efficacy of immunotherapy in certain contexts. Because of such complexity in the interaction between the RB pathway and innate immune signaling, continued research is needed to elucidate how RB impacts tumor immunogenicity through cell-autonomous and non-cell-autonomous functions.
6. Role of Other Tumor Suppressor Proteins in the TME
Based on the extensive analysis of datasets obtained from clinical studies, researchers have identified several markers that are highly predictive of favorable T-cell response. These include higher PD-L1 expression, the degree of tumor mutation burden (TMB), and the number of tumor-infiltrating lymphocytes (TILs) residing in the TME [83,84,85]. In addition, somatic mutations in cancer cells, including loss-of-function mutations in JAK1/2, APLNR, PTPN2, and PBRM1, significantly correlate with the efficacy of cancer immunotherapies [102,103,104,105,106,107,108].
As mentioned above, it is likely that RB mutation in cancer cells might predict the sensitivity to ICB for certain types of cancer. We here refer to an example that tumor suppressors other than RB can also impact the TME through the regulation of cytokine and chemokine secretion. Liver kinase B1 (LKB1) is a well-known tumor suppressor protein, which is mutated or deleted in Peutz–Jeghers syndrome (PJS) and in a variety of cancer cells, such as NSCLC, breast cancer, and pancreatic cancer [109]. LKB1 is a serine/threonine kinase, which phosphorylates 13 members of the AMP-activated protein kinase (AMPK) family, thereby affecting multiple cellular functions that control cellular metabolism, cell cycle progression, apoptosis, and cell polarity, especially under nutrient-deprived conditions [109]. In general, activation of the LKB1-AMPK pathway contributes to the maintenance of energy homeostasis through multiple mechanisms, such as inhibition of anabolic metabolism, to save ATP consumption and to induce autophagy-mediated degradation of organelles for recycling nutrients [110]. Furthermore, the LKB1-AMPK pathway maintains energy homeostasis by promoting the clearance of damaged mitochondria via selective autophagy, so called mitophagy [111]. Accordingly, LKB1-inactive cancer cells accumulate significant levels of metabolic stress and oxidative stress due to the accumulation of damaged mitochondria, resulting in vulnerability to excessive metabolic stress induced by treatment with mitochondrial inhibitors, such as phenformin [112]. Indeed, addition of the LKB1 mutation to KRAS mutant NSCLC cells is significantly correlated with the aberrant accumulation of cytoplasmic mitochondrial DNA and higher mitochondrial ROS production [113]. Similar to RB-depleted cells that exhibit higher pro-inflammatory cytokines production due to increased mitochondrial ROS, KRAS;LKB1-mutated NSCLC cells highly produce cytokines, such as IL-6 and CCL5 [114,115]. Higher secretion of these pro-inflammatory cytokines not only promotes cell growth and cell survival in a cell-autonomous manner but also contributes to the formation of immunosuppressive TME in a non-cell-autonomous manner [114,115]. On the other hand, aberrant accumulation of cytoplasmic DNA derived from mitochondria in KRAS;LKB1-mutated NSCLC cells stimulates the cGAS-STING pathway, as discussed above. KRAS;LKB1-mutated NSCLC cells seem to need to attenuate the STING pathway during tumor evolution to protect the cells from cell death caused by intrinsic signals or enhance the recruitment of cytotoxic T cells to remove pathogenic cells [116]. Indeed, expression of STING and its downstream cytokines are significantly lower in KRAS;LKB1-mutated NSCLC patients than in LKB1 intact ones [113]. Consistent with these findings, KRAS;LKB1-mutated NSCLC patients exhibited lower infiltration of cytotoxic T cells into the TME and resistance to anti-PD-1 therapy [117] possibly because the secretion of type I interferon and CXCL10 downstream of STING plays a critical role in the anti-tumorigenic response following ICB.
7. Conclusions
Cell cycle dysregulation is one of the most important features caused by RB inactivation. In addition, RB exerts multifaceted functions and regulates many other biological events, including genome integrity, lineage plasticity, cellular metabolism, and innate immune signaling. The malignant phenotypes induced by RB inactivation can be context-dependent and the molecular mechanisms underlying this aspect are not fully understood. It is not surprising that, even after more than 30 years of investigation, RB still defies our full understanding of its function. As mentioned above, RB may have more than 300 binding partners. RB may select its binding partners depending on cell type and cellular context. In this review, we focused on the interaction between the RB pathway and innate immune signaling. To date, many studies have revealed that the status of RB affects cytokines/chemokines secretion directly or indirectly in an E2F-dependent or -independent manner. This suggests a novel RB function in controlling intracellular signaling, which may be exerted through non-cell-autonomous regulation of innate immune signaling. Recently, we have reported a novel function of RB in controlling the CCL2-CCR2 function, which will enable us to predict the nature of the TME depending on the RB status in tumor cells. This finding may provoke a novel debate on the utility of blockers of the CCL2-CCR2 pathway with an aim toward enhancing the sensitivity to ICB.
Funding
This work was supported by Grant-in-Aid for Scientific Research 25830077 (S.K.), and Hokuriku Bank Research Grant for Young Scientist (S.K.).
Acknowledgments
We thank S. Sundararaman for critical reading of the manuscript. We thank the program of the Strategic Young Researcher Overseas Visit Program for Accelerating Brain Circulation, and JSPS Postdoctoral Fellowship for Research Abroad for supporting the project.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| CDK | Cyclin-dependent kinase |
| HER | Human EGFR-related |
| TCA | Tricarboxylic acid |
| KDM | Lysine (K) Demethylase |
| ATP | Adenosine triphosphate |
| AMP | Adenosine monophosphate |
| IL- | Interleukin- |
| CCL | C-C motif ligand |
| CCR | C-C chemokine receptor |
| CXCL | C-X-C motif ligand |
| CoA | Coenzyme A |
| UTR | Untranslated region |
| MMTV | Mouse mammary tumor virus |
| JNK | c-jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| IFN | Interferon |
References
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Takahashi, C. Intersection of retinoblastoma tumor suppressor function, stem cells, metabolism, and inflammation. Cancer Sci. 2017, 108, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Knudsen, K.E. Tailoring to RB: Tumour suppressor status and therapeutic response. Nat. Rev. Cancer 2008, 8, 714–724. [Google Scholar] [CrossRef]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Rubin, S.M. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 2013, 38, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, L.; Lukas, J.; Bartkova, J.; Kjerulff, A.A.; Strauss, M.; Bartek, J. Aberrations of p16Ink4 and retinoblastoma tumour-suppressor genes occur in distinct sub-sets of human cancer cell lines. Int. J. Cancer 1995, 61, 115–120. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16(ink4a) expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle. 2011, 10, 2497–2503. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Johnson, J.; Thijssen, B.; McDermott, U.; Garnett, M.; Wessels, L.F.; Bernards, R. Targeting the RB-E2F pathway in breast cancer. Oncogene 2016, 35, 4829–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.; O’Brien, M.; et al. Inhibition of pluripotency networks by the rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 2015, 16, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, R.; Gou, D.; Jawdekar, G.; Johnson, S.A.; Nava, M.; Su, T.; Yousef, A.F.; Zemke, N.R.; Pellegrini, M.; Kurdistani, S.K.; et al. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host. Microbe. 2014, 16, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Coschi, C.H.; Ishak, C.A.; Gallo, D.; Marshall, A.; Talluri, S.; Wang, J.; Cecchini, M.J.; Martens, A.L.; Percy, V.; Welch, I.; et al. Haploinsufficiency of an RB-E2F1-Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov. 2014, 4, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef] [Green Version]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef] [Green Version]
- Sage, J. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. 2012, 26, 1409–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolay, B.N.; Dyson, N.J. The multiple connections between pRB and cell metabolism. Curr. Opin. Cell. Biol. 2013, 25, 735–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benevolenskaya, E.V.; Frolov, M.V. Emerging Links between E2F Control and Mitochondrial Function. Cancer Res. 2015, 75, 619–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Zacksenhaus, E.; Shrestha, M.; Liu, J.C.; Vorobieva, I.; Chung, P.E.D.; Ju, Y.; Nir, U.; Jiang, Z. Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis. Trends Cancer 2017, 3, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.; Witkiewicz, A.K.; Knudsen, E.S. The RB tumor suppressor at the intersection of proliferation and immunity: Relevance to disease immune evasion and immunotherapy. Cell. Cycle 2015, 14, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Deng, T.; Jones, R.; Li, H.; Herschkowitz, J.I.; Liu, J.C.; Weigman, V.J.; Tsao, M.S.; Lane, T.F.; Perou, C.M.; et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J. Clin. Investig. 2010, 120, 3296–3309. [Google Scholar] [CrossRef] [Green Version]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Walter, D.M.; Yates, T.J.; Ruiz-Torres, M.; Kim-Kiselak, C.; Gudiel, A.A.; Deshpande, C.; Wang, W.Z.; Cicchini, M.; Stokes, K.L.; Tobias, J.W.; et al. RB constrains lineage fidelity and multiple stages of tumour progression and metastasis. Nature 2019, 569, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Inoue, Y.; Shibata, T.; Hayashi, H.; Nagano, O.; Saya, H.; Taya, Y. Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res. 2008, 68, 5104–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, E.E.; Wang, Y.; Xu, H.; Zilfou, J.T.; Knudsen, K.E.; Aronow, B.J.; Lowe, S.W.; Knudsen, E.S. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J. Clin. Investig. 2007, 117, 218–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Yeow, W.S.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morrissey, C.; Zhang, X.; Comstock, C.E.; Witkiewicz, A.K.; et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Clem, B.; Zuba-Surma, E.K.; El-Naggar, S.; Telang, S.; Jenson, A.B.; Wang, Y.; Shao, H.; Ratajczak, M.Z.; Chesney, J.; et al. Mouse fibroblasts lacking RB1 function form spheres and undergo reprogramming to a cancer stem cell phenotype. Cell Stem Cell 2009, 4, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Pajcini, K.V.; Corbel, S.Y.; Sage, J.; Pomerantz, J.H.; Blau, H.M. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell Stem Cell 2010, 7, 198–213. [Google Scholar]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Kohno, S.; Kondoh, A.; Sasaki, N.; Nishimoto, Y.; Li, F.; Abdallah Mohammed, M.S.; Muranaka, H.; Nagatani, N.; Suzuki, M.; et al. Undifferentiated State Induced by Rb-p53 Double Inactivation in Mouse Thyroid Neuroendocrine Cells and Embryonic Fibroblasts. Stem Cells 2015, 33, 1657–1669. [Google Scholar]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in life and death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef]
- Shamma, A.; Takegami, Y.; Miki, T.; Kitajima, S.; Noda, M.; Obara, T.; Okamoto, T.; Takahashi, C. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell 2009, 15, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, M.; Vogel, H.; Lee, E.Y.; Bradley, A.; Donehower, L.A. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res. 1995, 55, 1146–1151. [Google Scholar] [PubMed]
- Williams, B.O.; Remington, L.; Albert, D.M.; Mukai, S.; Bronson, R.T.; Jacks, T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat. Genet. 1994, 7, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell. Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, B.N.; Gameiro, P.A.; Tschop, K.; Korenjak, M.; Heilmann, A.M.; Asara, J.M.; Stephanopoulos, G.; Iliopoulos, O.; Dyson, N.J. Loss of RBF1 changes glutamine catabolism. Genes Dev. 2013, 27, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.Y.; Hung, C.L.; Chen, Y.R.; Yang, J.C.; Wang, J.; Campbell, M.; Izumiya, Y.; Chen, H.W.; Wang, W.C.; Ann, D.K.; et al. KDM4A Coactivates E2F1 to Regulate the PDK-Dependent Metabolic Switch between Mitochondrial Oxidation and Glycolysis. Cell Rep. 2016, 16, 3016–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Yoshida, A.; Kohno, S.; Li, F.; Suzuki, S.; Nagatani, N.; Nishimoto, Y.; Sasaki, N.; Muranaka, H.; Wan, Y.; et al. The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 2017, 36, 5145–5157. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; De Milito, A.; Demiroglu-Zergeroglu, A.; Gullbo, J.; D’Arcy, P.; Linder, S. Eradicating Quiescent Tumor Cells by Targeting Mitochondrial Bioenergetics. Trends Cancer 2016, 2, 657–663. [Google Scholar] [CrossRef]
- Jones, R.A.; Robinson, T.J.; Liu, J.C.; Shrestha, M.; Voisin, V.; Ju, Y.; Chung, P.E.; Pellecchia, G.; Fell, V.L.; Bae, S.; et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Investig. 2016, 126, 3739–3757. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Martin, V.; Gomez-Manzano, C.; Johnson, D.G.; Alonso, M.; White, E.; Xu, J.; McDonnell, T.J.; Shinojima, N.; Fueyo, J. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010, 70, 7882–7893. [Google Scholar] [CrossRef] [Green Version]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef] [Green Version]
- Ciavarra, G.; Zacksenhaus, E. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J. Cell Biol. 2010, 191, 291–301. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, B.N.; Danielian, P.S.; Kottakis, F.; Lapek, J.D., Jr.; Sanidas, I.; Miles, W.O.; Dehnad, M.; Tschop, K.; Gierut, J.J.; Manning, A.L.; et al. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev. 2015, 29, 1875–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, M.L.; Berman, H.K.; Miller, C.; Kozakeiwicz, K.; Chew, K.; Moore, D.; Rabban, J.; Chen, Y.Y.; Kerlikowske, K.; Tlsty, T.D. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell 2007, 12, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiz-Ladera, C.; Lara, M.F.; Garin, M.; Ruiz, S.; Santos, M.; Lorz, C.; Garcia-Escudero, R.; Martinez-Fernandez, M.; Bravo, A.; Fernandez-Capetillo, O.; et al. p21 suppresses inflammation and tumorigenesis on pRB-deficient stratified epithelia. Oncogene 2014, 33, 4599–4612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Hinohara, K.; Kobayashi, S.; Kanauchi, H.; Shimizu, S.; Nishioka, K.; Tsuji, E.; Tada, K.; Umezawa, K.; Mori, M.; Ogawa, T.; et al. ErbB receptor tyrosine kinase/NF-kappaB signaling controls mammosphere formation in human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 6584–6589. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [Green Version]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Aref, A.R.; Cohoon, T.J.; Barbie, T.U.; Imamura, Y.; Yang, S.; Moody, S.E.; Shen, R.R.; Schinzel, A.C.; Thai, T.C.; et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014, 4, 452–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Wruck, C.J.; Streetz, K.; Pavic, G.; Gotz, M.E.; Tohidnezhad, M.; Brandenburg, L.O.; Varoga, D.; Eickelberg, O.; Herdegen, T.; Trautwein, C.; et al. Nrf2 induces interleukin-6 (IL-6) expression via an antioxidant response element within the IL-6 promoter. J. Biol. Chem. 2011, 286, 4493–4499. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Yoshida, A.; Kitajima, S.; Li, F.; Cheng, C.; Takegami, Y.; Kohno, S.; Wan, Y.S.; Hayashi, N.; Muranaka, H.; Nishimoto, Y.; et al. MicroRNA-140 mediates RB tumor suppressor function to control stem cell-like activity through interleukin-6. Oncotarget 2017, 8, 13872–13885. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Kitajima, S.; Kohno, S.; Yoshida, A.; Tange, S.; Sasaki, S.; Okada, N.; Nishimoto, Y.; Muranaka, H.; Nagatani, N.; et al. Retinoblastoma Inactivation Induces a Protumoral Microenvironment via Enhanced CCL2 Secretion. Cancer Res. 2019, 79, 3903–3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.E.; Burke, K.P.; Van Allen, E.M. Genomic correlates of response to immune checkpoint blockade. Nat. Med. 2019, 25, 389–402. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Bhateja, P.; Chiu, M.; Wildey, G.; Lipka, M.B.; Fu, P.; Yang, M.C.L.; Ardeshir-Larijani, F.; Sharma, N.; Dowlati, A. Retinoblastoma mutation predicts poor outcomes in advanced non small cell lung cancer. Cancer Med. 2019, 8, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Taura, M.; Suico, M.A.; Koyama, K.; Komatsu, K.; Miyakita, R.; Matsumoto, C.; Kudo, E.; Kariya, R.; Goto, H.; Kitajima, S.; et al. Rb/E2F1 regulates the innate immune receptor Toll-like receptor 3 in epithelial cells. Mol. Cell. Biol. 2012, 32, 1581–1590. [Google Scholar]
- Zhu, X.; Pattenden, S.; Bremner, R. pRB is required for interferon-gamma-induction of the MHC class II abeta gene. Oncogene 1999, 18, 4940–4947. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell. Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell. 2019, 73, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Terai, H.; Kitajima, S.; Potter, D.S.; Matsui, Y.; Quiceno, L.G.; Chen, T.; Kim, T.J.; Rusan, M.; Thai, T.C.; Piccioni, F.; et al. ER Stress Signaling Promotes the Survival of Cancer "Persister Cells" Tolerant to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2018, 78, 1044–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Asahina, H.; Chen, T.; Guo, S.; Quiceno, L.G.; Cavanaugh, J.D.; Merlino, A.A.; Tange, S.; Terai, H.; Kim, J.W.; et al. Overcoming Resistance to Dual Innate Immune and MEK Inhibition Downstream of KRAS. Cancer Cell 2018, 34, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
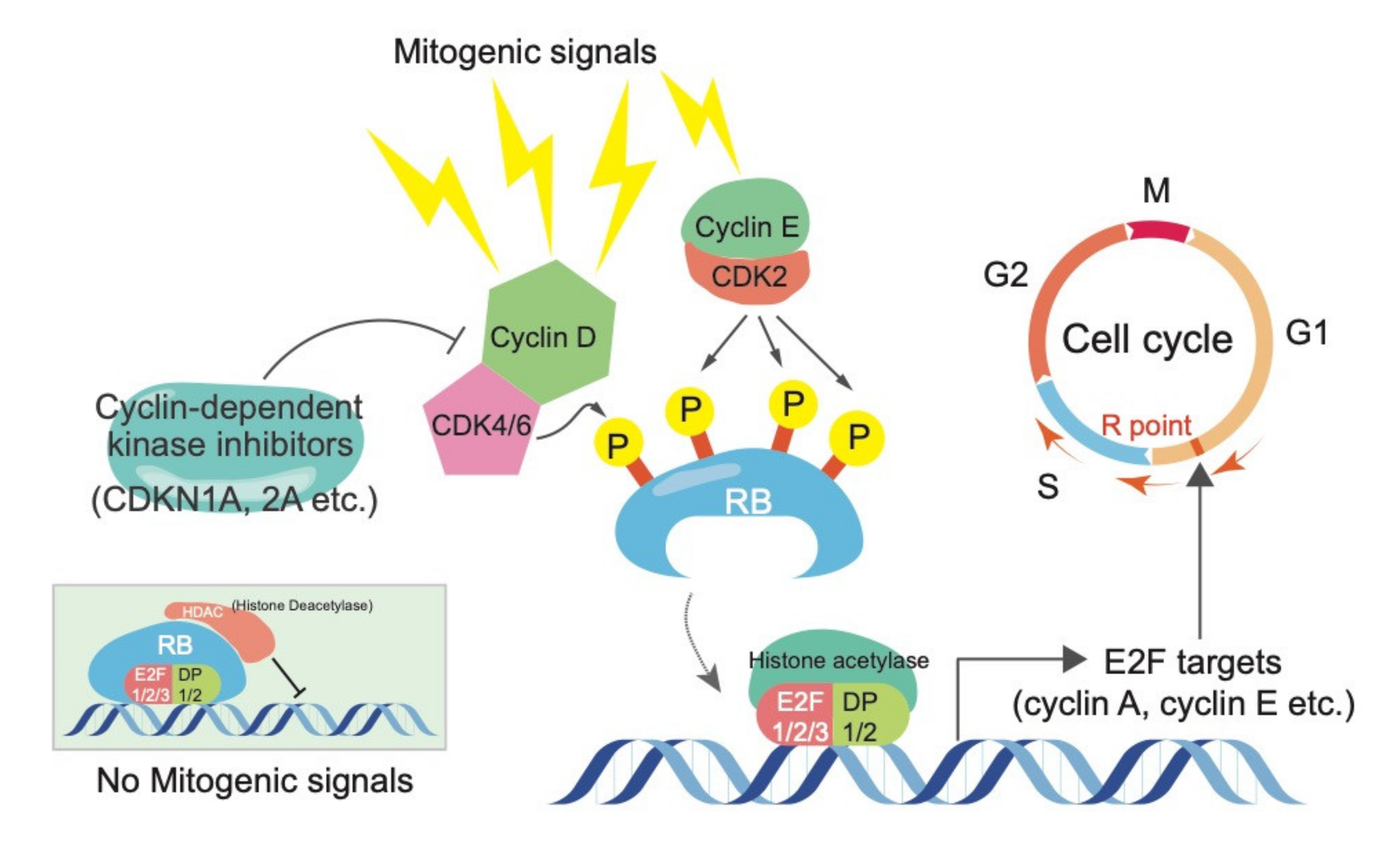
Figure 1.
The canonical RB function in controlling cell cycle progression. Cyclin-CDK complexes regulate RB activity via phosphorylation, while cyclin-dependent kinase inhibitors suppress the activity of Cyclin-CDK complexes. Phosphorylated RB releases E2Fs, which stimulate cells to enter to the S and M phases via induction of E2F targets, such as cyclin A and E.
Figure 1.
The canonical RB function in controlling cell cycle progression. Cyclin-CDK complexes regulate RB activity via phosphorylation, while cyclin-dependent kinase inhibitors suppress the activity of Cyclin-CDK complexes. Phosphorylated RB releases E2Fs, which stimulate cells to enter to the S and M phases via induction of E2F targets, such as cyclin A and E.

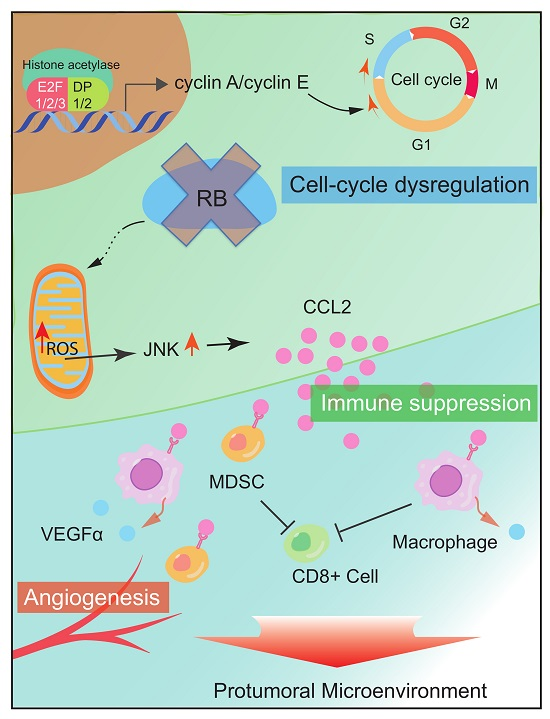
Figure 2.
RB impacts the TME via cytokine and chemokine secretion. RB inactivation upregulates CCL2 and VEGFα secretion, which recruits immune suppressive cells, such as MDSCs and tumor-associated macrophages and promotes angiogenesis.
Figure 2.
RB impacts the TME via cytokine and chemokine secretion. RB inactivation upregulates CCL2 and VEGFα secretion, which recruits immune suppressive cells, such as MDSCs and tumor-associated macrophages and promotes angiogenesis.

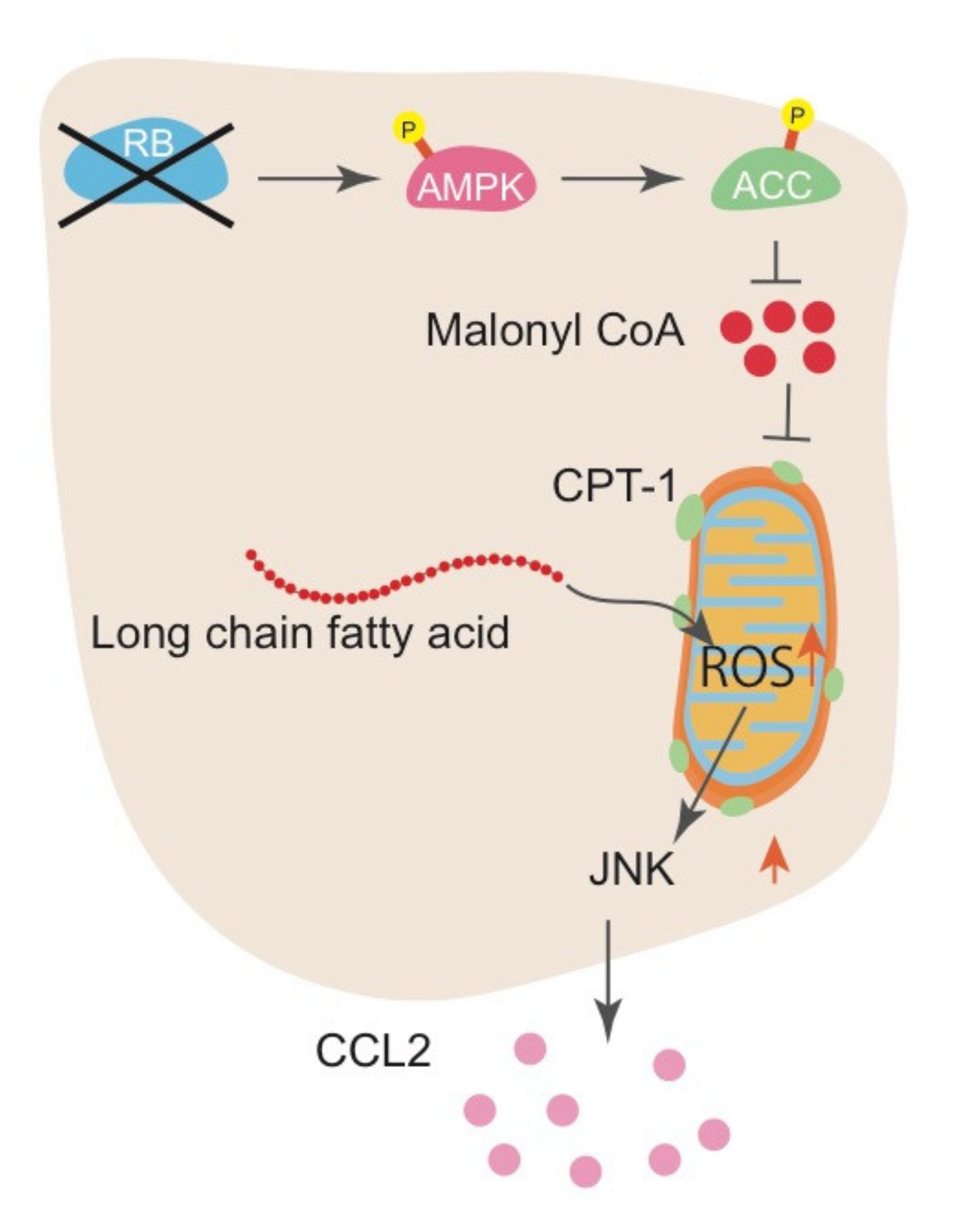
Figure 3.
The mechanism of CCL2 upregulation following RB inactivation. RB inactivation increases AMPK phosphorylation and subsequent ACC phosphorylation, resulting in downregulation of malonyl-CoA synthesis. A decrease in malonyl-CoA allows CPT1 to transport long-chain fatty acids into mitochondria for FAO, promoting mitochondrial ROS production.
Figure 3.
The mechanism of CCL2 upregulation following RB inactivation. RB inactivation increases AMPK phosphorylation and subsequent ACC phosphorylation, resulting in downregulation of malonyl-CoA synthesis. A decrease in malonyl-CoA allows CPT1 to transport long-chain fatty acids into mitochondria for FAO, promoting mitochondrial ROS production.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kitajima, S.; Li, F.; Takahashi, C. Tumor Milieu Controlled by RB Tumor Suppressor. Int. J. Mol. Sci. 2020, 21, 2450. https://doi.org/10.3390/ijms21072450
AMA Style
Kitajima S, Li F, Takahashi C. Tumor Milieu Controlled by RB Tumor Suppressor. International Journal of Molecular Sciences. 2020; 21(7):2450. https://doi.org/10.3390/ijms21072450
Chicago/Turabian StyleKitajima, Shunsuke, Fengkai Li, and Chiaki Takahashi. 2020. "Tumor Milieu Controlled by RB Tumor Suppressor" International Journal of Molecular Sciences 21, no. 7: 2450. https://doi.org/10.3390/ijms21072450
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.