Loss of ISWI ATPase SMARCA5 (SNF2H) in Acute Myeloid Leukemia Cells Inhibits Proliferation and Chromatid Cohesion

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. SMARCA5 Overexpression Marks the Hyperproliferation and Cytogenetically Abnormal AML Patients
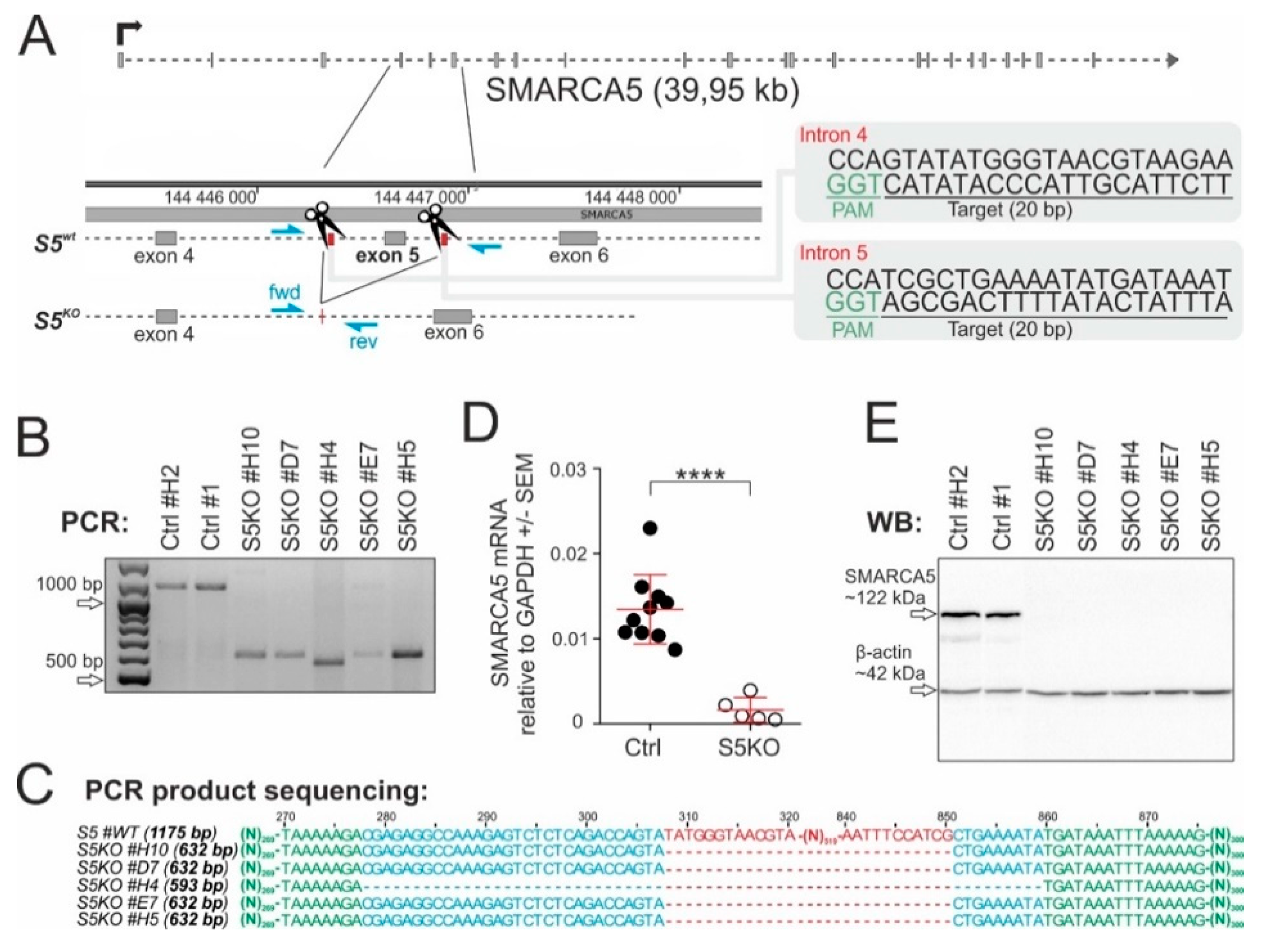
2.2. SMARCA5 Deletion Inhibits AML Cell Proliferation
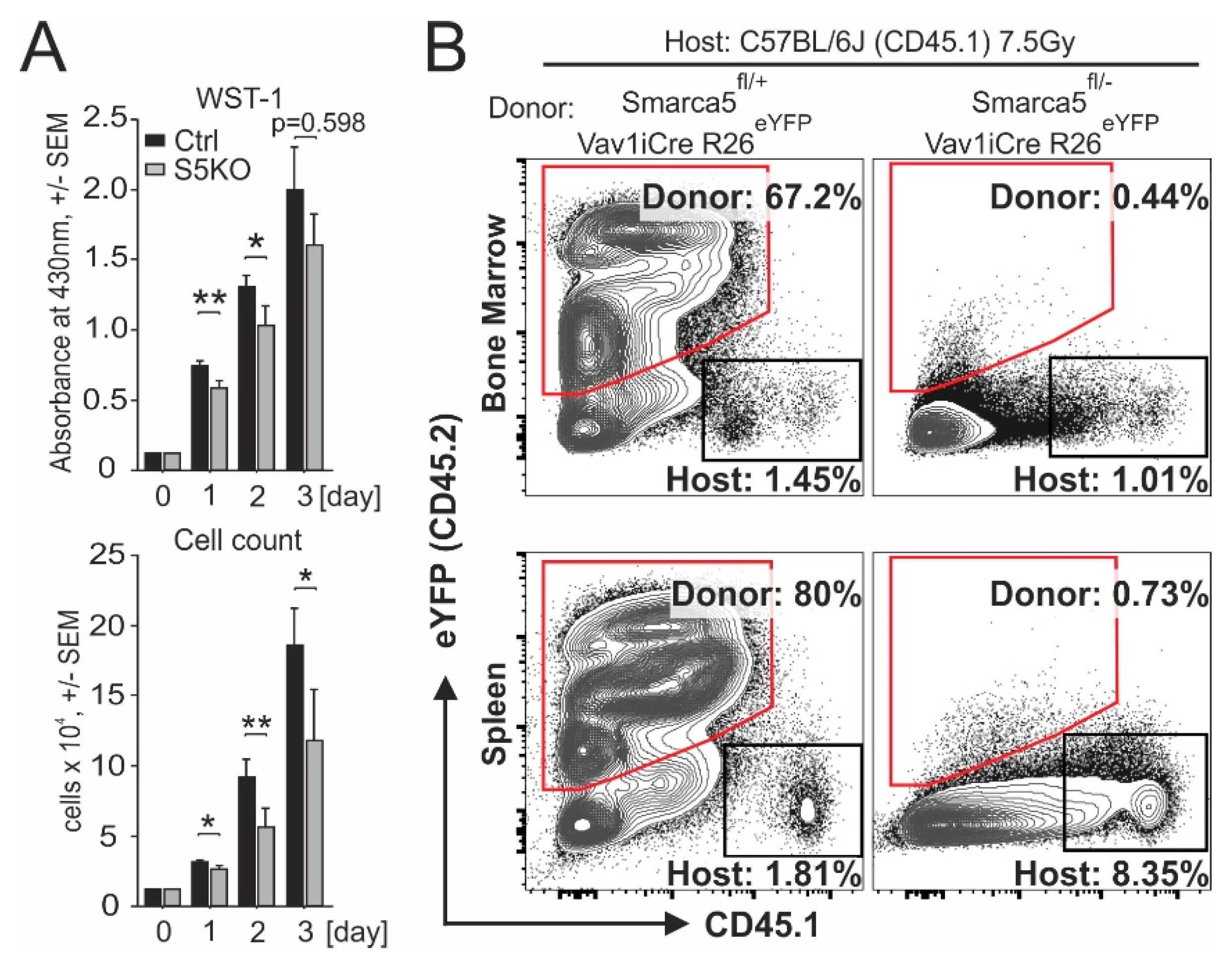
2.3. Smarca5 Deletion Inhibits Proliferation of Myeloblasts and Affects Function of Normal Stem Cells
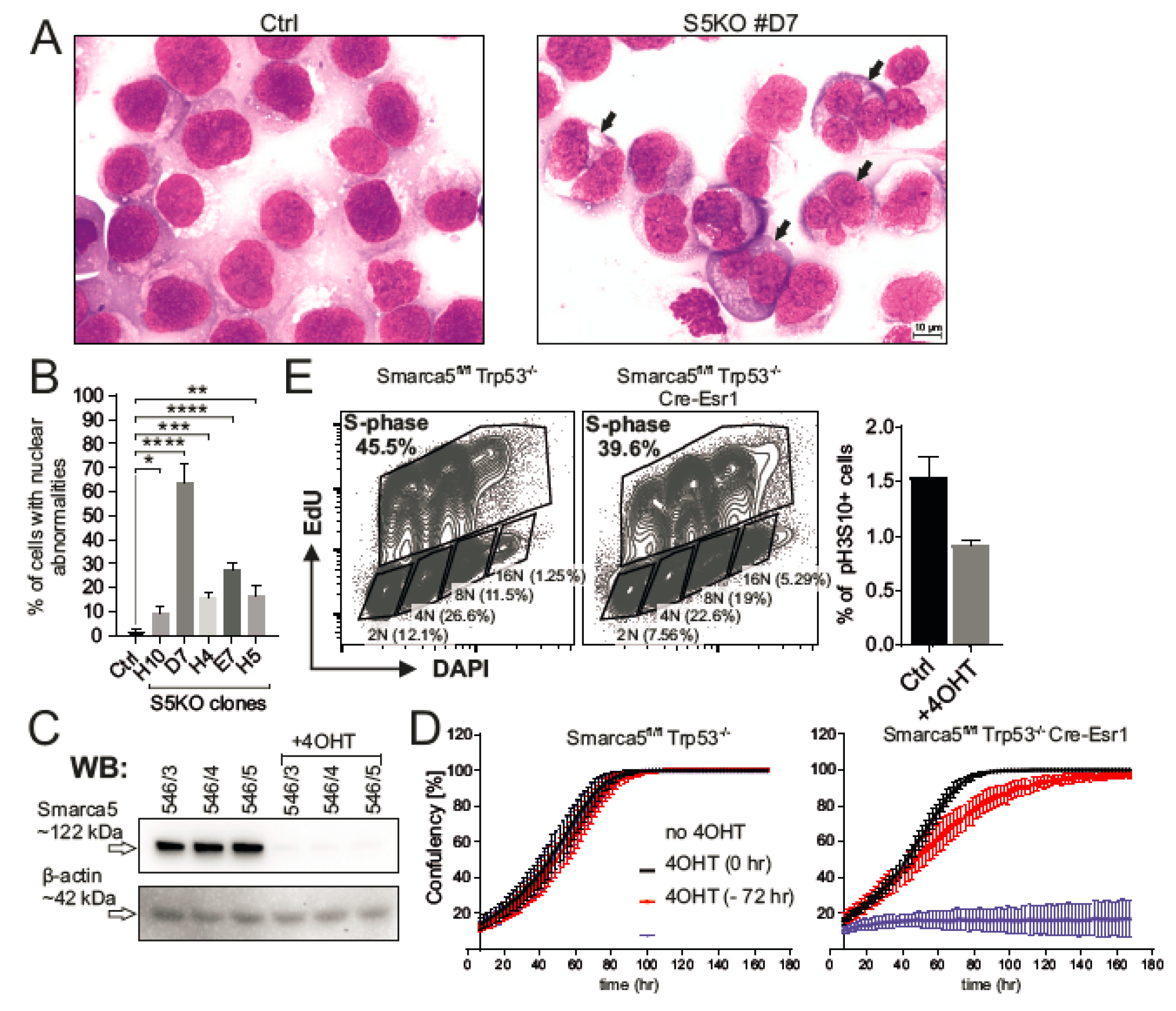
2.4. Inactivation of Smarca5 Causes Nuclear Abnormalities and Polyploidy
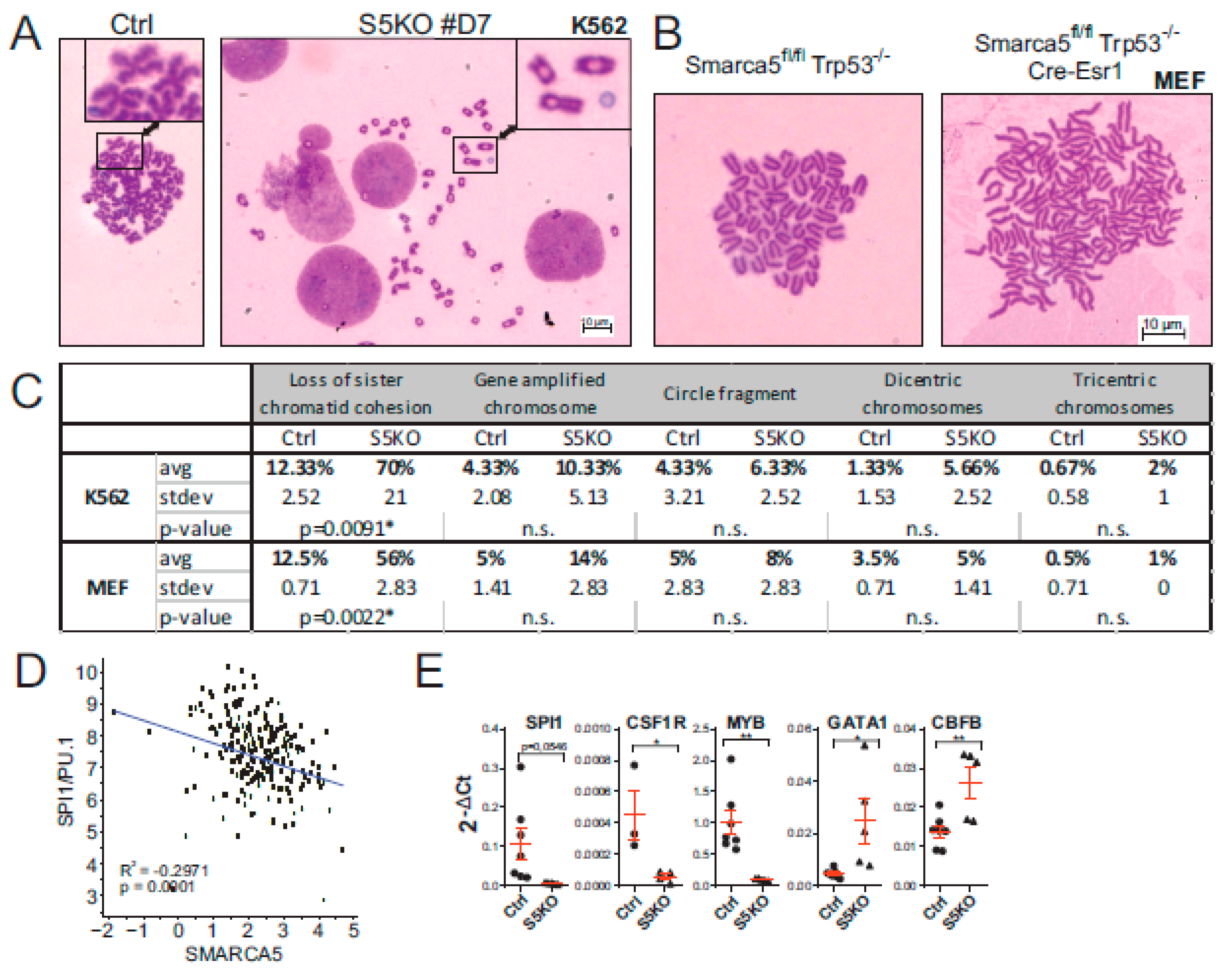
2.5. Cytogenetic Abnormalities and Gene Expression Dysregulation in the S5KO AML Cells
3. Discussion
4. Materials and Methods
4.1. CRISPR Vector Design
4.2. Cell Lines
4.3. AML Patients and Statistics
4.4. Real-Time qPCR
4.5. Western Blot
4.6. Cytogenetics
4.7. Hematopoietic Reconstitution
4.8. Analysis of S5KO MEF Cells
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SMARCA5 | SWI/SNF-Related, Matrix-Associated, Actin-Dependent Regulator of Chromatin, Subfamily A, Member 5 |
| SNF2H | Sucrose Nonfermenting Protein 2 Homolog |
| AML | Acute Myeloid Leukemia |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
References
- Kokavec, J.; Zikmund, T.; Savvulidi, F.; Kulvait, V.; Edelmann, W.; Skoultchi, A.I.; Stopka, T. The ISWI ATPase Smarca5 (Snf2h) Is Required for Proliferation and Differentiation of Hematopoietic Stem and Progenitor Cells. Stem Cells 2017, 35, 1614–1623. [Google Scholar] [CrossRef] [Green Version]
- Stopka, T.; Skoultchi, A.I. The ISWI ATPase Snf2h is required for early mouse development. Proc. Natl. Acad. Sci. USA 2003, 100, 14097–14102. [Google Scholar] [CrossRef] [Green Version]
- Zikmund, T.; Kokavec, J.; Turkova, T.; Savvulidi, F.; Paszekova, H.; Vodenkova, S.; Sedlacek, R.; Skoultchi, A.I.; Stopka, T. ISWI ATPase Smarca5 Regulates Differentiation of Thymocytes Undergoing beta-Selection. J. Immunol. 2019, 202, 3434–3446. [Google Scholar] [CrossRef]
- Alvarez-Saavedra, M.; De Repentigny, Y.; Lagali, P.S.; Raghu Ram, E.V.; Yan, K.; Hashem, E.; Ivanochko, D.; Huh, M.S.; Yang, D.; Mears, A.J.; et al. Snf2h-mediated chromatin organization and histone H1 dynamics govern cerebellar morphogenesis and neural maturation. Nat. Commun. 2014, 5, 4181. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Limi, S.; McGreal, R.S.; Xie, Q.; Brennan, L.A.; Kantorow, W.L.; Kokavec, J.; Majumdar, R.; Hou, H., Jr.; Edelmann, W.; et al. Chromatin remodeling enzyme Snf2h regulates embryonic lens differentiation and denucleation. Development 2016, 143, 1937–1947. [Google Scholar] [CrossRef] [Green Version]
- Barisic, D.; Stadler, M.B.; Iurlaro, M.; Schubeler, D. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 2019, 569, 136–140. [Google Scholar] [CrossRef]
- Dluhosova, M.; Curik, N.; Vargova, J.; Jonasova, A.; Zikmund, T.; Stopka, T. Epigenetic control of SPI1 gene by CTCF and ISWI ATPase SMARCA5. PLoS ONE 2014, 9, e87448. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.A.; Baek, S.; Sung, M.H.; John, S.; Wiench, M.; Johnson, T.A.; Schiltz, R.L.; Hager, G.L. Overlapping chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions. Nat. Struct. Mol. Biol. 2014, 21, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, L.R.; Picketts, D.J. The role of ISWI chromatin remodeling complexes in brain development and neurodevelopmental disorders. Mol. Cell Neurosci. 2018, 87, 55–64. [Google Scholar] [CrossRef]
- Erdel, F.; Rippe, K. Chromatin remodelling in mammalian cells by ISWI-type complexes—Where, when and why? FEBS J. 2011, 278, 3608–3618. [Google Scholar] [CrossRef]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [Green Version]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Sardiu, M.; Gogol, M.; Gilmore, J.; Zhang, D.; Florens, L.; Abmayr, S.M.; Washburn, M.P.; Workman, J.L. Composition and Function of Mutant Swi/Snf Complexes. Cell Rep. 2017, 18, 2124–2134. [Google Scholar] [CrossRef]
- Gigek, C.O.; Lisboa, L.C.; Leal, M.F.; Silva, P.N.; Lima, E.M.; Khayat, A.S.; Assumpcao, P.P.; Burbano, R.R.; Smith Mde, A. SMARCA5 methylation and expression in gastric cancer. Cancer Investig. 2011, 29, 162–166. [Google Scholar] [CrossRef]
- Reis, S.T.; Timoszczuk, L.S.; Pontes-Junior, J.; Viana, N.; Silva, I.A.; Dip, N.; Srougi, M.; Leite, K.R. The role of micro RNAs let7c, 100 and 218 expression and their target RAS, C-MYC, BUB1, RB, SMARCA5, LAMB3 and Ki-67 in prostate cancer. Clinics 2013, 68, 652–657. [Google Scholar] [CrossRef]
- Sheu, J.J.; Choi, J.H.; Yildiz, I.; Tsai, F.J.; Shaul, Y.; Wang, T.L.; Shih Ie, M. The roles of human sucrose nonfermenting protein 2 homologue in the tumor-promoting functions of Rsf-1. Cancer Res. 2008, 68, 4050–4057. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.; Mao, X.; Li, B.; Guan, S.; Yao, F.; Jin, F. Overexpression of SMARCA5 correlates with cell proliferation and migration in breast cancer. Tumour. Biol. 2015, 36, 1895–1902. [Google Scholar] [CrossRef]
- Zhao, X.C.; An, P.; Wu, X.Y.; Zhang, L.M.; Long, B.; Tian, Y.; Chi, X.Y.; Tong, D.Y. Overexpression of hSNF2H in glioma promotes cell proliferation, invasion, and chemoresistance through its interaction with Rsf-1. Tumour. Biol. 2016, 37, 7203–7212. [Google Scholar] [CrossRef]
- Stopka, T.; Zakova, D.; Fuchs, O.; Kubrova, O.; Blafkova, J.; Jelinek, J.; Necas, E.; Zivny, J. Chromatin remodeling gene SMARCA5 is dysregulated in primitive hematopoietic cells of acute leukemia. Leukemia 2000, 14, 1247–1252. [Google Scholar] [CrossRef] [Green Version]
- Rosenbauer, F.; Wagner, K.; Kutok, J.L.; Iwasaki, H.; Le Beau, M.M.; Okuno, Y.; Akashi, K.; Fiering, S.; Tenen, D.G. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet. 2004, 36, 624–630. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511. [Google Scholar] [CrossRef]
- Law, J.C.; Ritke, M.K.; Yalowich, J.C.; Leder, G.H.; Ferrell, R.E. Mutational inactivation of the p53 gene in the human erythroid leukemic K562 cell line. Leuk. Res. 1993, 17, 1045–1050. [Google Scholar] [CrossRef]
- Hakimi, M.A.; Bochar, D.A.; Schmiesing, J.A.; Dong, Y.; Barak, O.G.; Speicher, D.W.; Yokomori, K.; Shiekhattar, R. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 2002, 418, 994–998. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-S.; He, X.; Orr, B.; Wutz, G.; Hill, V.; Peters, J.-M.; Compton, D.A.; Waldman, T. Intact Cohesion, Anaphase, and Chromosome Segregation in Human Cells Harboring Tumor-Derived Mutations in STAG2. PLOS Genet. 2016, 12, e1005865. [Google Scholar] [CrossRef]
- Leman, A.R.; Noguchi, C.; Lee, C.Y.; Noguchi, E. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J. Cell Sci. 2010, 123, 660–670. [Google Scholar] [CrossRef] [Green Version]
- De Lange, J.; Faramarz, A.; Oostra, A.B.; de Menezes, R.X.; van der Meulen, I.H.; Rooimans, M.A.; Rockx, D.A.; Brakenhoff, R.H.; van Beusechem, V.W.; King, R.W.; et al. Defective sister chromatid cohesion is synthetically lethal with impaired APC/C function. Nat. Commun. 2015, 6, 8399. [Google Scholar] [CrossRef] [Green Version]
- Kishtagari, A.N.; Jarman, C.; Tiwari, A.D.; Phillips, J.G.; Schuerger, C.; Jha, B.K.; Saunthararajah, Y. A First-in-Class Inhibitor of ISWI-Mediated (ATP-Dependent) Transcription Repression Releases Terminal-Differentiation in AML Cells While Sparing Normal Hematopoiesis. Blood 2018, 132, 216. [Google Scholar] [CrossRef]
- Herold, T.; Jurinovic, V.; Batcha, A.M.N.; Bamopoulos, S.A.; Rothenberg-Thurley, M.; Ksienzyk, B.; Hartmann, L.; Greif, P.A.; Phillippou-Massier, J.; Krebs, S.; et al. A 29-gene and cytogenetic score for the prediction of resistance to induction treatment in acute myeloid leukemia. Haematologica 2018, 103, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Stief, S.M.; Hanneforth, A.L.; Weser, S.; Mattes, R.; Carlet, M.; Liu, W.H.; Bartoschek, M.D.; Dominguez Moreno, H.; Oettle, M.; Kempf, J.; et al. Loss of KDM6A confers drug resistance in acute myeloid leukemia. Leukemia 2020, 34, 50–62. [Google Scholar] [CrossRef]
- Batcha, A.M.N.; Bamopoulos, S.A.; Kerbs, P.; Kumar, A.; Jurinovic, V.; Rothenberg-Thurley, M.; Ksienzyk, B.; Philippou-Massier, J.; Krebs, S.; Blum, H.; et al. Allelic Imbalance of Recurrently Mutated Genes in Acute Myeloid Leukaemia. Sci. Rep. 2019, 9, 11796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zikmund, T.; Paszekova, H.; Kokavec, J.; Kerbs, P.; Thakur, S.; Turkova, T.; Tauchmanova, P.; Greif, P.A.; Stopka, T. Loss of ISWI ATPase SMARCA5 (SNF2H) in Acute Myeloid Leukemia Cells Inhibits Proliferation and Chromatid Cohesion. Int. J. Mol. Sci. 2020, 21, 2073. https://doi.org/10.3390/ijms21062073
Zikmund T, Paszekova H, Kokavec J, Kerbs P, Thakur S, Turkova T, Tauchmanova P, Greif PA, Stopka T. Loss of ISWI ATPase SMARCA5 (SNF2H) in Acute Myeloid Leukemia Cells Inhibits Proliferation and Chromatid Cohesion. International Journal of Molecular Sciences. 2020; 21(6):2073. https://doi.org/10.3390/ijms21062073
Chicago/Turabian StyleZikmund, Tomas, Helena Paszekova, Juraj Kokavec, Paul Kerbs, Shefali Thakur, Tereza Turkova, Petra Tauchmanova, Philipp A. Greif, and Tomas Stopka. 2020. "Loss of ISWI ATPase SMARCA5 (SNF2H) in Acute Myeloid Leukemia Cells Inhibits Proliferation and Chromatid Cohesion" International Journal of Molecular Sciences 21, no. 6: 2073. https://doi.org/10.3390/ijms21062073