Iron Metabolism at the Interface between Host and Pathogen: From Nutritional Immunity to Antibacterial Development

 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Iron and Nutritional Immunity
1.1. Iron: A Double-Edged Sword
1.2. The Mechanisms of Nutritional Immunity
1.3. Nutritional Immunity under Infection Conditions
2. Mechanisms of Iron Acquisition by Staphylococcus aureus
2.1. Siderophores
2.1.1. Endogenous Siderophores
Carboxylate-Type Siderophores: Staphyloferrin A and Staphyloferrin B
Other Endogenous Iron-Chelators
2.1.2. Exogenous Siderophores
2.2. Hemic Iron
2.2.1. Hemolysins and Leukocidins
2.2.2. Isd System
Isd in Heme Uptake
Isd Moonlighting Activities
2.2.3. Fep System
2.3. Heme and Iron Homeostasis inside S. aureus
3. Effects of Iron Restriction and Mutations of Assimilatory Pathways on Fitness and Virulence
4. Antibiotic Strategies Targeting Iron Uptake in S. aureus - Small Molecules
4.1. Targeting Pre-Iron-Uptake Systems
4.1.1. Quorum Sensing Inhibitors
4.1.2. Small Molecules as Hemolysin Inhibitors
4.1.3. Iron Chelators
4.2. Targeting the Iron-Uptake Systems
4.2.1. Exploiting the Iron-Uptake System
Trojan Horses
Gallium-Derivatives
4.2.2. Inhibiting the Iron-Uptake System
Inhibitors of Siderophore Biosynthesis
4.3. Targeting the Post-Iron-Uptake Metabolism
Heme Oxygenase Inhibitors
5. Biopharmaceutical Approaches Targeting the Iron System in S. aureus
5.1. Biologics Targeting the Staphylococcal Iron/Heme Uptake Systems
5.2. Biologics Targeting Staphylococcal Hemolysins
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hood, M.I.; Skaar, E.P. Nutritional immunity: Transition metals at the pathogen–host interface. Nat. Rev. Microbiol. 2012, 10, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Gherardini, F.C. Lack of a role for iron in the lyme disease pathogen. Science 2000, 288, 1651–1653. [Google Scholar] [CrossRef]
- Pandey, A.; Bringel, F.; Meyer, J.-M. Iron requirement and search for siderophores in lactic acid bacteria. Appl. Microbiol. Biotechnol. 1994, 40, 735–739. [Google Scholar] [CrossRef]
- Perutz, M.F. Stereochemistry of cooperative effects in haemoglobin: Haem–haem interaction and the problem of allostery. Nature 1970, 228, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.; Sabio, L.; Gálvez, N.; Capdevila, M.; Dominguez-Vera, J.M. Iron chemistry at the service of life. IUBMB Life 2017, 69, 382–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.T.; Shan Ho, J.Z.; Ho, B.; Ding, J.L. Iron-withholding strategy in innate immunity. Immunobiology 2006, 211, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Ilbert, M.; Bonnefoy, V. Insight into the evolution of the iron oxidation pathways. Biochim. Biophys. Acta - Bioenerg. 2013, 1827, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Golonka, R.; Yeoh, B.S.; Vijay-Kumar, M. The iron tug-of-war between bacterial siderophores and innate immunity. J. Innate Immun. 2019, 11, 249–262. [Google Scholar] [CrossRef]
- Fenton, H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Bullen, J.J.; Rogers, H.J.; Griffiths, E. Role of iron in bacterial infection. Curr. Top. Microbiol. Immunol. 1978, 80, 1–35. [Google Scholar] [CrossRef]
- Andrews, N.C. Forging a field: The golden age of iron biology. Blood 2008, 112, 219–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, E.D. Nutritional immunity. JAMA 1975, 231, 39. [Google Scholar] [CrossRef]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and natural iron chelators: Therapeutic potential and clinical use. Future Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef] [Green Version]
- Núñez, G.; Sakamoto, K.; Soares, M.P. Innate nutritional immunity. J. Immunol. 2018, 201, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamroz, R.C.; Gasdaska, J.R.; Bradfield, J.Y.; Law, J.H. Transferrin in a cockroach: Molecular cloning, characterization, and suppression by juvenile hormone. Proc. Natl. Acad. Sci. USA 1993, 90, 1320–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Janeway Jr, C.A. Innate immunity: Impact on the adaptive immune response. Curr. Opin. Immunol. 1997, 9, 4–9. [Google Scholar] [CrossRef]
- Wright, S.D. Multiple receptors for endotoxin. Curr. Opin. Immunol. 1991, 3, 83–90. [Google Scholar] [CrossRef]
- Theurl, I.; Fritsche, G.; Ludwiczek, S.; Garimorth, K.; Bellmann-Weiler, R.; Weiss, G. The macrophage: A cellular factory at the interphase between iron and immunity for the control of infections. BioMetals 2005, 18, 359–367. [Google Scholar] [CrossRef]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Canonne-Hergaux, F.; Gruenheid, S.; Ponka, P.; Gros, P. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood 1999, 93, 4406–4417. [Google Scholar] [CrossRef]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta - Bioenerg. 1996, 1275, 161–203. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.M.; Artymiuk, P.J.; Yewdall, S.J.; Smith, J.M.A.; Livingstone, J.C.; Treffry, A.; Luzzago, A.; Levi, S.; Arosio, P.; Cesareni, G.; et al. Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature 1991, 349, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, H.; Sadler, P.J. Transferrin as a metal ion mediator. Chem. Rev. 1999, 99, 2817–2842. [Google Scholar] [CrossRef] [PubMed]
- Wally, J.; Halbrooks, P.J.; Vonrhein, C.; Rould, M.A.; Everse, S.J.; Mason, A.B.; Buchanan, S.K. The crystal structure of iron-free human serum transferrin provides insight into inter-lobe communication and receptor binding. J. Biol. Chem. 2006, 281, 24934–24944. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Zhang, H.; Wang, M.; Hao, Q.; Sun, H. Iron and bismuth bound human serum transferrin reveals a partially-opened conformation in the N-lobe. Sci. Rep. 2012, 2, 999. [Google Scholar] [CrossRef] [Green Version]
- Rosa, L.; Cutone, A.; Lepanto, M.; Paesano, R.; Valenti, P. Lactoferrin: A natural glycoprotein involved in iron and inflammatory homeostasis. Int. J. Mol. Sci. 2017, 18, 1985. [Google Scholar] [CrossRef]
- Hwang, S.-A.; Kruzel, M.L.; Actor, J.K. Immunomodulatory effects of recombinant lactoferrin during MRSA infection. Int. Immunopharmacol. 2014, 20, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Ammons, M.C.; Copié, V. Mini-review: Lactoferrin: A bioinspired, anti-biofilm therapeutic. Biofouling 2013, 29, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Van Vlierberghe, H.; Langlois, M.; Delanghe, J. Haptoglobin polymorphisms and iron homeostasis in health and in disease. Clin. Chim. Acta 2004, 345, 35–42. [Google Scholar] [CrossRef]
- Delanghe, J.; Allcock, K.; Langlois, M.; Claeys, L.; De Buyzere, M. Fast determination of haptoglobin phenotype and calculation of hemoglobin binding capacity using high pressure gel permeation chromatography. Clin. Chim. Acta 2000, 291, 43–51. [Google Scholar] [CrossRef]
- Cahill, L.E.; El-Sohemy, A. Haptoglobin genotype modifies the association between dietary vitamin C and serum ascorbic acid deficiency. Am. J. Clin. Nutr. 2010, 92, 1494–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Hepcidin regulation: Ironing out the details. J. Clin. Investig. 2007, 117, 1755–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rishi, G.; Wallace, D.F.; Subramaniam, V.N. Hepcidin: Regulation of the master iron regulator. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flower, D.R. The lipocalin protein family: Structure and function. Biochem. J. 1996, 318, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Correnti, C.; Strong, R.K. Mammalian siderophores, siderophore-binding lipocalins, and the labile iron pool. J. Biol. Chem. 2012, 287, 13524–13531. [Google Scholar] [CrossRef] [Green Version]
- Hider, R.C.; Kong, X. Chemistry and biology of siderophores. Nat. Prod. Rep. 2010, 27, 637–657. [Google Scholar] [CrossRef]
- Wilson, M.K.; Abergel, R.J.; Raymond, K.N.; Arceneaux, J.E.L.; Byers, B.R. Siderophores of Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis. Biochem. Biophys. Res. Commun. 2006, 348, 320–325. [Google Scholar] [CrossRef]
- Ganz, T. Iron and infection. Int. J. Hematol. 2018, 107, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, E.D. Iron availability and infection. Biochim. Biophys. Acta - Gen. Subj. 2009, 1790, 600–605. [Google Scholar] [CrossRef] [Green Version]
- DePalma, R.G.; Hayes, V.W.; Zacharski, L.R. Bloodletting: Past and present. J. Am. Coll. Surg. 2007, 205, 132–144. [Google Scholar] [CrossRef]
- Melby, K.; Slordahl, S.; Gutteberg, T.J.; Nordbo, S.A. Septicaemia due to Yersinia enterocolitica after oral overdoses of iron. BMJ 1982, 285, 467–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddock, R.L.; Cousens, S.N.; Guzman, C.C. Infant diet and salmonellosis. Am. J. Public Health 1991, 81, 997–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Conroy, B.S.; Grigg, J.C.; Kolesnikov, M.; Morales, L.D.; Murphy, M.E.P. Staphylococcus aureus heme and siderophore-iron acquisition pathways. BioMetals 2019, 32, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Bilitewski, U.; Blodgett, J.A.V.; Duhme-Klair, A.-K.; Dallavalle, S.; Laschat, S.; Routledge, A.; Schobert, R. Chemical and biological aspects of nutritional immunity-perspectives for new anti-infectives that target iron uptake systems. Angew. Chemie Int. Ed. 2017, 56, 14360–14382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endicott, N.P.; Lee, E.; Wencewicz, T.A. Structural basis for xenosiderophore utilization by the human pathogen Staphylococcus aureus. ACS Infect. Dis. 2017, 3, 542–553. [Google Scholar] [CrossRef]
- Deane, C. Metal-pilfering pathogens. Nat. Chem. Biol. 2016, 12, 575. [Google Scholar] [CrossRef]
- Cheng, A.G.; DeDent, A.C.; Schneewind, O.; Missiakas, D.M. A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol. 2011, 19, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Cassat, J.E.; Moore, J.L.; Wilson, K.J.; Stark, Z.; Prentice, B.M.; Van de Plas, R.; Perry, W.J.; Zhang, Y.; Virostko, J.; Colvin, D.C.; et al. Integrated molecular imaging reveals tissue heterogeneity driving host-pathogen interactions. Sci. Transl. Med. 2018, 10, eaan6361. [Google Scholar] [CrossRef] [Green Version]
- Perry, W.J.; Spraggins, J.M.; Sheldon, J.R.; Grunenwald, C.M.; Heinrichs, D.E.; Cassat, J.E.; Skaar, E.P.; Caprioli, R.M. Staphylococcus aureus exhibits heterogeneous siderophore production within the vertebrate host. Proc. Natl. Acad. Sci. USA 2019, 116, 21980–21982. [Google Scholar] [CrossRef] [Green Version]
- Hammer, N.D.; Skaar, E.P. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu. Rev. Microbiol. 2011, 65, 129–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassat, J.E.; Skaar, E.P. Metal ion acquisition in Staphylococcus aureus: Overcoming nutritional immunity. Semin. Immunopathol. 2012, 34, 215–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaar, E.P.; Humayun, M.; Bae, T.; DeBord, K.L.; Schneewind, O. Iron-source preference of Staphylococcus aureus infections. Science 2004, 305, 1626–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldon, J.R.; Heinrichs, D.E. The iron-regulated staphylococcal lipoproteins. Front. Cell. Infect. Microbiol. 2012, 2, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazmanian, S.K. Passage of heme-iron across the envelope of Staphylococcus aureus. Science 2003, 299, 906–909. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.J.; Pishchany, G.; Humayun, M.; Schneewind, O.; Skaar, E.P. Staphylococcus aureus IsdB is a hemoglobin receptor required for heme iron utilization. J. Bacteriol. 2006, 188, 8421–8429. [Google Scholar] [CrossRef] [Green Version]
- Grigg, J.C.; Cooper, J.D.; Cheung, J.; Heinrichs, D.E.; Murphy, M.E.P. The Staphylococcus aureus siderophore receptor HtsA undergoes localized conformational changes to enclose staphyloferrin A in an arginine-rich binding pocket. J. Biol. Chem. 2010, 285, 11162–11171. [Google Scholar] [CrossRef] [Green Version]
- Beasley, F.C.; Vinés, E.D.; Grigg, J.C.; Zheng, Q.; Liu, S.; Lajoie, G.A.; Murphy, M.E.P.; Heinrichs, D.E. Characterization of staphyloferrin A biosynthetic and transport mutants in Staphylococcus aureus. Mol. Microbiol. 2009, 72, 947–963. [Google Scholar] [CrossRef]
- Kobylarz, M.J.; Grigg, J.C.; Liu, Y.; Lee, M.S.F.; Heinrichs, D.E.; Murphy, M.E.P. Deciphering the substrate specificity of SbnA, the enzyme catalyzing the first step in staphyloferrin B biosynthesis. Biochemistry 2016, 55, 927–939. [Google Scholar] [CrossRef]
- Kobylarz, M.J.; Grigg, J.C.; Takayama, S.J.; Rai, D.K.; Heinrichs, D.E.; Murphy, M.E.P. Synthesis of L-2,3-diaminopropionic acid, a siderophore and antibiotic precursor. Chem. Biol. 2014, 21, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Kobylarz, M.J.; Grigg, J.C.; Sheldon, J.R.; Heinrichs, D.E.; Murphy, M.E.P. SbnG, a citrate synthase in Staphylococcus aureus. J. Biol. Chem. 2014, 289, 33797–33807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstraete, M.M.; Perez-Borrajero, C.; Brown, K.L.; Heinrichs, D.E.; Murphy, M.E.P. SbnI is a free serine kinase that generates O -phospho-l-serine for staphyloferrin B biosynthesis in Staphylococcus aureus. J. Biol. Chem. 2018, 293, 6147–6160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Ju, Y.; Gu, Q.; Xu, J.; Zhou, H. Structural insights into substrate recognition and activity regulation of the key decarboxylase SbnH in staphyloferrin B biosynthesis. J. Mol. Biol. 2019, 431, 4868–4881. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J.C.; Murphy, M.E.P. Crystal structure of Staphylococcus aureus SirA Complexed with staphyloferrin B. 2010. Available online: https://www.rcsb.org/structure/3MWF (accessed on 17 March 2020). [CrossRef]
- Podkowa, K.J.; Briere, L.-A.K.; Heinrichs, D.E.; Shilton, B.H. Crystal and solution structure analysis of FhuD2 from Staphylococcus aureus in multiple unliganded conformations and bound to ferrioxamine-B. Biochemistry 2014, 53, 2017–2031. [Google Scholar] [CrossRef]
- Mariotti, P.; Malito, E.; Biancucci, M.; Lo Surdo, P.; Mishra, R.P.N.; Nardi-Dei, V.; Savino, S.; Nissum, M.; Spraggon, G.; Grandi, G.; et al. Structural and functional characterization of the Staphylococcus aureus virulence factor and vaccine candidate FhuD2. Biochem. J. 2013, 449, 683–693. [Google Scholar] [CrossRef]
- Kobylarz, M.J.; Heieis, G.A.; Loutet, S.A.; Murphy, M.E.P. Iron uptake oxidoreductase (IruO) uses a flavin adenine dinucleotide semiquinone intermediate for iron-siderophore reduction. ACS Chem. Biol. 2017, 12, 1778–1786. [Google Scholar] [CrossRef]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of Staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1865. [Google Scholar] [CrossRef]
- Yamashita, K.; Kawai, Y.; Tanaka, Y.; Hirano, N.; Kaneko, J.; Tomita, N.; Ohta, M.; Kamio, Y.; Yao, M.; Tanaka, I. Crystal structure of the octameric pore of staphylococcal gamma-hemolysin reveals the beta-barrel pore formation mechanism by two components. Proc. Natl. Acad. Sci. USA 2011, 108, 17314–17319. [Google Scholar] [CrossRef] [Green Version]
- Nocadello, S.; Minasov, G.; Shuvalova, L.; Dubrovska, I.; Sabini, E.; Bagnoli, F.; Grandi, G.; Anderson, W.F. Crystal structures of the components of the Staphylococcus aureus leukotoxin ED. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Huseby, M.J.; Shi, K.; Kruse, A.C.; Ohlendorf, D.H. Crystal structure of beta toxin from Staphylococcus aureus F277A, P278A mutant. 2010. Available online: https://www.rcsb.org/structure/3I41 (accessed on 17 March 2020). [CrossRef]
- Loureiro-Ferreira, N.; Rodrigues, J.; Brito, R.M.M. NMR structure of delta-toxin from Staphylococcus aureus in CD3OH. 2009. Available online: https://www.wwpdb.org/pdb?id=pdb_00002kam(accessed on 17 March 2020). [CrossRef]
- Zong, Y.; Bice, T.W.; Ton-That, H.; Schneewind, O.; Narayana, S.V.L. Crystal structures of Staphylococcus aureus sortase A and its substrate complex. J. Biol. Chem. 2004, 279, 31383–31389. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wu, R.; Joachimiak, G.; Mazmanian, S.K.; Missiakas, D.M.; Gornicki, P.; Schneewind, O.; Joachimiak, A. Structures of sortase B from Staphylococcus aureus and Bacillus anthracis reveal catalytic amino acid triad in the active site. Structure 2004, 12, 1147–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobitz, A.W.; Wereszczynski, J.; Yi, S.W.; Amer, B.R.; Huang, G.L.; Nguyen, A.V.; Sawaya, M.R.; Jung, M.E.; McCammon, J.A.; Clubb, R.T. Structural and computational studies of the Staphylococcus aureus sortase B-substrate complex reveal a substrate-stabilized oxyanion hole. J. Biol. Chem. 2014, 289, 8891–8902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, C.F.; Jacques, D.A.; Clubb, R.T.; Guss, J.M.; Gell, D.A. The structure of haemoglobin bound to the haemoglobin receptor IsdH from Staphylococcus aureus shows disruption of the native α-globin haem pocket. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1295–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowden, C.F.M.; Chan, A.C.K.; Li, E.J.W.; Arrieta, A.L.; Eltis, L.D.; Murphy, M.E.P. Structure–function analyses reveal key features in Staphylococcus aureus IsdB-associated unfolding of the heme-binding pocket of human hemoglobin. J. Biol. Chem. 2018, 293, 177–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, J.H.; Runager, K.S.; Andersen, C.B.F. The human protein haptoglobin inhibits IsdH-mediated heme-sequestering by Staphylococcus aureus. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Grigg, J.C.; Vermeiren, C.L.; Heinrichs, D.E.; Murphy, M.E.P. Haem recognition by a Staphylococcus aureus NEAT domain. Mol. Microbiol. 2007, 63, 139–149. [Google Scholar] [CrossRef]
- Sharp, K.H.; Schneider, S.; Cockayne, A.; Paoli, M. Crystal structure of the heme-IsdC complex, the central conduit of the Isd iron/heme uptake system in Staphylococcus aureus. J. Biol. Chem. 2007, 282, 10625–10631. [Google Scholar] [CrossRef] [Green Version]
- Grigg, J.C.; Vermeiren, C.L.; Heinrichs, D.E.; Murphy, M.E.P. Heme coordination by Staphylococcus aureus IsdE. J. Biol. Chem. 2007, 282, 28815–28822. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Skaar, E.P.; Zhang, R.; Joachimiak, G.; Gornicki, P.; Schneewind, O.; Joachimiak, A. Staphylococcus aureus IsdG and IsdI, heme-degrading enzymes with structural similarity to monooxygenases. J. Biol. Chem. 2005, 280, 2840–2846. [Google Scholar] [CrossRef] [Green Version]
- Reniere, M.L.; Ukpabi, G.N.; Harry, S.R.; Stec, D.F.; Krull, R.; Wright, D.W.; Bachmann, B.O.; Murphy, M.E.P.; Skaar, E.P. The IsdG-family of haem oxygenases degrades haem to a novel chromophore. Mol. Microbiol. 2010, 75, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Challis, G.L. A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. ChemBioChem 2005, 6, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Cotton, J.L.; Tao, J.; Balibar, C.J. Identification and characterization of the Staphylococcus aureus gene cluster coding for staphyloferrin A. Biochemistry 2009, 48, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.M.; Lascelles, J. The effect of growth conditions on oxidative and dehydrogenase activity in Staphylococcus aureus. J. Gen. Microbiol. 1962, 29, 531–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldon, J.R.; Marolda, C.L.; Heinrichs, D.E. TCA cycle activity in Staphylococcus aureus is essential for iron-regulated synthesis of staphyloferrin A, but not staphyloferrin B: The benefit of a second citrate synthase. Mol. Microbiol. 2014, 92, 824–839. [Google Scholar] [CrossRef]
- Hannauer, M.; Sheldon, J.R.; Heinrichs, D.E. Involvement of major facilitator superfamily proteins SfaA and SbnD in staphyloferrin secretion in Staphylococcus aureus. FEBS Lett. 2015, 589, 730–737. [Google Scholar] [CrossRef] [Green Version]
- Nakaminami, H.; Chen, C.; Truong-Bolduc, Q.C.; Kim, E.S.; Wang, Y.; Hooper, D.C. Efflux transporter of siderophore staphyloferrin A in Staphylococcus aureus contributes to bacterial fitness in abscesses and epithelial cells. Infect. Immun. 2017, 85, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Dale, S.E.; Doherty-Kirby, A.; Lajoie, G.A.; Heinrichs, D.E. Role of siderophore biosynthesis in virulence of Staphylococcus aureus: Identification and characterization of genes involved in production of a siderophore. Infect. Immun. 2004, 72, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Madsen, J.L.H.; Johnstone, T.C.; Nolan, E.M. Chemical synthesis of staphyloferrin B affords insight into the molecular structure, iron chelation, and biological activity of a polycarboxylate siderophore deployed by the human pathogen Staphylococcus aureus. J. Am. Chem. Soc. 2015, 137, 9117–9127. [Google Scholar] [CrossRef] [Green Version]
- Beasley, F.C.; Cheung, J.; Heinrichs, D.E. Mutation of L-2,3-diaminopropionic acid synthase genes blocks staphyloferrin B synthesis in Staphylococcus aureus. BMC Microbiol. 2011, 11, 199. [Google Scholar] [CrossRef] [Green Version]
- Cheung, J.; Murphy, M.E.P.; Heinrichs, D.E. Discovery of an iron-regulated citrate synthase in Staphylococcus aureus. Chem. Biol. 2012, 19, 1568–1578. [Google Scholar] [CrossRef] [Green Version]
- Cheung, J.; Beasley, F.C.; Liu, S.; Lajoie, G.A.; Heinrichs, D.E. Molecular characterization of staphyloferrin B biosynthesis in Staphylococcus aureus. Mol. Microbiol. 2009, 74, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Laakso, H.A.; Marolda, C.L.; Pinter, T.B.; Stillman, M.J.; Heinrichs, D.E. A heme-responsive regulator controls synthesis of staphyloferrin B in Staphylococcus aureus. J. Biol. Chem. 2016, 291, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, S.E.; Sebulsky, M.T.; Heinrichs, D.E. Involvement of SirABC in iron-siderophore import in Staphylococcus aureus. J. Bacteriol. 2004, 186, 8356–8362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrichs, J.H.; Gatlin, L.E.; Kunsch, C.; Choi, G.H.; Hanson, M.S. Identification and characterization of SirA, an iron-regulated protein from Staphylococcus aureus. J. Bacteriol. 1999, 181, 1436–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannauer, M.; Arifin, A.J.; Heinrichs, D.E. Involvement of reductases IruO and NtrA in iron acquisition by Staphylococcus aureus. Mol. Microbiol. 2015, 96, 1192–1210. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, J.R.; Heinrichs, D.E. Recent developments in understanding the iron acquisition strategies of gram positive pathogens. FEMS Microbiol. Rev. 2015, 39, 592–630. [Google Scholar] [CrossRef] [Green Version]
- Courcol, R.J.; Trivier, D.; Bissinger, M.C.; Martin, G.R.; Brown, M.R.W. Siderophore production by Staphylococcus aureus and identification of iron-regulated proteins. Infect. Immun. 1997, 65, 1944–1948. [Google Scholar] [CrossRef] [Green Version]
- Sebulsky, M.T.; Hohnstein, D.; Hunter, M.D.; Heinrichs, D.E. Identification and characterization of a membrane permease involved in iron-hydroxamate transport in Staphylococcus aureus. J. Bacteriol. 2000, 182, 4394–4400. [Google Scholar] [CrossRef] [Green Version]
- Ghssein, G.; Brutesco, C.; Ouerdane, L.; Fojcik, C.; Izaute, A.; Wang, S.; Hajjar, C.; Lobinski, R.; Lemaire, D.; Richaud, P.; et al. Biosynthesis of a broad-spectrum nicotianamine-like metallophore in Staphylococcus aureus. Science 2016, 352, 1105–1109. [Google Scholar] [CrossRef]
- Fojcik, C.; Arnoux, P.; Ouerdane, L.; Aigle, M.; Alfonsi, L.; Borezée-Durant, E. Independent and cooperative regulation of staphylopine biosynthesis and trafficking by Fur and Zur. Mol. Microbiol. 2018, 108, 159–177. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Zhang, Y.; Chen, W.; Gu, T.; Zhang, S.-Y.; Ji, Q. Mechanistic insights into staphylopine-mediated metal acquisition. Proc. Natl. Acad. Sci. USA 2018, 115, 3942–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Hooper, D.C. Intracellular accumulation of staphylopine impairs the fitness of Staphylococcus aureus cntE mutant. FEBS Lett. 2019, 593, 1213–1222. [Google Scholar] [CrossRef]
- Neumann, W.; Gulati, A.; Nolan, E.M. Metal homeostasis in infectious disease: Recent advances in bacterial metallophores and the human metal-withholding response. Curr. Opin. Chem. Biol. 2017, 37, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, J.H.; Ng, J. The effect of desferrioxamine on the growth of Staphylococcus aureus, Yersinia enterocolitica and Streptococcus faecalis in human serum: Uptake of desferrioxamine-bound iron. FEMS Microbiol. Lett. 1983, 20, 439–442. [Google Scholar] [CrossRef]
- Sebulsky, M.T.; Shilton, B.H.; Speziali, C.D.; Heinrichs, D.E. The role of FhuD2 in iron(III)-hydroxamate transport in Staphylococcus aureus. J. Biol. Chem. 2003, 278, 49890–49900. [Google Scholar] [CrossRef] [Green Version]
- Beasley, F.C.; Marolda, C.L.; Cheung, J.; Buac, S.; Heinrichs, D.E. Staphylococcus aureus transporters Hts, Sir, and Sst capture iron liberated from human transferrin by staphyloferrin A, staphyloferrin B, and catecholamine stress hormones, respectively, and contribute to virulence. Infect. Immun. 2011, 79, 2345–2355. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.P.N.; Mariotti, P.; Fiaschi, L.; Nosari, S.; Maccari, S.; Liberatori, S.; Fontana, M.R.; Pezzicoli, A.; De Falco, M.G.; Falugi, F.; et al. Staphylococcus aureus FhuD2 Is involved in the early phase of staphylococcal dissemination and generates protective immunity in mice. J. Infect. Dis. 2012, 206, 1041–1049. [Google Scholar] [CrossRef] [Green Version]
- Sebulsky, M.T.; Heinrichs, D.E. Identification and characterization of fhuD1 and fhuD2, two genes involved in iron-hydroxamate uptake in Staphylococcus aureus. J. Bacteriol. 2001, 183, 4994–5000. [Google Scholar] [CrossRef] [Green Version]
- Sandrini, S.M.; Shergill, R.; Woodward, J.; Muralikuttan, R.; Haigh, R.D.; Lyte, M.; Freestone, P.P. Elucidation of the mechanism by which catecholamine stress hormones liberate iron from the innate immune defense proteins transferrin and lactoferrin. J. Bacteriol. 2010, 192, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, J.A.; Cockayne, A.; Hill, P.J.; Williams, P. Molecular cloning and analysis of a putative siderophore ABC transporter from Staphylococcus aureus. Infect. Immun. 2000, 68, 6281–6288. [Google Scholar] [CrossRef]
- Miethke, M.; Klotz, O.; Linne, U.; May, J.J.; Beckering, C.L.; Marahiel, M.A. Ferri-bacillibactin uptake and hydrolysis in Bacillus subtilis. Mol. Microbiol. 2006, 61, 1413–1427. [Google Scholar] [CrossRef] [PubMed]
- Abergel, R.J.; Zawadzka, A.M.; Hoette, T.M.; Raymond, K.N. Enzymatic hydrolysis of trilactone siderophores: Where chiral recognition occurs in enterobactin and bacillibactin iron transport. J. Am. Chem. Soc. 2009, 131, 12682–12692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Fischbach, M.A.; Liu, D.R.; Walsh, C.T. In vitro characterization of salmochelin and enterobactin trilactone hydrolases IroD, IroE, and Fes. J. Am. Chem. Soc. 2005, 127, 11075–11084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, A.L.; Ying, P. Regulation of alpha- and beta-hemolysins by the sar locus of Staphylococcus aureus. J. Bacteriol. 1994, 176, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novick, R.P. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 2003, 48, 1429–1449. [Google Scholar] [CrossRef] [PubMed]
- Venkatasubramaniam, A.; Kanipakala, T.; Ganjbaksh, N.; Mehr, R.; Mukherjee, I.; Krishnan, S.; Bae, T.; Aman, M.J.; Adhikari, R.P. A critical role for HlgA in Staphylococcus aureus pathogenesis revealed by a switch in the SaeRS two-component regulatory system. Toxins (Basel). 2018, 10, 377. [Google Scholar] [CrossRef] [Green Version]
- Bhakdil, S.; Tranum-Jensen, J. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar] [CrossRef]
- Callegan, M.C.; Engel, L.S.; Hill, J.M.; O’Callaghan, R.J. Corneal virulence of Staphylococcus aureus: Roles of alpha-toxin and protein A in pathogenesis. Infect. Immun. 1994, 62, 2478–2482. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, I.-M.; Hartford, O.; Foster, T.J.; Tarkowski, A. Alpha-toxin and gamma-toxin jointly promote Staphylococcus aureus virulence in murine septic arthritis. Infect. Immun. 1999, 67, 1045–1049. [Google Scholar] [CrossRef] [Green Version]
- Wardenburg, J.B.; Patel, R.J.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- Kielian, T.; Cheung, A.L.; Hickey, W.F. Diminished virulence of an alpha-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect. Immun. 2001, 69, 6902–6911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaan, A.N.; Reyes-Robles, T.; Badiou, C.; Cochet, S.; Boguslawski, K.M.; Yoong, P.; Day, C.J.; de Haas, C.J.C.; van Kessel, K.P.M.; Vandenesch, F.; et al. Staphylococcus aureus targets the duffy antigen receptor for chemokines (DARC) to lyse erythrocytes. Cell Host Microbe 2015, 18, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malachowa, N.; Whitney, A.R.; Kobayashi, S.D.; Sturdevant, D.E.; Kennedy, A.D.; Braughton, K.R.; Shabb, D.W.; Diep, B.A.; Chambers, H.F.; Otto, M.; et al. Global changes in Staphylococcus aureus gene expression in human blood. PLoS ONE 2011, 6, e18617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaan, A.N.; Vrieling, M.; Wallet, P.; Badiou, C.; Reyes-Robles, T.; Ohneck, E.A.; Benito, Y.; de Haas, C.J.C.; Day, C.J.; Jennings, M.P.; et al. The staphylococcal toxins γ-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat. Commun. 2014, 5, 5438. [Google Scholar] [CrossRef] [Green Version]
- Alonzo III, F.; Benson, M.A.; Chen, J.; Novick, R.P.; Shopsin, B.; Torres, V.J. Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol. Microbiol. 2012, 83, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Robles, T.; Alonzo III, F.; Kozhaya, L.; Lacy, D.B.; Unutmaz, D.; Torres, V.J. Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 2013, 14, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Yoong, P.; Torres, V.J. Counter inhibition between leukotoxins attenuates Staphylococcus aureus virulence. Nat. Commun. 2015, 6, 8125. [Google Scholar] [CrossRef] [Green Version]
- Huseby, M.J.; Kruse, A.C.; Digre, J.; Kohler, P.L.; Vocke, J.A.; Mann, E.E.; Bayles, K.W.; Bohach, G.A.; Schlievert, P.M.; Ohlendorf, D.H.; et al. Beta toxin catalyzes formation of nucleoprotein matrix in staphylococcal biofilms. Proc. Natl. Acad. Sci. USA 2010, 107, 14407–14412. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Braughton, K.R.; Kretschmer, D.; Bach, T.-H.L.; Queck, S.Y.; Li, M.; Kennedy, A.D.; Dorward, D.W.; Klebanoff, S.J.; Peschel, A.; et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 2007, 13, 1510–1514. [Google Scholar] [CrossRef]
- Cogen, A.L.; Yamasaki, K.; Sanchez, K.M.; Dorschner, R.A.; Lai, Y.; MacLeod, D.T.; Torpey, J.W.; Otto, M.; Nizet, V.; Kim, J.E.; et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J. Investig. Dermatol. 2010, 130, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Oscherwitz, J.; Cease, K.B.; Chan, S.M.; Muñoz-Planillo, R.; Hasegawa, M.; Villaruz, A.E.; Cheung, G.Y.C.; McGavin, M.J.; Travers, J.B.; et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature 2013, 503, 397–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenesch, F.; Lina, G.; Henry, T. Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: A redundant arsenal of membrane-damaging virulence factors? Front. Cell. Infect. Microbiol. 2012, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraunholz, M.J.; Sinha, B. Intracellular staphylococcus aureus: Live-in and let die. Front. Cell. Infect. Microbiol. 2012, 2, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldovan, A.; Fraunholz, M.J. In or out: Phagosomal escape of Staphylococcus aureus. Cell. Microbiol. 2019, 21, e12997. [Google Scholar] [CrossRef] [Green Version]
- Strobel, M.; Pförtner, H.; Tuchscherr, L.; Völker, U.; Schmidt, F.; Kramko, N.; Schnittler, H.-J.; Fraunholz, M.J.; Löffler, B.; Peters, G.; et al. Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin. Microbiol. Infect. 2016, 22, 799–809. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, A.J.; Lindsay, J.A. Staphylococcus aureus innate immune evasion is lineage-specific: A bioinfomatics study. Infect. Genet. Evol. 2013, 19, 7–14. [Google Scholar] [CrossRef]
- Schneewind, O.; Fowler, A.; Faull, K.F. Structure of the cell wall anchor of surface proteins in Staphylococcus aureus. Science 1995, 268, 103–106. [Google Scholar] [CrossRef]
- Ton-That, H.; Liu, G.; Mazmanian, S.K.; Faull, K.F.; Schneewind, O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA 1999, 96, 12424–12429. [Google Scholar] [CrossRef] [Green Version]
- Mazmanian, S.K.; Ton-That, H.; Su, K.; Schneewind, O. An iron-regulated sortase anchors a class of surface protein during Staphylococcus aureus pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 2293–2298. [Google Scholar] [CrossRef] [Green Version]
- Dryla, A.; Gelbmann, D.; Von Gabain, A.; Nagy, E. Identification of a novel iron regulated staphylococcal surface protein with haptoglobin-haemoglobin binding activity. Mol. Microbiol. 2003, 49, 37–53. [Google Scholar] [CrossRef]
- Taylor, J.M.; Heinrichs, D.E. Transferrin binding in Staphylococcus aureus: Involvement of a cell wall-anchored protein. Mol. Microbiol. 2002, 43, 1603–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, S.R.; Wiltshire, M.D.; Foster, S.J. IsdA of Staphylococcus aureus is a broad spectrum, iron-regulated adhesin. Mol. Microbiol. 2004, 51, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.A.; Ciccarelli, F.D.; Perez-Iratxeta, C.; Bork, P. NEAT: A domain duplicated in genes near the components of a putative Fe3+ siderophore transporter from Gram-positive pathogenic bacteria. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pishchany, G.; Sheldon, J.R.; Dickson, C.F.; Alam, M.T.; Read, T.D.; Gell, D.A.; Heinrichs, D.E.; Skaar, E.P. IsdB-dependent hemoglobin binding is required for acquisition of heme by Staphylococcus aureus. J. Infect. Dis. 2014, 209, 1764–1772. [Google Scholar] [CrossRef] [Green Version]
- Pishchany, G.; McCoy, A.L.; Torres, V.J.; Krause, J.C.; Crowe, J.E.; Fabry, M.E.; Skaar, E.P. Specificity for human hemoglobin enhances Staphylococcus aureus infection. Cell Host Microbe 2010, 8, 544–550. [Google Scholar] [CrossRef] [Green Version]
- Pishchany, G.; Dickey, S.E.; Skaar, E.P. Subcellular localization of the Staphylococcus aureus heme iron transport components IsdA and IsdB. Infect. Immun. 2009, 77, 2624–2634. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.G.; Kim, H.K.; Burts, M.L.; Krausz, T.; Schneewind, O.; Missiakas, D.M. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 2009, 23, 3393–3404. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; DeDent, A.C.; Cheng, A.G.; McAdow, M.; Bagnoli, F.; Missiakas, D.M.; Schneewind, O. IsdA and IsdB antibodies protect mice against Staphylococcus aureus abscess formation and lethal challenge. Vaccine 2010, 28, 6382–6392. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Xie, G.; Liu, M.; Olson, J.S.; Fabian, M.; Dooley, D.M.; Lei, B. Pathway for heme uptake from human methemoglobin by the iron-regulated surface determinants system of Staphylococcus aureus. J. Biol. Chem. 2008, 283, 18450–18460. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Tanaka, W.N.; Zhu, H.; Xie, G.; Dooley, D.M.; Lei, B. Direct hemin transfer from IsdA to IsdC in the iron-regulated surface determinant (Isd) heme acquisition system of Staphylococcus aureus. J. Biol. Chem. 2008, 283, 6668–6676. [Google Scholar] [CrossRef] [Green Version]
- Abe, R.; Caaveiro, J.M.M.; Kozuka-Hata, H.; Oyama, M.; Tsumoto, K. Mapping ultra-weak protein-protein interactions between heme transporters of Staphylococcus aureus. J. Biol. Chem. 2012, 287, 16477–16487. [Google Scholar] [CrossRef] [Green Version]
- Tiedemann, M.T.; Heinrichs, D.E.; Stillman, M.J. Multiprotein heme shuttle pathway in Staphylococcus aureus: Iron-regulated surface determinant cog-wheel kinetics. J. Am. Chem. Soc. 2012, 134, 16578–16585. [Google Scholar] [CrossRef] [PubMed]
- Skaar, E.P.; Gaspar, A.H.; Schneewind, O. IsdG and IsdI, heme-degrading enzymes in the cytoplasm of Staphylococcus aureus. J. Biol. Chem. 2004, 279, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.C.; Reniere, M.L.; Skaar, E.P.; Murphy, M.E.P. Ruffling of metalloporphyrins bound to IsdG and IsdI, two heme-degrading enzymes in Staphylococcus aureus. J. Biol. Chem. 2008, 283, 30957–30963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, S.J.; Ukpabi, G.N.; Murphy, M.E.P.; Mauk, A.G. Electronic properties of the highly ruffled heme bound to the heme degrading enzyme IsdI. Proc. Natl. Acad. Sci. USA 2011, 108, 13071–13076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reniere, M.L.; Skaar, E.P. Staphylococcus aureus haem oxygenases are differentially regulated by iron and haem. Mol. Microbiol. 2008, 69, 1304–1315. [Google Scholar] [CrossRef] [Green Version]
- Verstraete, M.M.; Morales, L.D.; Kobylarz, M.J.; Loutet, S.A.; Laakso, H.A.; Pinter, T.B.; Stillman, M.J.; Heinrichs, D.E.; Murphy, M.E.P. The heme-sensitive regulator SbnI has a bifunctional role in staphyloferrin B production by Staphylococcus aureus. J. Biol. Chem. 2019, 294, 11622–11636. [Google Scholar] [CrossRef]
- Loutet, S.A.; Kobylarz, M.J.; Chau, C.H.T.; Murphy, M.E.P. IruO is a reductase for heme degradation by IsdI and IsdG proteins in Staphylococcus aureus. J. Biol. Chem. 2013, 288, 25749–25759. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.R.; Mohamed, R.; Bian, L.; Routh, A.F.; Kokai-Kun, J.F.; Mond, J.J.; Tarkowski, A.; Foster, S.J. The Staphylococcus aureus surface protein IsdA mediates resistance to innate defenses of human skin. Cell Host Microbe 2007, 1, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.R.; Foster, S.J. IsdA protects Staphylococcus aureus against the bactericidal protease activity of apolactoferrin. Infect. Immun. 2008, 76, 1518–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visai, L.; Yanagisawa, N.; Josefsson, E.; Tarkowski, A.; Pezzali, I.; Rooijakkers, S.H.M.; Foster, T.J.; Speziale, P. Immune evasion by Staphylococcus aureus conferred by iron-regulated surface determinant protein IsdH. Microbiology 2009, 155, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speziale, P.; Pietrocola, G.; Foster, T.J.; Geoghegan, J.A. Protein-based biofilm matrices in Staphylococci. Front. Cell. Infect. Microbiol. 2014, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Missineo, A.; Di Poto, A.; Geoghegan, J.A.; Rindi, S.; Heilbronner, S.; Gianotti, V.; Arciola, C.R.; Foster, T.J.; Speziale, P.; Pietrocola, G. IsdC from Staphylococcus lugdunensis induces biofilm formation under low-iron growth conditions. Infect. Immun. 2014, 82, 2448–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miajlovic, H.; Zapotoczna, M.; Geoghegan, J.A.; Kerrigan, S.W.; Speziale, P.; Foster, T.J. Direct interaction of iron-regulated surface determinant IsdB of Staphylococcus aureus with the GPIIb/IIIa receptor on platelets. Microbiology 2010, 156, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapotoczna, M.; Jevnikar, Z.; Miajlovic, H.; Kos, J.; Foster, T.J. Iron-regulated surface determinant B (IsdB) promotes Staphylococcus aureus adherence to and internalization by non-phagocytic human cells. Cell. Microbiol. 2013, 15, 1026–1041. [Google Scholar] [CrossRef] [Green Version]
- Biswas, L.; Biswas, R.; Nerz, C.; Ohlsen, K.; Schlag, M.; Schafer, T.; Lamkemeyer, T.; Ziebandt, A.-K.; Hantke, K.; Rosenstein, R.; et al. Role of the twin-arginine translocation pathway in Staphylococcus. J. Bacteriol. 2009, 191, 5921–5929. [Google Scholar] [CrossRef] [Green Version]
- Turlin, E.; Débarbouillé, M.; Augustyniak, K.; Gilles, A.-M.; Wandersman, C. Staphylococcus aureus FepA and FepB proteins drive heme iron utilization in Escherichia coli. PLoS ONE 2013, 8, e56529. [Google Scholar] [CrossRef]
- Létoffé, S.; Heuck, G.; Delepelaire, P.; Lange, N.; Wandersman, C. Bacteria capture iron from heme by keeping tetrapyrrol skeleton intact. Proc. Natl. Acad. Sci. USA 2009, 106, 11719–11724. [Google Scholar] [CrossRef] [Green Version]
- Reniere, M.L.; Torres, V.J.; Skaar, E.P. Intracellular metalloporphyrin metabolism in Staphylococcus aureus. BioMetals 2007, 20, 333–345. [Google Scholar] [CrossRef]
- Tiburzi, F.; Imperi, F.; Visca, P. Is the host heme incorporated in microbial heme-proteins? IUBMB Life 2009, 61, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; Stauff, D.L.; Pishchany, G.; Bezbradica, J.S.; Gordy, L.E.; Iturregui, J.; Anderson, K.L.; Dunman, P.M.; Joyce, S.; Skaar, E.P. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe 2007, 1, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauff, D.L.; Torres, V.J.; Skaar, E.P. Signaling and DNA-binding activities of the Staphylococcus aureus HssR-HssS two-component system required for heme sensing. J. Biol. Chem. 2007, 282, 26111–26121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauff, D.L.; Bagaley, D.; Torres, V.J.; Joyce, R.; Anderson, K.L.; Kuechenmeister, L.; Dunman, P.M.; Skaar, E.P. Staphylococcus aureus HrtA is an ATPase required for protection against heme toxicity and prevention of a transcriptional heme stress response. J. Bacteriol. 2008, 190, 3588–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horsburgh, M.J.; Clements, M.O.; Crossley, H.; Ingham, E.; Foster, S.J. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect. Immun. 2001, 69, 3744–3754. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.C.; Robinson, A.K.; Rodríguez-Quiñones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Zühlke, D.; Dörries, K.; Bernhardt, J.; Maaß, S.; Muntel, J.; Liebscher, V.; Pané-Farré, J.; Riedel, K.; Lalk, M.; Völker, U.; et al. Costs of life - dynamics of the protein inventory of Staphylococcus aureus during anaerobiosis. Sci. Rep. 2016, 6, 28172. [Google Scholar] [CrossRef]
- Morrissey, J.A.; Cockayne, A.; Brummell, K.J.; Williams, P. The staphylococcal ferritins are differentially regulated in response to iron and manganese and via PerR and Fur. Infect. Immun. 2004, 72, 972–979. [Google Scholar] [CrossRef] [Green Version]
- Nobre, L.S.; Saraiva, L.M. Effect of combined oxidative and nitrosative stresses on Staphylococcus aureus transcriptome. Appl. Microbiol. Biotechnol. 2013, 97, 2563–2573. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, G.; Jensen, E.R.; Lenoy, E.; Schneewind, O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. USA 2000, 97, 5510–5515. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, I.; Mazmanian, S.K.; Schneewind, O.; Verdrengh, M.; Bremell, T.; Tarkowski, A. On the role of Staphylococcus aureus sortase and sortase-catalyzed surface protein anchoring in murine septic arthritis. J. Infect. Dis. 2002, 185, 1417–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Liu, B.; Wang, D.; Wang, L.; Deng, X.; Bi, C.; Xiong, Y.; Wu, Q.; Cui, Y.; Zhang, Y.; et al. Role of sortase A in the pathogenesis of Staphylococcus aureus-induced mastitis in mice. FEMS Microbiol. Lett. 2014, 351, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, W.J.; Lenoy, E.; Murphy, T.; Tardio, L.; Brugio, P.; Projan, S.J.; Schnewind, O.; Alksne, L. Effect of srtA and srtB gene expression on the virulence of Staphylococcus aureus in animal models of infection. J. Antimicrob. Chemother. 2004, 53, 480–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, I.; Mazmanian, S.K.; Schneewind, O.; Bremell, T.; Tarkowski, A. The role of Staphylococcus aureus sortase A and sortase B in murine arthritis. Microbes Infect. 2003, 5, 775–780. [Google Scholar] [CrossRef]
- Mason, W.J.; Skaar, E.P. Assessing the contribution of heme-iron acquisition to Staphylococcus aureus pneumonia using computed tomography. PLoS ONE 2009, 4, e6668. [Google Scholar] [CrossRef] [Green Version]
- Speziali, C.D.; Dale, S.E.; Henderson, J.A.; Vinés, E.D.; Heinrichs, D.E.; Vines, E.D.; Heinrichs, D.E. Requirement of Staphylococcus aureus ATP-binding cassette-ATPase FhuC for iron-restricted growth and evidence that it functions with more than one iron transporter. J. Bacteriol. 2006, 188, 2048–2055. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Cockayne, A.; Morrissey, J.A. Iron-regulated biofilm formation in Staphylococcus aureus Newman requires ica and the secreted protein Emp. Infect. Immun. 2008, 76, 1756–1765. [Google Scholar] [CrossRef] [Green Version]
- Horsburgh, M.J.; Ingham, E.; Foster, S.J. In Staphylococcus aureus, Fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J. Bacteriol. 2001, 183, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Sengupta, M.; Purves, J.; Tarrant, E.; Williams, P.H.; Cockayne, A.; Muthaiyan, A.; Stephenson, R.; Ledala, N.; Wilkinson, B.J.; et al. Fur is required for the activation of virulence gene expression through the induction of the sae regulatory system in Staphylococcus aureus. Int. J. Med. Microbiol. 2011, 301, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.J.; Attia, A.S.; Mason, W.J.; Hood, M.I.; Corbin, B.D.; Beasley, F.C.; Anderson, K.L.; Stauff, D.L.; McDonald, W.H.; Zimmerman, L.J.; et al. Staphylococcus aureus Fur regulates the expression of virulence factors that contribute to the pathogenesis of pneumonia. Infect. Immun. 2010, 78, 1618–1628. [Google Scholar] [CrossRef] [Green Version]
- Méhi, O.; Bogos, B.; Csörgő, B.; Pál, F.; Nyerges, Á.; Papp, B.; Pál, C. Perturbation of iron homeostasis promotes the evolution of antibiotic resistance. Mol. Biol. Evol. 2014, 31, 2793–2804. [Google Scholar] [CrossRef] [Green Version]
- Xiong, A.; Singh, V.K.; Cabrera, G.; Jayaswal, R.K. Molecular characterization of the ferric-uptake regulator, Fur, from Staphylococcus aureus. Microbiology 2000, 146, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, M.; Moisan, H.; Brouillette, É.; Gervais, A.L.; Jacques, M.; Lacasse, P.; Diarra, M.S.; Malouin, F. Transcriptional modulation of some Staphylococcus aureus iron-regulated genes during growth in vitro and in a tissue cage model in vivo. Microbes Infect. 2006, 8, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Lojek, L.J.; Farrand, A.J.; Weiss, A.; Skaar, E.P. Fur regulation of Staphylococcus aureus heme oxygenases is required for heme homeostasis. Int. J. Med. Microbiol. 2018, 308, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Porcheron, G.; Dozois, C.M. Interplay between iron homeostasis and virulence: Fur and RyhB as major regulators of bacterial pathogenicity. Vet. Microbiol. 2015, 179, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Rochat, T.; Toffano-Nioche, C.; Le Lam, T.N.; Bouloc, P.; Morvan, C. Assessment of bona fide sRNAs in Staphylococcus aureus. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Mäder, U.; Nicolas, P.; Depke, M.; Pané-Farré, J.; Debarbouille, M.; van der Kooi-Pol, M.M.; Guérin, C.; Dérozier, S.; Hiron, A.; Jarmer, H.; et al. Staphylococcus aureus transcriptome architecture: From laboratory to infection-mimicking conditions. PLOS Genet. 2016, 12, e1005962. [Google Scholar] [CrossRef]
- Shahmirzadi, S.V.; Nguyen, M.-T.; Götz, F. Evaluation of Staphylococcus aureus lipoproteins: Role in nutritional acquisition and pathogenicity. Front. Microbiol. 2016, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bubeck Wardenburg, J.; Williams, W.A.; Missiakas, D.M. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc. Natl. Acad. Sci. USA 2006, 103, 13831–13836. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Tawaratsumida, K.; Kariya, H.; Aoyama, K.; Tamura, T.; Suda, Y. Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int. Immunol. 2006, 18, 355–362. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Pang, K.C.; Masters, S.L. Intercellular communication for innate immunity. Mol. Immunol. 2017, 86, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Schmaler, M.; Jann, N.J.; Ferracin, F.; Landolt, L.Z.; Biswas, L.; Götz, F.; Landmann, R. Lipoproteins in Staphylococcus aureus mediate inflammation by TLR2 and iron-dependent growth in vivo. J. Immunol. 2009, 182, 7110–7118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recsei, P.; Kreiswirth, B.; O’Reilly, M.; Schlievert, P.M.; Gruss, A.; Novick, R.P. Regulation of exoprotein gene expression in Staphylococcus aureus by agr. Mol. Gen. Genet. MGG 1986, 202, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.Q.; Willard, J.; Yeaman, M.R.; Cheung, A.L.; Bayer, A.S. Regulation of Staphylococcus aureus α-toxin gene (hla) expression by agr, sarA, and sae in vitro and in experimental infective endocarditis. J. Infect. Dis. 2006, 194, 1267–1275. [Google Scholar] [CrossRef] [Green Version]
- Novick, R.P.; Projan, S.J.; Kornblum, J.; Ross, H.F.; Ji, G.; Kreiswirth, B.; Vandenesch, F.; Moghazeh, S.; Novick, R.P. Theagr P2 operon: An autocatalytic sensory transduction system in Staphylococcus aureus. Mol. Gen. Genet. MGG 1995, 248, 446–458. [Google Scholar] [CrossRef]
- Koenig, R.L.; Ray, J.L.; Maleki, S.J.; Smeltzer, M.S.; Hurlburt, B.K. Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region. J. Bacteriol. 2004, 186, 7549–7555. [Google Scholar] [CrossRef] [Green Version]
- Reyes, D.; Andrey, D.O.; Monod, A.; Kelley, W.L.; Zhang, G.; Cheung, A.L. Coordinated regulation by AgrA, SarA, and SarR to control agr expression in Staphylococcus aureus. J. Bacteriol. 2011, 193, 6020–6031. [Google Scholar] [CrossRef] [Green Version]
- Sidote, D.J.; Barbieri, C.M.; Wu, T.; Stock, A.M. Structure of the Staphylococcus aureus AgrA LytTR domain bound to DNA reveals a beta fold with an unusual mode of binding. Structure 2008, 16, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Leonard, P.G.; Bezar, I.F.; Sidote, D.J.; Stock, A.M. Identification of a hydrophobic cleft in the LytTR domain of AgrA as a locus for small molecule interactions that inhibit DNA binding. Biochemistry 2012, 51, 10035–10043. [Google Scholar] [CrossRef] [Green Version]
- Bezar, I.F.; Mashruwala, A.A.; Boyd, J.M.; Stock, A.M. Drug-like fragments inhibit agr-mediated virulence expression in Staphylococcus aureus. Sci. Rep. 2019, 9, 6786. [Google Scholar] [CrossRef] [Green Version]
- Khodaverdian, V.; Pesho, M.; Truitt, B.; Bollinger, L.; Patel, P.; Nithianantham, S.; Yu, G.; Delaney, E.; Jankowsky, E.; Shoham, M. Discovery of antivirulence agents against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 3645–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Kuo, D.; Shoham, M.; Viswanathan, R. Combinatorial synthesis and in vitro evaluation of a biaryl hydroxyketone library as antivirulence agents against MRSA. ACS Comb. Sci. 2014, 16, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Kuo, D.; Yu, G.; Hoch, W.; Gabay, D.; Long, L.; Ghannoum, M.A.; Nagy, N.; Harding, C.V.; Viswanathan, R.; Shoham, M. Novel quorum-quenching agents promote methicillin-resistant Staphylococcus aureus (MRSA) wound healing and sensitize MRSA to β-lactam antibiotics. Antimicrob. Agents Chemother. 2015, 59, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.; Kuo, D.; Jankowsky, E.; Long, L.; Hager, C.; Bandi, K.; Ma, D.; Manoharan, D.; Shoham, Y.; Harte, W.; et al. Small-molecule AgrA inhibitors F12 and F19 act as antivirulence agents against gram-positive pathogens. Sci. Rep. 2018, 8, 14578. [Google Scholar] [CrossRef]
- Shoham, M.; Viswanathan, R.; Yu, G. Anti-virulence compositions and methods. U.S. Patent 8,859,626, 25 January 2018. [Google Scholar]
- Sully, E.K.; Malachowa, N.; Elmore, B.O.; Alexander, S.M.; Femling, J.K.; Gray, B.M.; DeLeo, F.R.; Otto, M.; Cheung, A.L.; Edwards, B.S.; et al. Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog. 2014, 10, e1004174. [Google Scholar] [CrossRef]
- Figueroa, M.; Jarmusch, A.K.; Raja, H.A.; El-Elimat, T.; Kavanaugh, J.S.; Horswill, A.R.; Cooks, R.G.; Cech, N.B.; Oberlies, N.H. Polyhydroxyanthraquinones as quorum sensing inhibitors from the guttates of Penicillium restrictum and their analysis by desorption electrospray ionization mass spectrometry. J. Nat. Prod. 2014, 77, 1351–1358. [Google Scholar] [CrossRef] [Green Version]
- Daly, S.M.; Elmore, B.O.; Kavanaugh, J.S.; Triplett, K.D.; Figueroa, M.; Raja, H.A.; El-Elimat, T.; Crosby, H.A.; Femling, J.K.; Cech, N.B.; et al. ω-Hydroxyemodin limits Staphylococcus aureus quorum sensing-mediated pathogenesis and inflammation. Antimicrob. Agents Chemother. 2015, 59, 2223–2235. [Google Scholar] [CrossRef] [Green Version]
- Parlet, C.P.; Kavanaugh, J.S.; Crosby, H.A.; Raja, H.A.; El-Elimat, T.; Todd, D.A.; Pearce, C.J.; Cech, N.B.; Oberlies, N.H.; Horswill, A.R. Apicidin attenuates MRSA virulence through quorum-sensing inhibition and enhanced host defense. Cell Rep. 2019, 27, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Ji, G.; Beavis, R.; Novick, R.P. Bacterial interference caused by autoinducing peptide variants. Science 1997, 276, 2027–2030. [Google Scholar] [CrossRef]
- Mayville, P.; Ji, G.; Beavis, R.; Yang, H.; Goger, M.; Novick, R.P.; Muir, T.W. Structure-activity analysis of synthetic autoinducing thiolactone peptides from Staphylococcus aureus responsible for virulence. Proc. Natl. Acad. Sci. USA 1999, 96, 1218–1223. [Google Scholar] [CrossRef] [Green Version]
- Tal-Gan, Y.; Stacy, D.M.; Foegen, M.K.; Koenig, D.W.; Blackwell, H.E. Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide. J. Am. Chem. Soc. 2013, 135, 7869–7882. [Google Scholar] [CrossRef] [PubMed]
- Tal-Gan, Y.; Ivancic, M.; Cornilescu, G.; Cornilescu, C.C.; Blackwell, H.E. Structural characterization of native autoinducing peptides and abiotic analogues reveals key features essential for activation and inhibition of an AgrC quorum sensing receptor in Staphylococcus aureus. J. Am. Chem. Soc. 2013, 135, 18436–18444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, J.K.; Tal-Gan, Y.; Cornilescu, G.; Tyler, K.A.; Blackwell, H.E. Simplified AIP-II peptidomimetics are potent inhibitors of Staphylococcus aureus AgrC quorum sensing receptors. ChemBioChem 2017, 18, 413–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, J.K.; Blackwell, H.E. Simplified autoinducing peptide mimetics with single-nanomolar activity against the Staphylococcus aureus AgrC quorum sensing receptor. ACS Infect. Dis. 2019, 5, 484–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwell, H.E.; Vasquez, J.K.; Tal-Gan, Y. Simplified structural mimetics of AIPS as quorum sensing inhibitors. U.S. Patent 15,850,300, 21 June 2018. [Google Scholar]
- Niu, X.; Qiu, J.; Wang, X.; Gao, X.; Dong, J.; Wang, J.; Li, H.; Zhang, Y.; Dai, X.; Lu, C.; et al. Molecular insight into the inhibition mechanism of cyrtominetin to α-hemolysin by molecular dynamics simulation. Eur. J. Med. Chem. 2013, 62, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Qiu, J.; Zhang, Y.; Lu, C.; Dai, X.; Wang, J.; Li, H.; Wang, X.; Tan, W.; Luo, M.; et al. Oroxylin a inhibits hemolysis via hindering the self-assembly of α-hemolysin heptameric transmembrane pore. PLoS Comput. Biol. 2013, 9, e1002869. [Google Scholar] [CrossRef]
- Qiu, J.; Wang, D.; Zhang, Y.; Dong, J.; Wang, J.; Niu, X. Molecular modeling reveals the novel inhibition mechanism and binding mode of three natural compounds to staphylococcal α-hemolysin. PLoS ONE 2013, 8, e80197. [Google Scholar] [CrossRef]
- Qiu, P.; Li, Y.; Shiloach, J.; Cui, X.; Sun, J.; Trinh, L.; Kubler-Kielb, J.; Vinogradov, E.; Mani, H.; Al-Hamad, M.; et al. Bacillus anthracis cell wall peptidoglycan but not lethal or edema toxins produces changes consistent with disseminated intravascular coagulation in a rat model. J. Infect. Dis. 2013, 208, 978–989. [Google Scholar] [CrossRef] [Green Version]
- Rani, N.; Saravanan, V.; Lakshmi, P.T.V.; Annamalai, A. Inhibition of pore formation by blocking the assembly of Staphylococcus aureus α-hemolysin through a novel peptide inhibitor: An in silco approach. Int. J. Pept. Res. Ther. 2014, 20, 575–583. [Google Scholar] [CrossRef]
- Ragle, B.E.; Karginov, V.A.; Bubeck Wardenburg, J. Prevention and treatment of Staphylococcus aureus pneumonia with beta-cyclodextrin derivative. Antimicrob. Agents Chemother. 2010, 54, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Karginov, V.A.; Nestorovich, E.M.; Schmidtmann, F.; Robinson, T.M.; Yohannes, A.; Fahmi, N.E.; Bezrukov, S.M.; Hecht, S.M. Inhibition of S. aureus α-hemolysin and B. anthracis lethal toxin by β-cyclodextrin derivatives. Bioorg. Med. Chem. 2007, 15, 5424–5431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, C.C.; Caballero, A.R.; Balzli, C.L.; Tang, A.; O’Callaghan, R.J. Chemical inhibition of alpha-toxin, a key corneal virulence factor of Staphylococcus aureus. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2848–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, L.R.; Merzlyak, P.G.; Valeva, A.; Krasilnikov, O.V. Interaction of heparins and dextran sulfates with a mesoscopic protein nanopore. Biophys. J. 2009, 97, 2894–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, M.C.A.; Teixeira, L.R.; Pol-Fachin, L.; Rodrigues, C.G. Inhibition of the hemolytic activity caused by Staphylococcus aureus alpha-hemolysin through isatin-Schiff copper(II) complexes. FEMS Microbiol. Lett. 2016, 363, fnv207. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.-M.; Shin, S.-H. Effect of iron-chelator deferiprone on the in vitro growth of staphylococci. J. Korean Med. Sci. 2009, 24, 289–295. [Google Scholar] [CrossRef]
- Thompson, M.G.; Corey, B.W.; Si, Y.; Craft, D.W.; Zurawski, D.V. Antibacterial activities of iron chelators against common nosocomial pathogens. Antimicrob. Agents Chemother. 2012, 56, 5419–5421. [Google Scholar] [CrossRef] [Green Version]
- Richter, K.; Thomas, N.; Claeys, J.; McGuane, J.; Prestidge, C.A.; Coenye, T.; Wormald, P.-J.; Vreugde, S. A topical hydrogel with deferiprone and gallium-protoporphyrin targets bacterial iron metabolism and has antibiofilm activity. Antimicrob. Agents Chemother. 2017, 61, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Richter, K.; Thomas, N.; Zhang, G.; Prestidge, C.A.; Coenye, T.; Wormald, P.-J.; Vreugde, S. Deferiprone and gallium-protoporphyrin have the capacity to potentiate the activity of antibiotics in Staphylococcus aureus small colony variants. Front. Cell. Infect. Microbiol. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.-J.; Zhang, M.-X.; Hider, R.C.; Zhou, T. In vitro antimicrobial activity of hydroxypyridinone hexadentate-based dendrimeric chelators alone and in combination with norfloxacin. FEMS Microbiol. Lett. 2014, 355, 124–130. [Google Scholar] [CrossRef]
- Zhou, Y.-J.; Liu, M.-S.; Osamah, A.R.; Kong, X.-L.; Alsam, S.; Battah, S.; Xie, Y.-Y.; Hider, R.C.; Zhou, T. Hexadentate 3-hydroxypyridin-4-ones with high iron(III) affinity: Design, synthesis and inhibition on methicillin resistant Staphylococcus aureus and Pseudomonas strains. Eur. J. Med. Chem. 2015, 94, 8–21. [Google Scholar] [CrossRef]
- Ang, M.T.C.; Gumbau-Brisa, R.; Allan, D.S.; McDonald, R.; Ferguson, M.J.; Holbein, B.E.; Bierenstiel, M. DIBI, a 3-hydroxypyridin-4-one chelator iron-binding polymer with enhanced antimicrobial activity. Medchemcomm 2018, 9, 1206–1212. [Google Scholar] [CrossRef]
- Holbein, B.E.; Minhua, F.; Huber, A.L.; Kidby, D.K. Metal chelating compositions and methods for controlling the growth or activities of a living cell or organism. WO Patent 2012/167368, 13 December 2012. [Google Scholar]
- Li, J.; Olaleye, E.D.; Kong, X.; Zhou, T.; Ma, Y.; Jurach, J.; Al Rugaie, O.; Hider, R.C.; Zhang, G.; Alsam, S.; et al. Macromolecular iron-chelators via RAFT-polymerization for the inhibition of methicillin-resistant Staphylococcus aureus growth. Polymer (Guildf). 2016, 87, 64–72. [Google Scholar] [CrossRef]
- Del Carmen Parquet, M.; Savage, K.A.; Allan, D.S.; Davidson, R.J.; Holbein, B.E. Novel iron-chelator DIBI inhibits Staphylococcus aureus growth, suppresses experimental MRSA infection in mice and enhances the activities of diverse antibiotics in vitro. Front. Microbiol. 2018, 9, 1811. [Google Scholar] [CrossRef]
- Workman, D.G.; Hunter, M.; Dover, L.G.; Tétard, D. Synthesis of novel iron(III) chelators based on triaza macrocycle backbone and 1-hydroxy-2(H)-pyridin-2-one coordinating groups and their evaluation as antimicrobial agents. J. Inorg. Biochem. 2016, 160, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, V.; Pramanik, A.; Gwinner, T.; Köberle, M.; Bohn, E. Sideromycins: Tools and antibiotics. BioMetals 2009, 22, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Górska, A.; Sloderbach, A.; Marszałł, M.P. Siderophore–drug complexes: Potential medicinal applications of the ‘Trojan horse’ strategy. Trends Pharmacol. Sci. 2014, 35, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Möllmann, U.; Heinisch, L.; Bauernfeind, A.; Köhler, T.; Ankel-Fuchs, D. Siderophores as drug delivery agents: Application of the “Trojan Horse” strategy. BioMetals 2009, 22, 615–624. [Google Scholar] [CrossRef]
- Reynolds, D.M.; Schatz, A.; Waksman, S.A. Grisein, a new antibiotic produced by a strain of Streptomyces griseus. Exp. Biol. Med. 1947, 64, 50–54. [Google Scholar] [CrossRef]
- Benz, G.; Schröder, T.; Kurz, J.; Wünsche, C.; Karl, W.; Steffens, G.; Pfitzner, J.; Schmidt, D. Constitution of the deferriform of the albomycins δ1, δ2, and ε. Angew. Chemie Int. Ed. English 1982, 21, 527–528. [Google Scholar] [CrossRef]
- Gause, G.F. Recent studies on albomycin, a new antibiotic. Br. Med. J. 1955, 2, 1177–1179. [Google Scholar] [CrossRef] [Green Version]
- Vértesy, L.; Aretz, W.; Fehlhaber, H.-W.; Kogler, H. Salmycin A-D, antibiotika aus Streptomyces violaceus, DSM 8286, mit siderophor-aminoglycosid-struktur. Helv. Chim. Acta 1995, 78, 46–60. [Google Scholar] [CrossRef]
- Bunet, R.; Brock, A.; Rexer, H.-U.; Takano, E. Identification of genes involved in siderophore transport in Streptomyces coelicolor A3(2). FEMS Microbiol. Lett. 2006, 262, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vértesy, L.; Aretz, W.; Fehlhaber, H.-W.; Ganguli, B.N. Salmycins, a process for their preparation and their use as a pharmaceutical. U.S. patent 5,475,094, 12 December 1995. [Google Scholar]
- Vértesy, L.; Aretz, W.; Fehlhaber, H.-W. Chelating agents, their preparation from the antibiotics salmycin A, B, C or D, and their use. U.S. patent 5,519,123, 21 May 1996. [Google Scholar]
- Ballouche, M.; Cornelis, P.; Baysse, C. Iron metabolism: A promising target for antibacterial strategies. Recent Pat. Antiinfect. Drug Discov. 2009, 4, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Wencewicz, T.A.; Long, T.E.; Möllmann, U.; Miller, M.J. Trihydroxamate siderophore–fluoroquinolone conjugates are selective sideromycin antibiotics that target Staphylococcus aureus. Bioconjug. Chem. 2013, 24, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Braun, V. Active transport of siderophore-mimicking antibacterials across the outer membrane. Drug Resist. Updat. 1999, 2, 363–369. [Google Scholar] [CrossRef]
- Pugsley, A.P.; Zimmerman, W.; Wehrli, W. Highly efficient uptake of a rifamycin derivative via the FhuA-TonB-dependent uptake route in Escherichia coli. Microbiology 1987, 133, 3505–3511. [Google Scholar] [CrossRef] [Green Version]
- Negash, K.H.; Norris, J.K.S.; Hodgkinson, J.T. Siderophore–antibiotic conjugate design: New drugs for bad bugs? Molecules 2019, 24, 3314. [Google Scholar] [CrossRef] [Green Version]
- Neumann, W.; Sassone-Corsi, M.; Raffatellu, M.; Nolan, E.M. Esterase-catalyzed siderophore hydrolysis activates an enterobactin–ciprofloxacin conjugate and confers targeted antibacterial activity. J. Am. Chem. Soc. 2018, 140, 5193–5201. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-M.; Ghosh, M.; Miller, P.A.; Möllmann, U.; Miller, M.J. Synthetic sideromycins (skepticism and optimism): Selective generation of either broad or narrow spectrum gram-negative antibiotics. BioMetals 2019, 32, 425–451. [Google Scholar] [CrossRef]
- Kong, H.; Cheng, W.; Wei, H.; Yuan, Y.; Yang, Z.; Zhang, X. An overview of recent progress in siderophore-antibiotic conjugates. Eur. J. Med. Chem. 2019, 182, 111615. [Google Scholar] [CrossRef]
- Zähner, H.; Diddens, H.; Keller-Schierlein, W.; Nägeli, H.U. Some experiments with semisynthetic sideromycins. Jpn. J. Antibiot. 1977, 30 Suppl, 201–206. Available online: https://europepmc.org/article/med/612705 (accessed on 17 March 2020).
- Ghosh, M.; Miller, M.J. Synthesis and in vitro antibacterial activity of spermidine-based mixed catechol- and hydroxamate-containing siderophore-vancomycin conjugates. Bioorganic Med. Chem. 1996, 4, 43–48. [Google Scholar] [CrossRef]
- Ghosh, M.; Miller, M.J. Design, synthesis, and biological evaluation of isocyanurate-based antifungal and macrolide antibiotic conjugates: Iron transport-mediated drug delivery. Bioorg. Med. Chem. 1995, 3, 1519–1525. [Google Scholar] [CrossRef]
- Milner, S.J.; Seve, A.; Snelling, A.M.; Thomas, G.H.; Kerr, K.G.; Routledge, A.; Duhme-Klair, A.-K. Staphyloferrin A as siderophore-component in fluoroquinolone-based Trojan horse antibiotics. Org. Biomol. Chem. 2013, 11, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Miller, M.J. Chemical syntheses and in vitro antibacterial activity of two desferrioxamine B-ciprofloxacin conjugates with potential esterase and phosphatase triggered drug release linkers. Bioorg. Med. Chem. 2012, 20, 3828–3836. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.; Miller, M.J. Siderophore–fluoroquinolone conjugates containing potential reduction-triggered linkers for drug release: Synthesis and antibacterial activity. BioMetals 2015, 28, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, L.S.; Britigan, B.E. Gallium-containing compounds for the treatment of infections caused by intracellular pathogens and pathogens causing chronic pulmunary infection. U.S. Patent 6,203,822, 20 March 2001. [Google Scholar]
- DeLeon, K.; Balldin, F.; Watters, C.; Hamood, A.; Griswold, J.; Sreedharan, S.; Rumbaugh, K.P. Gallium maltolate treatment eradicates Pseudomonas aeruginosa infection in thermally injured mice. Antimicrob. Agents Chemother. 2009, 53, 1331–1337. [Google Scholar] [CrossRef] [Green Version]
- Bonchi, C.; Imperi, F.; Minandri, F.; Visca, P.; Frangipani, E. Repurposing of gallium-based drugs for antibacterial therapy. BioFactors 2014, 40, 303–312. [Google Scholar] [CrossRef]
- Rangel-Vega, A.; Bernstein, L.R.; Mandujano-Tinoco, E.A.; Garcí a-Contreras, S.J.; Garcí a-Contreras, R. Drug repurposing as an alternative for the treatment of recalcitrant bacterial infections. Front. Microbiol. 2015, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Baldoni, D.; Steinhuber, A.; Zimmerli, W.; Trampuz, A. In vitro activity of gallium maltolate against staphylococci in logarithmic, stationary, and biofilm growth phases: Comparison of conventional and calorimetric susceptibility testing methods. Antimicrob. Agents Chemother. 2010, 54, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Kelson, A.B.; Carnevali, M.; Truong-Le, V. Gallium-based anti-infectives: Targeting microbial iron-uptake mechanisms. Curr. Opin. Pharmacol. 2013, 13, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, L.R. Mechanisms of therapeutic activity for gallium. Pharmacol. Rev. 1998, 50, 665–682. [Google Scholar] [PubMed]
- Emery, T. Exchange of iron by gallium in siderophores. Biochemistry 1986, 25, 4629–4633. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Thoendel, M.; Olakanmi, O.; Britigan, B.E.; Singh, P.K. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J. Clin. Investig. 2007, 117, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Van den Driessche, F.; Coenye, T. Innovative approaches to treat Staphylococcus aureus biofilm-related infections. Essays Biochem. 2017, 61, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minandri, F.; Bonchi, C.; Frangipani, E.; Imperi, F.; Visca, P. Promises and failures of gallium as an antibacterial agent. Future Microbiol. 2014, 9, 379–397. [Google Scholar] [CrossRef]
- Bernstein, L.R.; Tanner, T.; Godfrey, C.; Noll, B. Chemistry and pharmacokinetics of gallium maltolate, a compound with high oral gallium bioavailability. Met. Based. Drugs 2000, 7, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.M.; Gardner, R.; Kaur, N.; Phanstiel, O. Utilization of Fe3+-acinetoferrin analogs as an iron source by Mycobacterium tuberculosis. BioMetals 2008, 21, 93–103. [Google Scholar] [CrossRef]
- Stojiljkovic, I.; Kumar, V.; Srinivasan, N. Non-iron metalloporphyrins: Potent antibacterial compounds that exploit haem/Hb uptake systems of pathogenic bacteria. Mol. Microbiol. 1999, 31, 429–442. [Google Scholar] [CrossRef]
- Stojiljkovic, I.; Churchward, G.G. Non-iron metalloporphyrins and methods of use. U.S. Patent 6,066,628, 23 May 2000. [Google Scholar]
- Richter, K.; Ramezanpour, M.; Thomas, N.; Prestidge, C.A.; Wormald, P.-J.; Vreugde, S. Mind “De GaPP”: In vitro efficacy of deferiprone and gallium-protoporphyrin against Staphylococcus aureus biofilms. Int. Forum Allergy Rhinol. 2016, 6, 737–743. [Google Scholar] [CrossRef]
- Chang, D.; Garcia, R.; Akers, K.; Mende, K.; Murray, C.; Wenke, J.; Sanchez Jr., C. J. Activity of gallium meso- and protoporphyrin IX against biofilms of multidrug-resistant Acinetobacter baumannii isolates. Pharmaceuticals 2016, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Grunenwald, C.M.; Bennett, M.R.; Skaar, E.P. Nonconventional therapeutics against Staphylococcus aureus. Microbiol. Spectr. 2018, 6, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, S.; Visaggio, D.; Pirolo, M.; Frangipani, E.; Bernstein, L.R.; Visca, P. Antimicrobial activity of gallium compounds on ESKAPE pathogens. Front. Cell. Infect. Microbiol. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.G.; Truong-Le, V.; Alamneh, Y.A.; Black, C.C.; Anderl, J.; Honnold, C.L.; Pavlicek, R.L.; Abu-Taleb, R.; Wise, M.C.; Hall, E.R.; et al. Evaluation of gallium citrate formulations against a multidrug-resistant strain of Klebsiella pneumoniae in a murine wound model of infection. Antimicrob. Agents Chemother. 2015, 59, 6484–6493. [Google Scholar] [CrossRef] [Green Version]
- Hijazi, S.; Visca, P.; Frangipani, E. Gallium-protoporphyrin IX inhibits Pseudomonas aeruginosa growth by targeting cytochromes. Front. Cell. Infect. Microbiol. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wakeman, C.A.; Stauff, D.L.; Zhang, Y.; Skaar, E.P. Differential activation of Staphylococcus aureus heme detoxification machinery by heme analogues. J. Bacteriol. 2014, 196, 1335–1342. [Google Scholar] [CrossRef] [Green Version]
- Soares Lopes, L.Q.; Ramos, A.P.; Copetti, P.M.; Acunha, T.V.; Iglesias, B.A.; Vianna Santos, R.C.; Machado, A.K.; Sagrillo, M.R. Antimicrobial activity and safety applications of meso-tetra(4-pyridyl)platinum(II) porphyrin. Microb. Pathog. 2019, 128, 47–54. [Google Scholar] [CrossRef]
- Managa, M.; Nyokong, T. Photodynamic antimicrobial chemotherapy activity of gallium tetra-(4-carboxyphenyl) porphyrin when conjugated to differently shaped platinum nanoparticles. J. Mol. Struct. 2015, 1099, 432–440. [Google Scholar] [CrossRef]
- Gianquinto, E.; Moscetti, I.; De Bei, O.; Campanini, B.; Marchetti, M.; Luque, F.J.; Cannistraro, S.; Ronda, L.; Bizzarri, A.R.; Spyrakis, F.; et al. Interaction of human hemoglobin and semi-hemoglobins with the Staphylococcus aureus hemophore IsdB: A kinetic and mechanistic insight. Sci. Rep. 2019, 9, 18629. [Google Scholar] [CrossRef] [Green Version]
- Ellis-Guardiola, K.; Clayton, J.; Pham, C.; Mahoney, B.J.; Wereszczynski, J.; Clubb, R.T. The Staphylococcus aureus IsdH receptor forms a dynamic complex with human hemoglobin that triggers heme release via two distinct hot spots. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Conger, M.A.; Pokhrel, D.; Liptak, M.D. Tight binding of heme to Staphylococcus aureus IsdG and IsdI precludes design of a competitive inhibitor. Metallomics 2017, 9, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascioferro, S.; Raffa, D.; Maggio, B.; Raimondi, M.V.; Schillaci, D.; Daidone, G. Sortase A inhibitors: Recent advances and future perspectives. J. Med. Chem. 2015, 58, 9108–9123. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, X.; Sun, L.; Gao, Y.; Niu, X.; Wang, H. Novel inhibitor discovery of Staphylococcus aureus sortase B and the mechanism confirmation via molecular modeling. Molecules 2018, 23, 977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Gao, Y.; Wang, H.; Niu, X.; Wang, J. Baicalin weakens Staphylococcus aureus pathogenicity by targeting sortase B. Front. Cell. Infect. Microbiol. 2018, 8, 418. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Schofield, M.M.; Chlipala, G.E.; Schultz, P.J.; Yim, I.; Newmister, S.A.; Nusca, T.D.; Scaglione, J.B.; Hanna, P.C.; Tamayo-Castillo, G.; et al. Baulamycins A and B, broad-spectrum antibiotics identified as inhibitors of siderophore biosynthesis in Staphylococcus aureus and Bacillus anthracis. J. Am. Chem. Soc. 2014, 136, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Neres, J.; Labello, N.P.; Somu, R.V.; Boshoff, H.I.; Wilson, D.J.; Vannada, J.; Chen, L.; Barry, C.E.; Bennett, E.M.; Aldrich, C.C. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: Structure−activity relationships of the nucleobase domain of 5′-O-[N -(Salicyl)sulfamoyl]adenosine. J. Med. Chem. 2008, 51, 5349–5370. [Google Scholar] [CrossRef] [Green Version]
- Ferreras, J.A.; Ryu, J.-S.; Di Lello, F.; Tan, D.S.; Quadri, L.E.N. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 2005, 1, 29–32. [Google Scholar] [CrossRef]
- Gupte, A.; Boshoff, H.I.; Wilson, D.J.; Neres, J.; Labello, N.P.; Somu, R.V.; Xing, C.; Barry, C.E.; Aldrich, C.C. Inhibition of siderophore biosynthesis by 2-triazole substituted analogues of 5′-O-[N-(Salicyl)sulfamoyl]adenosine: Antibacterial nucleosides effective against Mycobacterium tuberculosis. J. Med. Chem. 2008, 51, 7495–7507. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, C.L.; Conger, M.A.; Pittman, D.S.; Liptak, M.D. Hydrogen bond donation to the heme distal ligand of Staphylococcus aureus IsdG tunes the electronic structure. JBIC J. Biol. Inorg. Chem. 2015, 20, 757–770. [Google Scholar] [CrossRef]
- Redi, D.; Raffaelli, C.S.; Rossetti, B.; De Luca, A.; Montagnani, F. Staphylococcus aureus vaccine preclinical and clinical development: Current state of the art. New Microbiol. 2018, 41, 208–213. [Google Scholar]
- Kuklin, N.A.; Clark, D.J.; Secore, S.; Cook, J.; Cope, L.D.; McNeely, T.B.; Noble, L.; Brown, M.J.; Zorman, J.K.; Wang, X.M.; et al. A novel Staphylococcus aureus vaccine: Iron surface determinant B induces rapid antibody responses in Rhesus macaques and specific increased survival in a murine S. aureus sepsis model. Infect. Immun. 2006, 74, 2215–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etz, H.; Minh, D.B.; Henics, T.; Dryla, A.; Winkler, B.; Triska, C.; Boyd, A.P.; Sollner, J.; Schmidt, W.; Von Ahsen, U.; et al. Identification of in vivo expressed vaccine candidate antigens from Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2002, 99, 6573–6578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harro, C.D.; Betts, R.F.; Orenstein, W.; Kwak, E.-J.; Greenberg, H.E.; Onorato, M.T.; Hartzel, J.S.; Lipka, J.; DiNubile, M.J.; Kartsonis, N.A. Safety and immunogenicity of a novel Staphylococcus aureus vaccine: Results from the first study of the vaccine dose range in humans. Clin. Vaccine Immunol. 2010, 17, 1868–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harro, C.D.; Betts, R.F.; Hartzel, J.S.; Onorato, M.T.; Lipka, J.; Smugar, S.S.; Kartsonis, N.A. The immunogenicity and safety of different formulations of a novel Staphylococcus aureus vaccine (V710): Results of two phase I studies. Vaccine 2012, 30, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, M.; Aronoff, G.R.; Chandran, C.; Hartzel, J.S.; Smugar, S.S.; Galphin, C.M.; Mailloux, L.U.; Brown, E.; DiNubile, M.J.; Kartsonis, N.A.; et al. Phase IIa study of the immunogenicity and safety of the novel Staphylococcus aureus vaccine V710 in adults with end-stage renal disease receiving hemodialysis. Clin. Vaccine Immunol. 2012, 19, 1509–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, V.G.; Allen, K.B.; Moreira, E.D.; Moustafa, M.; Isgro, F.; Boucher, H.W.; Corey, G.R.; Carmeli, Y.; Betts, R.F.; Hartzel, J.S.; et al. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery. JAMA 2013, 309, 1368–1378. [Google Scholar] [CrossRef]
- Anderson, A.S.; Miller, A.A.; Donald, R.G.K.; Scully, I.L.; Nanra, J.S.; Cooper, D.; Jansen, K.U. Development of a multicomponent Staphylococcus aureus vaccine designed to counter multiple bacterial virulence factors. Hum. Vaccin. Immunother. 2012, 8, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- McNeely, T.B.; Shah, N.A.; Fridman, A.; Joshi, A.; Hartzel, J.S.; Keshari, R.S.; Lupu, F.; DiNubile, M.J. Mortality among recipients of the Merck V710 Staphylococcus aureus vaccine after postoperative S. aureus infections: An analysis of possible contributing host factors. Hum. Vaccin. Immunother. 2014, 10, 3513–3516. [Google Scholar] [CrossRef] [Green Version]
- Paling, F.P.; Olsen, K.; Ohneberg, K.; Wolkewitz, M.; Fowler, V.G.; DiNubile, M.J.; Jafri, H.S.; Sifakis, F.; Bonten, M.J.M.; Harbarth, S.J.; et al. Risk prediction for Staphylococcus aureus surgical site infection following cardiothoracic surgery; a secondary analysis of the V710-P003 trial. PLoS ONE 2018, 13, e0193445. [Google Scholar] [CrossRef]
- O’Brien, E.C.; McLoughlin, R.M. Considering the ‘alternatives’ for next-generation anti-Staphylococcus aureus vaccine development. Trends Mol. Med. 2019, 25, 171–184. [Google Scholar] [CrossRef]
- Stranger-Jones, Y.K.; Bae, T.; Schneewind, O. Vaccine assembly from surface proteins of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2006, 103, 16942–16947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.R.; Bombardi, R.G.; Kose, N.; Parrish, E.H.; Nagel, M.B.; Petit, R.A.; Read, T.D.; Schey, K.L.; Thomsen, I.P.; Skaar, E.P.; et al. Human mAbs to Staphylococcus aureus IsdA provide protection through both heme-blocking and Fc-mediated mechanisms. J. Infect. Dis. 2019, 219, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.A.; Foletti, D.; Deng, X.; Abdiche, Y.; Strop, P.; Glanville, J.; Pitts, S.; Lindquist, K.; Sundar, P.D.; Sirota, M.; et al. Germline-encoded neutralization of a Staphylococcus aureus virulence factor by the human antibody repertoire. Nat. Commun. 2016, 7, 13376. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Dong, J.; Bombardi, R.G.; Soto, C.; Parrington, H.M.; Nargi, R.S.; Schoeder, C.T.; Nagel, M.B.; Schey, K.L.; Meiler, J.; et al. Human VH1-69 gene-encoded human monoclonal antibodies against Staphylococcus aureus IsdB use at least three distinct modes of binding to inhibit bacterial growth and pathogenesis. MBio 2019, 10, e02473-19. [Google Scholar] [CrossRef] [Green Version]
- Bacconi, M.; Haag, A.F.; Chiarot, E.; Donato, P.; Bagnoli, F.; Delany, I.; Bensi, G. In vivo analysis of Staphylococcus aureus-infected mice reveals differential temporal and spatial expression patterns of fhuD2. Infect. Immun. 2017, 85, e00270-17. [Google Scholar] [CrossRef] [Green Version]
- Irene, C.; Fantappiè, L.; Caproni, E.; Zerbini, F.; Anesi, A.; Tomasi, M.; Zanella, I.; Stupia, S.; Prete, S.; Valensin, S.; et al. Bacterial outer membrane vesicles engineered with lipidated antigens as a platform for Staphylococcus aureus vaccine. Proc. Natl. Acad. Sci. USA 2019, 116, 21780–21788. [Google Scholar] [CrossRef] [Green Version]
- Bagnoli, F.; Fiaschi, L.; Grandi, G.; Scarselli, M. Immunising against Staphylococcus aureus. WO Patent 2015/144655, 1 October 2015. [Google Scholar]
- Ragle, B.E.; Bubeck Wardenburg, J. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 2009, 77, 2712–2718. [Google Scholar] [CrossRef] [Green Version]
- Hua, L.; Hilliard, J.J.; Shi, Y.; Tkaczyk, C.; Cheng, L.I.; Yu, X.; Datta, V.; Ren, S.; Feng, H.; Zinsou, R.; et al. Assessment of an anti-alpha-toxin monoclonal antibody for prevention and treatment of Staphylococcus aureus-induced pneumonia. Antimicrob. Agents Chemother. 2014, 58, 1108–1117. [Google Scholar] [CrossRef] [Green Version]
- Tkaczyk, C.; Hua, L.; Varkey, R.; Shi, Y.; Dettinger, L.; Woods, R.; Barnes, A.; MacGill, R.S.; Wilson, S.; Chowdhury, P.; et al. Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin. Vaccine Immunol. 2012, 19, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Foletti, D.; Strop, P.; Shaughnessy, L.; Hasa-Moreno, A.; Casas, M.G.; Russell, M.; Bee, C.; Wu, S.; Pham, A.; Zeng, Z.; et al. Mechanism of action and in vivo efficacy of a human-derived antibody against Staphylococcus aureus α-hemolysin. J. Mol. Biol. 2013, 425, 1641–1654. [Google Scholar] [CrossRef]
- Magyarics, Z.; Leslie, F.; Bartko, J.; Rouha, H.; Luperchio, S.; Schörgenhofer, C.; Schwameis, M.; Derhaschnig, U.; Lagler, H.; Stiebellehner, L.; et al. Randomized, double-blind, placebo-controlled, single-ascending-dose study of the penetration of a monoclonal antibody combination (ASN100) targeting Staphylococcus aureus cytotoxins in the lung epithelial lining fluid of healthy volunteers. Antimicrob. Agents Chemother. 2019, 63, e00350-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aman, M.J. Integrated biotherapeutics. Hum. Vaccin. Immunother. 2018, 14, 1308–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Combination effects of antimicrobial peptides. Antimicrob. Agents Chemother. 2016, 60, 1717–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- François, B.; Mercier, E.; Gonzalez, C.; Asehnoune, K.; Nseir, S.; Fiancette, M.; Desachy, A.; Plantefève, G.; Meziani, F.; de Lame, P.-A.; et al. Safety and tolerability of a single administration of AR-301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: First-in-human trial. Intensive Care Med. 2018, 44, 1787–1796. [Google Scholar] [CrossRef]
- Rouha, H.; Badarau, A.; Visram, Z.C.; Battles, M.B.; Prinz, B.; Magyarics, Z.; Nagy, G.; Mirkina, I.; Stulik, L.; Zerbs, M.; et al. Five birds, one stone: Neutralization of α-hemolysin and 4 bi-component leukocidins of Staphylococcus aureus with a single human monoclonal antibody. MAbs 2015, 7, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Jangra, P.; Singh, A. Staphylococcus aureus β-hemolysin-neutralizing single-domain antibody isolated from phage display library of Indian desert camel. Asian Pac. J. Trop. Med. 2010, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Laventie, B.-J.; Rademaker, H.J.; Saleh, M.; de Boer, E.; Janssens, R.; Bourcier, T.; Subilia, A.; Marcellin, L.; van Haperen, R.; Lebbink, J.H.G.; et al. Heavy chain-only antibodies and tetravalent bispecific antibody neutralizing Staphylococcus aureus leukotoxins. Proc. Natl. Acad. Sci. USA 2011, 108, 16404–16409. [Google Scholar] [CrossRef] [Green Version]






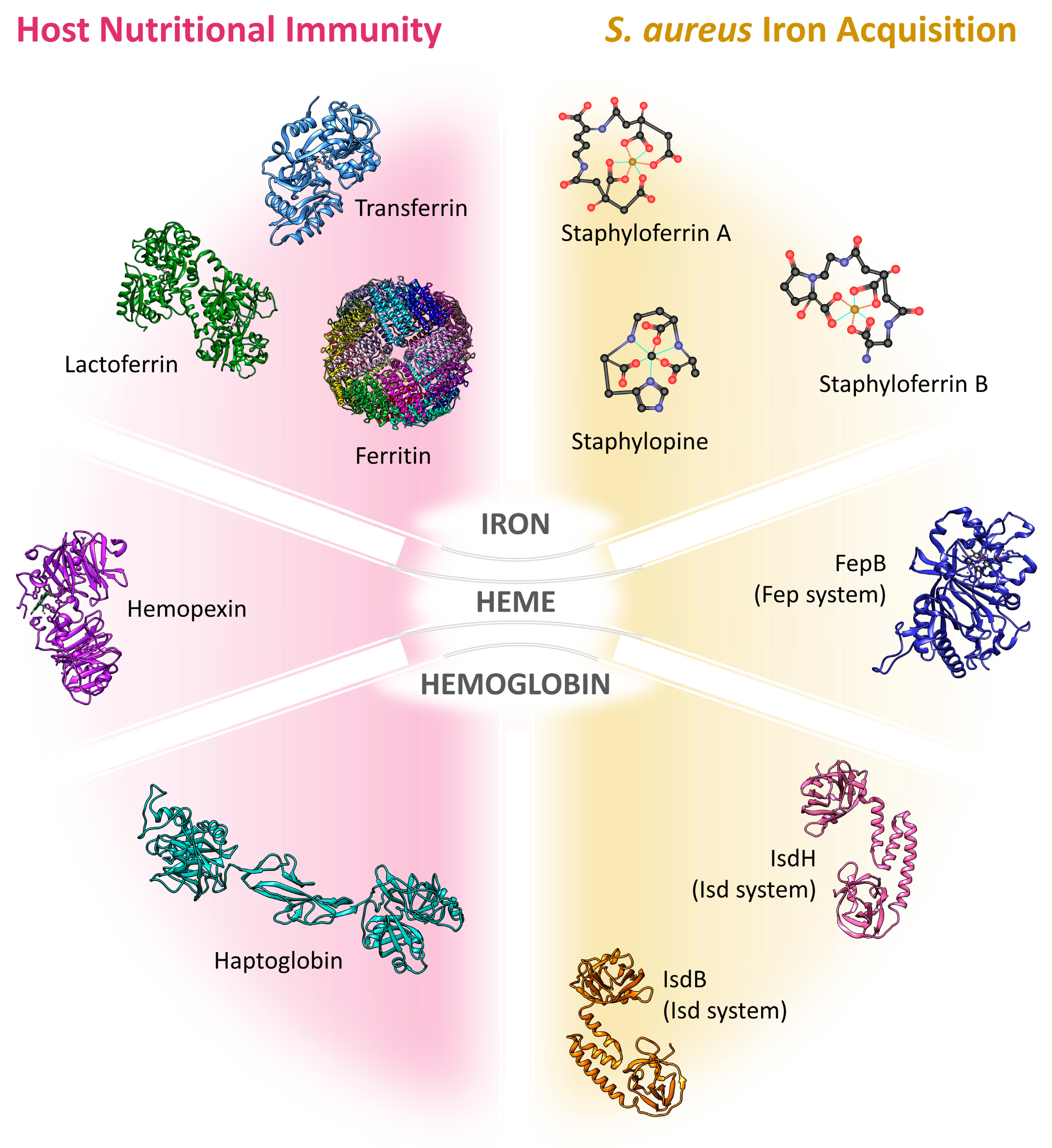{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Codes | Protein | Reference |
|---|---|---|
| 3lhs, 3li2 | HtsA | [57] |
| 3eiw, 3eix | HtsA | [58] |
| 5d84, 5d85 | SbnA | [59] |
| 4m54, 4mp3, 4mp6, 4mp8, 4mpd | SbnB | [60] |
| 4tv5 | SbnG | [61] |
| 5uje | SbnI | [62] |
| 6knh and 6kni | SbnH | [63] |
| 3mwf, 3mwg | SirA | [64] |
| 4fna, 4fil | FhuD2 | [65] |
| 4b8y | FhuD2 | [66] |
| 5twb, 5twc | IruO | [67] |
| 7ahl | Hla | [68] |
| 3b07 | Hlg | [69] |
| 4q7g | LukD | [70] |
| 3roh | LukE | [70] |
| 3i41 | β-toxin | [71] |
| 2kam | δ-toxin | [72] |
| 1t2p, 1t2w | SrtA | [73] |
| 1ng5 | SrtB | [74] |
| 4lfd | SrtB | [75] |
| 4xs0 | IsdH-Hb | [76] |
| 5vmm | IsdB-Hb | [77] |
| 6tb2 | IsdH-Hb-Hp | [78] |
| 2ite, 2itf | IsdA | [79] |
| 2o6p | IsdC | [80] |
| 2q8q | IsdE | [81] |
| 1xbw | IsdG | [82] |
| 3lgn | IsdI | [83] |
| System | Gene Cluster | Regulation | Strain | Deletion | Gene Contribution to Virulence in Vivo | Mouse Model of Infection | Evaluated District of Infection | Reference |
|---|---|---|---|---|---|---|---|---|
| Heme acquisition | ||||||||
| Sortases | srtA | Constitutive | Newman | ΔsrtA | Yes | C57BL/6, Swiss-Webster (intravenous) | Kidney | [183] |
| isdC-FsrtBisdG | Fur | Newman | ΔsrtA | Yes | NMRI (intra-articular) | Joints, kidney, blood | [184] | |
| Newman, USA300 | ΔsrtA | Yes | BALB/c (intravenous) | Kidney | [150] | |||
| Newman D2C | ΔsrtA | Yes | BALB/c (mammary injection) | Mammary glands | [185] | |||
| Newman | ΔsrtA, ΔsrtA-srtB | Yes | CD-1 (intraperitoneal), | Systemic, joints, | [186] | |||
| NMRI (intravenous), C3H/HeJ (bladder), Sprague-Dawley rats (intravenous) | kidney, heart | |||||||
| Newman | ΔsrtA, ΔsrtA-srtB | Yes | NMRI (intravenous) | Joints, kidney | [187] | |||
| Newman | ΔsrtB | Mild | Swiss-Webster (intravenous) | Kidney | [142] | |||
| Newman | ΔsrtB | Mild | NMRI (intravenous) | Joints, kidney | [187] | |||
| Newman | ΔsrtB | Mild | CD-1 (intraperitoneal), NMRI (intravenous), C3H/HeJ (bladder), Sprague-Dawley rats (intravenous) | Systemic, joints, kidney, heart | [186] | |||
| Isd | isdA | Fur | Newman | ΔisdBH, ΔhtsA-isdE | No | BALB/c (intranasal) | Lung | [188] |
| isdB | Fur | Newman | ΔhtsA-isdE | Yes | BALB/c (retro-orbital) | Lung, heart, kidney | [188] | |
| isdC-FsrtBisdG | Fur | Newman | ΔisdB, ΔisdA | Yes | BALB/c (intravenous) | Kidney | [150] | |
| orfXisdI | Fur | Newman | ΔisdC | Mild | BALB/c (intravenous) | Kidney | [150] | |
| isdH | Fur | Newman | ΔisdH | No | BALB/c (intravenous) | Kidney | [150] | |
| Newman | ΔisdB | Yes | C57BL/6J (retro-orbital) | Heart | [149] | |||
| Newman | ΔisdB | No | C57BL/6J (retro-orbital) | Liver | [149] | |||
| Newman | ΔisdA, ΔisdB, ΔisdC | Yes | BALB/c (retro-orbital) | Kidney | [151] | |||
| Newman | ΔisdH | No | BALB/c (retro-orbital) | Kidney | [151] | |||
| Newman | ΔisdG, ΔisdI, | Yes | BALB/c (retro-orbital) | Heart | [159] | |||
| Newman | ΔisdG-I | No | BALB/c (retro-orbital) | Kidney | [159] | |||
| Newman | ΔisdI | Yes | BALB/c (retro-orbital) | Kidney | [159] | |||
| Newman | ΔisdG, ΔisdG-I | No | BALB/c (retro-orbital) | Liver | [159] | |||
| Newman | ΔisdG, ΔisdI, | Mild | BALB/c (retro-orbital) | Kidney, spleen | [56] | |||
| Newman | ΔisdG-I | Yes | BALB/c (retro-orbital) | Kidney, spleen | [56] | |||
| 8325-4 | ΔisdH | Yes | NMRI (intravenous) | Blood | [165] | |||
| ΔisdB, ΔisdB-isdH | ||||||||
| ΔisdH | ||||||||
| Hss | hssRS | Constitutive, activated by heme | Newman | ΔhssR | Mutation increase virulence | BALB/c (retro-orbital) | Liver | [175] |
| Newman | ΔhssR | No | BALB/c (retro-orbital) | Spleen, kidney | [175] | |||
| Hrt | hrtAB | HssRS | Newman | ΔhrtA | Mutation increase virulence | BALB/c (retro-orbital) | Liver | [175] |
| Newman | ΔhrtA | No | BALB/c (retro-orbital) | Spleen, kidney | [175] | |||
| Endogenous siderophores | ||||||||
| Staphyloferrin A | sfaABC | Fur | Newman | Δsfa | Yes | BALB/c (subcutaneous) | Skin | [88] |
| sfaD | Fur | Newman | Δsfa-sbn | Yes | BALB/c (subcutaneous) | Skin | [88] | |
| htsABC | Fur | |||||||
| MW2 | ΔsfaA | Yes | Swiss-Webster (intravenous) | Kidney | [89] | |||
| Newman | ΔhtsA-isdE | No | BALB/c (intranasal) | Lung | [188] | |||
| Newman | ΔhtsA-isdE | Yes | BALB/c (retro-orbital) | Lung, heart, kidney | [188] | |||
| Newman | ΔhtsB, ΔhtsC | Yes | BALB/c (intravenous) | Kidney, liver | [53] | |||
| Staphyloferrin B | sbnA-I | Fur, SbnI | Newman | ΔsbnE | Yes | Swiss-Webster (intravenous) | Kidney | [90] |
| sirABC | Fur | Newman | Δsbn | No | BALB/c (subcutaneous) | Skin | [99] | |
| MW2 | ΔsbnD | Yes | Swiss-Webster (intravenous) | Kidney | [89] | |||
| Newman | ΔsbnG | No | BALB/c (intravenous) | Heart, kidney, liver | [87] | |||
| Newman | ΔsbnG-citZ | Yes | BALB/c (intravenous) | Heart, kidney, liver | [87] | |||
| Newman | Δsfa-sbn, Δhts-sir, Δsfa-sbn-sst, | Yes | BALB/c (intravenous) | Heart, kidney, liver | [109] | |||
| Δhts-sir-sst | ||||||||
| Xenosiderophores | ||||||||
| Hydroxamate | fhuBGC | Fur | Newman | ΔfhuD2 | Yes | CD1 (intravenous) | Kidney, blood | [110] |
| fhuD1 | Fur | Newman | ΔfhuBGC | Yes | Swiss-Webster (intravenous) | Kidney | [189] | |
| fhuD2 | Fur | |||||||
| Catecholate | sstABCD | Fur | Newman | Δsst | Yes | BALB/c (intravenous) | Heart | [109] |
| Inorganic iron acquisition | ||||||||
| Fep | tatAC | Constitutive | RN1HG | ΔtatAC, Δtat-fep | Yes | BALB/c (intravenous) | Kidney | [170] |
| fepABC | Fur | |||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchetti, M.; De Bei, O.; Bettati, S.; Campanini, B.; Kovachka, S.; Gianquinto, E.; Spyrakis, F.; Ronda, L. Iron Metabolism at the Interface between Host and Pathogen: From Nutritional Immunity to Antibacterial Development. Int. J. Mol. Sci. 2020, 21, 2145. https://doi.org/10.3390/ijms21062145
Marchetti M, De Bei O, Bettati S, Campanini B, Kovachka S, Gianquinto E, Spyrakis F, Ronda L. Iron Metabolism at the Interface between Host and Pathogen: From Nutritional Immunity to Antibacterial Development. International Journal of Molecular Sciences. 2020; 21(6):2145. https://doi.org/10.3390/ijms21062145
Chicago/Turabian StyleMarchetti, Marialaura, Omar De Bei, Stefano Bettati, Barbara Campanini, Sandra Kovachka, Eleonora Gianquinto, Francesca Spyrakis, and Luca Ronda. 2020. "Iron Metabolism at the Interface between Host and Pathogen: From Nutritional Immunity to Antibacterial Development" International Journal of Molecular Sciences 21, no. 6: 2145. https://doi.org/10.3390/ijms21062145
APA StyleMarchetti, M., De Bei, O., Bettati, S., Campanini, B., Kovachka, S., Gianquinto, E., Spyrakis, F., & Ronda, L. (2020). Iron Metabolism at the Interface between Host and Pathogen: From Nutritional Immunity to Antibacterial Development. International Journal of Molecular Sciences, 21(6), 2145. https://doi.org/10.3390/ijms21062145