Metabolic and Redox Signaling of the Nucleoredoxin-Like-1 Gene for the Treatment of Genetic Retinal Diseases


Abstract
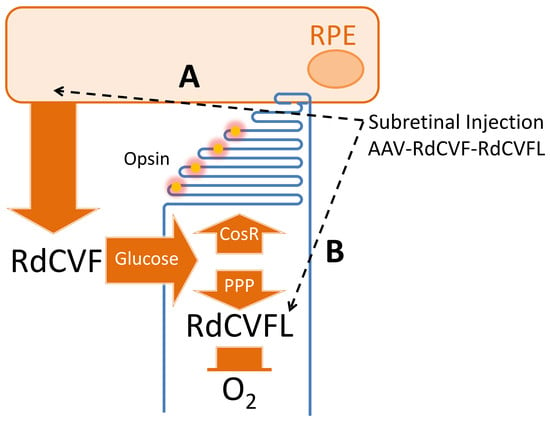
:
{kind=link}
{kind=link}
1. Introduction
2. Rod-Derived Cone Viability Factor
3. The Thioredoxin RdCVFL
4. Design of the Therapeutic Molecule
5. Chemical Manufacturing
6. Preclinical Studies
7. Clinical Trial
8. Conclusions
9. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated viral vector |
| ATP | Adenosine triphosphate |
| BSG | Basigin |
| BSG1 | Basigin-1 with three immunoglobulin domains |
| BSG2 | Basigin-2 with two immunoglobulin domains |
| CBA | Chicken beta-actin |
| cGMP | Cyclic guanosine monophosphate |
| CMV | Cytomegalovirus |
| CNTF | Ciliary neurotrophic factor |
| COS-1 | Immortalizing CV-1 cell line |
| CsCl | Cesium chloride |
| CSNB | Congenital stationary night blindness |
| CX3CL1 | C-X3-C motif chemokine ligand 1 or fractalkine |
| DHAP | Dihydroxyacetone phosphate |
| EMA | European Medicines Agency |
| ERG | Electroretinogram |
| FDA | Food and Drug Administration |
| G3P | Glycerol-3-phosphate |
| G3PDH | Glycerol-3-phosphate dehydrogenase |
| G6P | Glucose-6-phosphate |
| GFP | Green fluorescent protein |
| GLP | Good laboratory practices |
| GLUT1 | Glucose transporter SLC2A1 |
| GMP | Good manufacturing practice |
| GNAT1 | rod transducin alpha subunit |
| HEK293 | Human embryonic kidney 293 cell line |
| HDAC4 | Histone deacetylase 4 |
| Ig | Immunoglobulin domain |
| ITRs | Invert terminal repeats |
| KLF15 | Krüppel-like factor 15 |
| LACT | Lactate |
| LCA | Leber congenital amaurosis |
| LCA2 | Leber congenital amaurosis with a mutation in the RPE65 gene |
| LCR | Locus control region |
| LDH | Lactate dehydrogenase |
| LRAT | Lecithin-retinol acyltransferase |
| MTCs | Lactate transporters |
| MPC | Mitochondrial pyruvate carrier |
| NADPH | Nicotinamide adenine dinucleotide phosphate, reduced |
| NHPs | Non-human primates |
| NXNL1 | Nucleoredoxin-like 1gene |
| OPN1L/MW | Long and middle wave opsin gene |
| OXPHO | Oxidative phosphorylation |
| PDE6B | Phosphodiesterase 6B |
| PPP | Pentose phosphate pathway |
| PRPF31 | Pre-mRNA processing factor 31 |
| PUFA | Poly-unsaturated fatty acids |
| PYR | Pyruvate |
| qPCR | Quantitative polymerase chain reaction |
| RB1 | Tumor suppressor retinoblastoma 1 |
| rd1 | Retinal degeneration 1 mutant |
| RdCVF | Rod-derived cone viability factor |
| RdCVFL | Thioredoxin enzyme rod-derived cone viability factor long |
| ROS | Reactive oxygen species |
| RP | Retinitis pigmentosa |
| RPE | Retinal pigmented epithelium |
| RPE65 | Retinal pigment epithelium protein of 65 kDa |
| SD-OCT | Spectral domain optical coherence tomography |
| SV40T | Simian virus 40 large T antigen |
| TCA | Tricarboxylic acid cycle |
| TXNRD | Thioredoxin reductase |
References
- Takahashi, V.K.L.; Takiuti, J.T.; Jauregui, R.; Tsang, S.H. Gene therapy in inherited retinal degenerative diseases, a review. Ophthalmic Genet. 2018, 39, 560–568. [Google Scholar] [CrossRef]
- Kumaran, N.; Michaelides, M.; Smith, A.J.; Ali, R.R.; Bainbridge, J.W.B. Retinal gene therapy. Br. Med. Bull. 2018, 126, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Trapani, I.; Auricchio, A. Has retinal gene therapy come of age? From bench to bedside and back to bench. Hum. Mol. Genet. 2019, 28, R108–R118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, R.S. Gene Therapy for Retinal Degeneration. Cell 2018, 173, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, J. Clinical research. Gene therapists celebrate a decade of progress. Science 2011, 334, 29–30. [Google Scholar] [CrossRef]
- Hamel, C.P.; Tsilou, E.; Pfeffer, B.A.; Hooks, J.J.; Detrick, B.; Redmond, T.M. Molecular cloning and expression of RPE65, a novel retinal pigment epithelium-specific microsomal protein that is post-transcriptionally regulated in vitro. J. Biol. Chem. 1993, 268, 15751–15757. [Google Scholar]
- Marlhens, F.; Bareil, C.; Griffoin, J.M.; Zrenner, E.; Amalric, P.; Eliaou, C.; Liu, S.Y.; Harris, E.; Redmond, T.M.; Arnaud, B.; et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat. Genet. 1997, 17, 139–141. [Google Scholar] [CrossRef]
- Redmond, T.M.; Yu, S.; Lee, E.; Bok, D.; Hamasaki, D.; Chen, N.; Goletz, P.; Ma, J.X.; Crouch, R.K.; Pfeifer, K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 1998, 20, 344–351. [Google Scholar] [CrossRef]
- Aguirre, G.D.; Baldwin, V.; Pearce-Kelling, S.; Narfstrom, K.; Ray, K.; Acland, G.M. Congenital stationary night blindness in the dog: Common mutation in the RPE65 gene indicates founder effect. Mol. Vis. 1998, 4, 23. [Google Scholar]
- Acland, G.M.; Aguirre, G.D.; Ray, J.; Zhang, Q.; Aleman, T.S.; Cideciyan, A.V.; Pearce-Kelling, S.E.; Anand, V.; Zeng, Y.; Maguire, A.M.; et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001, 28, 92–95. [Google Scholar] [CrossRef]
- Lehrman, S. Virus treatment questioned after gene therapy death. Nature 1999, 401, 517–518. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E. Gene therapy death prompts review of adenovirus vector. Science 1999, 286, 2244–2245. [Google Scholar] [CrossRef] [PubMed]
- Barbour, V. The balance of risk and benefit in gene-therapy trials. Lancet 2000, 355, 384. [Google Scholar] [CrossRef]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N., Jr.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Aleman, T.S.; Boye, S.L.; Schwartz, S.B.; Kaushal, S.; Roman, A.J.; Pang, J.J.; Sumaroka, A.; Windsor, E.A.; Wilson, J.M.; et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA 2008, 105, 15112–15117. [Google Scholar] [CrossRef] [Green Version]
- Ledford, H. Success against blindness encourages gene therapy researchers. Nature 2015, 526, 487–488. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.; Ashtari, M.; Wellman, J.; Marshall, K.A.; Cyckowski, L.L.; Chung, D.C.; McCague, S.; Pierce, E.A.; Chen, Y.; Bennicelli, J.L.; et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci. Transl. Med. 2012, 4, 120ra15. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Jacobson, S.G.; Beltran, W.A.; Sumaroka, A.; Swider, M.; Iwabe, S.; Roman, A.J.; Olivares, M.B.; Schwartz, S.B.; Komaromy, A.M.; et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc. Natl. Acad. Sci. USA 2013, 110, E517–E525. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, S.G.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Heon, E.; Hauswirth, W.W. Improvement and decline in vision with gene therapy in childhood blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boye, S.E.; Huang, W.C.; Roman, A.J.; Sumaroka, A.; Boye, S.L.; Ryals, R.C.; Olivares, M.B.; Ruan, Q.; Tucker, B.A.; Stone, E.M.; et al. Natural history of cone disease in the murine model of Leber congenital amaurosis due to CEP290 mutation: Determining the timing and expectation of therapy. PLoS ONE 2014, 9, e92928. [Google Scholar] [CrossRef] [PubMed]
- Beltran, W.A.; Cideciyan, A.V.; Iwabe, S.; Swider, M.; Kosyk, M.S.; McDaid, K.; Martynyuk, I.; Ying, G.S.; Shaffer, J.; Deng, W.T.; et al. Successful arrest of photoreceptor and vision loss expands the therapeutic window of retinal gene therapy to later stages of disease. Proc. Natl. Acad. Sci. USA 2015, 112, E5844–E5853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.B.; Redmond, T.M.; Nickerson, J.M. A History of the Classical Visual Cycle. Prog. Mol. Biol. Transl. Sci. 2015, 134, 433–448. [Google Scholar]
- Caruso, R.C.; Aleman, T.S.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Mullins, C.L.; Boye, S.L.; Hauswirth, W.W.; Jacobson, S.G. Retinal disease in Rpe65-deficient mice: Comparison to human leber congenital amaurosis due to RPE65 mutations. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5304–5313. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, N.; Baehr, W.; Fu, Y. Cone opsin determines the time course of cone photoreceptor degeneration in Leber congenital amaurosis. Proc. Natl. Acad. Sci. USA 2011, 108, 8879–8884. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Frison-Roche, R. Premier de Cordée: Roman; J’ai Lu: Paris, France, 1941. [Google Scholar]
- Darrow, J.J. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov. Today 2019, 24, 949–954. [Google Scholar] [CrossRef]
- Blond, F.; Leveillard, T. Functional Genomics of the Retina to Elucidate its Construction and Deconstruction. Int. J. Mol. Sci. 2019, 20, 4922. [Google Scholar] [CrossRef] [Green Version]
- Vithana, E.N.; Abu-Safieh, L.; Allen, M.J.; Carey, A.; Papaioannou, M.; Chakarova, C.; Al-Maghtheh, M.; Ebenezer, N.D.; Willis, C.; Moore, A.T.; et al. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol. Cell 2001, 8, 375–381. [Google Scholar]
- Rose, A.M.; Bhattacharya, S.S. Variant haploinsufficiency and phenotypic non-penetrance in PRPF31-associated retinitis pigmentosa. Clin. Genet. 2016, 90, 118–126. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Hahn, L.B.; Cowley, G.S.; Olsson, J.E.; Reichel, E.; Sandberg, M.A.; Berson, E.L. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 1990, 323, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Mendes, H.F.; van der Spuy, J.; Chapple, J.P.; Cheetham, M.E. Mechanisms of cell death in rhodopsin retinitis pigmentosa: Implications for therapy. Trends Mol. Med. 2005, 11, 177–185. [Google Scholar] [CrossRef]
- Gorbatyuk, M.S.; Knox, T.; LaVail, M.M.; Gorbatyuk, O.S.; Noorwez, S.M.; Hauswirth, W.W.; Lin, J.H.; Muzyczka, N.; Lewin, A.S. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. USA 2010, 107, 5961–5966. [Google Scholar] [CrossRef] [Green Version]
- Lewin, A.S.; Drenser, K.A.; Hauswirth, W.W.; Nishikawa, S.; Yasumura, D.; Flannery, J.G.; LaVail, M.M. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat. Med. 1998, 4, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Phylactou, L.A.; Kilpatrick, M.W.; Wood, M.J. Ribozymes as therapeutic tools for genetic disease. Hum. Mol. Genet. 1998, 7, 1649–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauswirth, W.W.; Lewin, A.S. Ribozyme uses in retinal gene therapy. Prog. Retin. Eye Res. 2000, 19, 689–710. [Google Scholar] [CrossRef]
- Malanson, K.M.; Lem, J. Rhodopsin-mediated retinitis pigmentosa. Prog. Mol. Biol. Transl. Sci. 2009, 88, 1–31. [Google Scholar]
- Audo, I.; Manes, G.; Mohand-Said, S.; Friedrich, A.; Lancelot, M.E.; Antonio, A.; Moskova-Doumanova, V.; Poch, O.; Zanlonghi, X.; Hamel, C.P.; et al. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3687–3700. [Google Scholar] [CrossRef]
- Fernandez-San Jose, P.; Blanco-Kelly, F.; Corton, M.; Trujillo-Tiebas, M.J.; Gimenez, A.; Avila-Fernandez, A.; Garcia-Sandoval, B.; Lopez-Molina, M.I.; Hernan, I.; Carballo, M.; et al. Prevalence of Rhodopsin mutations in autosomal dominant Retinitis Pigmentosa in Spain: Clinical and analytical review in 200 families. Acta Ophthalmol. 2015, 93, e38–e44. [Google Scholar] [CrossRef]
- Oh, K.T.; Longmuir, R.; Oh, D.M.; Stone, E.M.; Kopp, K.; Brown, J.; Fishman, G.A.; Sonkin, P.; Gehrs, K.M.; Weleber, R.G. Comparison of the clinical expression of retinitis pigmentosa associated with rhodopsin mutations at codon 347 and codon 23. Am. J. Ophthalmol. 2003, 136, 306–313. [Google Scholar] [CrossRef]
- Bonilha, V.L.; Rayborn, M.E.; Bell, B.A.; Marino, M.J.; Beight, C.D.; Pauer, G.J.; Traboulsi, E.I.; Hollyfield, J.G.; Hagstrom, S.A. Retinal histopathology in eyes from patients with autosomal dominant retinitis pigmentosa caused by rhodopsin mutations. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 2161–2169. [Google Scholar]
- O’Reilly, M.; Palfi, A.; Chadderton, N.; Millington-Ward, S.; Ader, M.; Cronin, T.; Tuohy, T.; Auricchio, A.; Hildinger, M.; Tivnan, A.; et al. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am. J. Hum. Genet. 2007, 81, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiang, A.S.; Palfi, A.; Ader, M.; Kenna, P.F.; Millington-Ward, S.; Clark, G.; Kennan, A.; O’Reilly, M.; Tam, L.C.; Aherne, A.; et al. Toward a gene therapy for dominant disease: Validation of an RNA interference-based mutation-independent approach. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Botta, S.; de Prisco, N.; Marrocco, E.; Renda, M.; Sofia, M.; Curion, F.; Bacci, M.L.; Ventrella, D.; Wilson, C.; Gesualdo, C.; et al. Targeting and silencing of rhodopsin by ectopic expression of the transcription factor KLF15. JCI Insight 2017, 2, e96560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Vagni, P.; Perlini, L.E.; Chenais, N.A.L.; Marchetti, T.; Parrini, M.; Contestabile, A.; Cancedda, L.; Ghezzi, D. Gene Editing Preserves Visual Functions in a Mouse Model of Retinal Degeneration. Front. Neurosci. 2019, 13, 945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.-R.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef] [Green Version]
- Samardzija, M.; Wenzel, A.; Thiersch, M.; Frigg, R.; Reme, C.; Grimm, C. Caspase-1 ablation protects photoreceptors in a model of autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5181–5190. [Google Scholar] [CrossRef]
- Marigo, V. Programmed cell death in retinal degeneration: Targeting apoptosis in photoreceptors as potential therapy for retinal degeneration. Cell Cycle 2007, 6, 652–655. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Wang, S.B.; Xu, H.; Ribic, A.; Mohns, E.J.; Zhou, Y.; Zhu, X.; Biederer, T.; Crair, M.C.; Chen, B. A short N-terminal domain of HDAC4 preserves photoreceptors and restores visual function in retinitis pigmentosa. Nat. Commun. 2015, 6, 8005. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, D.M.; Barnard, A.R.; Singh, M.S.; Martin, C.; Lee, E.J.; Davies, W.I.L.; MacLaren, R.E. CNTF Gene Therapy Confers Lifelong Neuroprotection in a Mouse Model of Human Retinitis Pigmentosa. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1308–1319. [Google Scholar] [CrossRef] [Green Version]
- Birch, D.G.; Bennett, L.D.; Duncan, J.L.; Weleber, R.G.; Pennesi, M.E. Long-term Follow-up of Patients with Retinitis Pigmentosa Receiving Intraocular Ciliary Neurotrophic Factor Implants. Am. J. Ophthalmol. 2016, 170, 10–14. [Google Scholar] [CrossRef] [Green Version]
- Karali, M.; Guadagnino, I.; Marrocco, E.; De Cegli, R.; Carissimo, A.; Pizzo, M.; Casarosa, S.; Conte, I.; Surace, E.M.; Banfi, S. AAV-miR-204 Protects from Retinal Degeneration by Attenuation of Microglia Activation and Photoreceptor Cell Death. Mol. Ther. Nucleic Acids 2019, 19, 144–156. [Google Scholar] [CrossRef]
- Wang, S.K.; Xue, Y.; Rana, P.; Hong, C.M.; Cepko, C.L. Soluble CX3CL1 gene therapy improves cone survival and function in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2019, 116, 10140–10149. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; MacColl Garfinkel, A.E.; Li, Y.; Benowitz, L.I.; Cepko, C.L. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Investig. 2015, 125, 1433–1445. [Google Scholar] [CrossRef] [Green Version]
- Picard, E.; Jonet, L.; Sergeant, C.; Vesvres, M.-H.; Behar-Cohen, F.; Courtois, Y.; Jeanny, J.-C. Overexpressed or intraperitoneally injected human transferrin prevents photoreceptor degeneration in rd10 mice. Mol. Vis. 2010, 16, 2612–2625. [Google Scholar]
- Petit, L.; Punzo, C. mTORC1 sustains vision in retinitis pigmentosa. Oncotarget 2015, 6, 16786–16787. [Google Scholar] [CrossRef]
- Zhang, L.; Du, J.; Justus, S.; Hsu, C.-W.; Bonet-Ponce, L.; Wu, W.-H.; Tsai, Y.-T.; Wu, W.-P.; Jia, Y.; Duong, J.K.; et al. Reprogramming metabolism by targeting sirtuin 6 attenuates retinal degeneration. J. Clin. Investig. 2016, 126, 4659–4673. [Google Scholar] [CrossRef] [Green Version]
- Portera-Cailliau, C.; Sung, C.H.; Nathans, J.; Adler, R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1994, 91, 974–978. [Google Scholar] [CrossRef] [Green Version]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Snyder, C. Jean Nougaret, the butcher from Provence, and his family. Arch. Ophthalmol. 1963, 69, 676–678. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; Hahn, L.B.; Reboul, T.; Arnaud, B. Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat. Genet. 1996, 13, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, T.; Haim, M.; Piczenik, Y.; Simonsen, S.E. Autosomal Dominant Stationary Night-Blindness—A Large Family Rediscovered. Acta Ophthalmol. 1991, 69, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef]
- Wright, A.F. A searchlight through the fog. Nat. Genet. 1997, 17, 132–134. [Google Scholar] [CrossRef]
- Kaplan, H.J.; Wang, W.; Dean, D.C. Restoration of Cone Photoreceptor Function in Retinitis Pigmentosa. Transl. Vis. Sci. Technol. 2017, 6, 5. [Google Scholar] [CrossRef]
- Geller, A.M.; Sieving, P.A. Assessment of foveal cone photoreceptors in Stargardt’s macular dystrophy using a small dot detection task. Vis. Res. 1993, 33, 1509–1524. [Google Scholar] [CrossRef] [Green Version]
- Petersen-Jones, S.M.; Occelli, L.M.; Winkler, P.A.; Lee, W.; Sparrow, J.R.; Tsukikawa, M.; Boye, S.L.; Chiodo, V.; Capasso, J.E.; Becirovic, E.; et al. Patients and animal models of CNGβ1-deficient retinitis pigmentosa support gene augmentation approach. J. Clin. Investig. 2017, 128, 190–206. [Google Scholar] [CrossRef]
- Cronin, T.; Leveillard, T.; Sahel, J.A. Retinal degenerations: From cell signaling to cell therapy; pre-clinical and clinical issues. Curr. Gene Ther. 2007, 7, 121–129. [Google Scholar] [CrossRef]
- Applebury, M.L. Molecular-Genetics—Insight into Blindness. Nature 1990, 343, 316–317. [Google Scholar] [CrossRef]
- Daiger, S.P.; Humphries, M.M.; Giesenschlag, N.; Sharp, E.; McWilliam, P.; Farrer, J.; Bradley, D.; Kenna, P.; McConnell, D.J.; Sparkes, R.S.; et al. Linkage analysis of human chromosome 4: Exclusion of autosomal dominant retinitis pigmentosa (ADRP) and detection of new linkage groups. Cytogenet. Cell Genet. 1989, 50, 181–187. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Reichel, E.; Hahn, L.B.; Cowley, G.S.; Yandell, D.W.; Sandberg, M.A.; Berson, E.L. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990, 343, 364–366. [Google Scholar] [CrossRef]
- Kolb, H.; Gouras, P. Electron microscopic observations of human retinitis pigmentosa, dominantly inherited. Investig. Ophthalmol. 1974, 13, 487–498. [Google Scholar]
- Berson, E.L. Ocular findings in a form of retinitis pigmentosa with a rhodopsin gene defect. Trans. Am. Ophthalmol. Soc. 1990, 88, 355–388. [Google Scholar]
- Cideciyan, A.V.; Hood, D.C.; Huang, Y.; Banin, E.; Li, Z.Y.; Stone, E.M.; Milam, A.H.; Jacobson, S.G. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc. Natl. Acad. Sci. USA 1998, 95, 7103–7108. [Google Scholar] [CrossRef] [Green Version]
- Carter-Dawson, L.D.; LaVail, M.M.; Sidman, R.L. Differential effect of the rd mutation on rods and cones in the mouse retina. Investig. Ophthalmol. Vis. Sci. 1978, 17, 489–498. [Google Scholar]
- Bowes, C.; Li, T.; Danciger, M.; Baxter, L.C.; Applebury, M.L.; Farber, D.B. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature 1990, 347, 677–680. [Google Scholar] [CrossRef]
- McLaughlin, M.E.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat. Genet. 1993, 4, 130–134. [Google Scholar] [CrossRef]
- Usukura, J.; Khoo, W.; Abe, T.; Breitman, M.L.; Shinohara, T. Cone cells fail to develop normally in transgenic mice showing ablation of rod photoreceptor cells. Cell Tissue Res. 1994, 275, 79–90. [Google Scholar] [CrossRef]
- McCall, M.A.; Gregg, R.G.; Merriman, K.; Goto, Y.; Peachey, N.S.; Stanford, L.R. Morphological and physiological consequences of the selective elimination of rod photoreceptors in transgenic mice. Exp. Eye Res. 1996, 63, 35–50. [Google Scholar] [CrossRef]
- Scott, P.A.; de Castro, J.P.; DeMarco, P.J.; Ross, J.W.; Njoka, J.; Walters, E.; Prather, R.S.; McCall, M.A.; Kaplan, H.J. Progression of Pro23His Retinopathy in a Miniature Swine Model of Retinitis Pigmentosa. Transl. Vis. Sci. Technol. 2017, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mieziewska, K.; Van Veen, T.; Aguirre, G.D. Development and fate of interphotoreceptor matrix components during dysplastic photoreceptor differentiation: A lectin cytochemical study of rod-cone dysplasia 1. Exp. Eye Res. 1993, 56, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Choi, R.Y.; Engbretson, G.A.; Solessio, E.C.; Jones, G.A.; Coughlin, A.; Aleksic, I.; Zuber, M.E. Cone degeneration following rod ablation in a reversible model of retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Mohand-Said, S.; Hicks, D.; Simonutti, M.; Tran-Minh, D.; Deudon-Combe, A.; Dreyfus, H.; Silverman, M.S.; Ogilvie, J.M.; Tenkova, T.; Sahel, J. Photoreceptor transplants increase host cone survival in the retinal degeneration (rd) mouse. Ophthalmic Res. 1997, 29, 290–297. [Google Scholar] [CrossRef]
- Mohand-Said, S.; Hicks, D.; Dreyfus, H.; Sahel, J.A. Selective transplantation of rods delays cone loss in a retinitis pigmentosa model. Arch. Ophthalmol. 2000, 118, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Mohand-Said, S.; Deudon-Combe, A.; Hicks, D.; Simonutti, M.; Forster, V.; Fintz, A.C.; Leveillard, T.; Dreyfus, H.; Sahel, J.A. Normal retina releases a diffusible factor stimulating cone survival in the retinal degeneration mouse. Proc. Natl. Acad. Sci. USA 1998, 95, 8357–8362. [Google Scholar] [CrossRef] [Green Version]
- Fintz, A.C.; Audo, I.; Hicks, D.; Mohand-Said, S.; Leveillard, T.; Sahel, J. Partial characterization of retina-derived cone neuroprotection in two culture models of photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2003, 44, 818–825. [Google Scholar] [CrossRef] [Green Version]
- Adler, R.; Hatlee, M. Plasticity and differentiation of embryonic retinal cells after terminal mitosis. Science 1989, 243, 391–393. [Google Scholar] [CrossRef]
- Hewitt, A.T.; Lindsey, J.D.; Carbott, D.; Adler, R. Photoreceptor survival-promoting activity in interphotoreceptor matrix preparations: Characterization and partial purification. Exp. Eye Res. 1990, 50, 79–88. [Google Scholar] [CrossRef]
- Weleber, R.G.; Gregory-Evans, K. Retinitis Pigmentosa and Allied Disorders A2—Ryan, Stephen, J. In Retina, 4th ed.; Chapter 17; Hinton, D.R., Schachat, A.P., Wilkinson, C.P., Eds.; Mosby: Edinburgh, UK, 2006; pp. 395–498. [Google Scholar]
- Leveillard, T.; Mohand-Said, S.; Lorentz, O.; Hicks, D.; Fintz, A.C.; Clerin, E.; Simonutti, M.; Forster, V.; Cavusoglu, N.; Chalmel, F.; et al. Identification and characterization of rod-derived cone viability factor. Nat. Genet. 2004, 36, 755–759. [Google Scholar] [CrossRef]
- Ait-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clerin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef] [Green Version]
- Lambard, S.; Reichman, S.; Berlinicke, C.; Niepon, M.L.; Goureau, O.; Sahel, J.A.; Leveillard, T.; Zack, D.J. Expression of rod-derived cone viability factor: Dual role of CRX in regulating promoter activity and cell-type specificity. PLoS ONE 2010, 5, e13075. [Google Scholar] [CrossRef] [Green Version]
- Reichman, S.; Kalathur, R.K.; Lambard, S.; Ait-Ali, N.; Yang, Y.; Lardenois, A.; Ripp, R.; Poch, O.; Zack, D.J.; Sahel, J.A.; et al. The homeobox gene CHX10/VSX2 regulates RdCVF promoter activity in the inner retina. Hum. Mol. Genet. 2010, 19, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Delyfer, M.N.; Raffelsberger, W.; Mercier, D.; Korobelnik, J.F.; Gaudric, A.; Charteris, D.G.; Tadayoni, R.; Metge, F.; Caputo, G.; Barale, P.O.; et al. Transcriptomic analysis of human retinal detachment reveals both inflammatory response and photoreceptor death. PLoS ONE 2011, 6, e28791. [Google Scholar] [CrossRef]
- Yang, Y.; Mohand-Said, S.; Danan, A.; Simonutti, M.; Fontaine, V.; Clerin, E.; Picaud, S.; Leveillard, T.; Sahel, J.A. Functional cone rescue by RdCVF protein in a dominant model of retinitis pigmentosa. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 787–795. [Google Scholar] [CrossRef]
- Leveillard, T.; Sahel, J.A. Rod-derived cone viability factor for treating blinding diseases: From clinic to redox signaling. Sci. Transl. Med. 2010, 2, 26ps16. [Google Scholar] [CrossRef] [Green Version]
- Sahel, J.A.; Leveillard, T.; Picaud, S.; Dalkara, D.; Marazova, K.; Safran, A.; Paques, M.; Duebel, J.; Roska, B.; Mohand-Said, S. Functional rescue of cone photoreceptors in retinitis pigmentosa. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 1669–1677. [Google Scholar]
- Byrne, L.C.; Dalkara, D.; Luna, G.; Fisher, S.K.; Clerin, E.; Sahel, J.A.; Leveillard, T.; Flannery, J.G. Viral-mediated RdCVF and RdCVFL expression protects cone and rod photoreceptors in retinal degeneration. J. Clin. Investig. 2015, 125, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Ochrietor, J.D.; Moroz, T.P.; van Ekeris, L.; Clamp, M.F.; Jefferson, S.C.; deCarvalho, A.C.; Fadool, J.M.; Wistow, G.; Muramatsu, T.; Linser, P.J. Retina-specific expression of 5A11/Basigin-2, a member of the immunoglobulin gene superfamily. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4086–4096, RetNet Retinal Information Network. Available online: https://sph.uth.edu/retnet/sum-dis.htm (accessed on 10 October 2019).
- Leveillard, T. Cancer metabolism of cone photoreceptors. Oncotarget 2015, 6, 32285–32286. [Google Scholar] [CrossRef]
- Chinchore, Y.; Begaj, T.; Wu, D.; Drokhlyansky, E.; Cepko, C.L. Glycolytic reliance promotes anabolism in photoreceptors. eLife 2017, 6, e25946. [Google Scholar] [CrossRef]
- Chalmel, F.; Leveillard, T.; Jaillard, C.; Lardenois, A.; Berdugo, N.; Morel, E.; Koehl, P.; Lambrou, G.; Holmgren, A.; Sahel, J.A.; et al. Rod-derived Cone Viability Factor-2 is a novel bifunctional-thioredoxin-like protein with therapeutic potential. BMC Mol. Biol. 2007, 8, 74. [Google Scholar] [CrossRef]
- Brennan, L.A.; Lee, W.; Kantorow, M. TXNL6 is a novel oxidative stress-induced reducing system for methionine sulfoxide reductase a repair of alpha-crystallin and cytochrome C in the eye lens. PLoS ONE 2010, 5, e15421. [Google Scholar] [CrossRef] [Green Version]
- Cronin, T.; Raffelsberger, W.; Lee-Rivera, I.; Jaillard, C.; Niepon, M.L.; Kinzel, B.; Clerin, E.; Petrosian, A.; Picaud, S.; Poch, O.; et al. The disruption of the rod-derived cone viability gene leads to photoreceptor dysfunction and susceptibility to oxidative stress. Cell Death Differ. 2010, 17, 1199–1210. [Google Scholar] [CrossRef]
- Sahel, J. Metabolic and redox signaling in the retina. Cell. Mol. Life Sci. CMLS 2016, 74, 3649–3665. [Google Scholar]
- Mei, X.; Chaffiol, A.; Kole, C.; Yang, Y.; Millet-Puel, G.; Clerin, E.; Ait-Ali, N.; Bennett, J.; Dalkara, D.; Sahel, J.A.; et al. The Thioredoxin Encoded by the Rod-Derived Cone Viability Factor Gene Protects Cone Photoreceptors Against Oxidative Stress. Antioxid. Redox Signal. 2016, 24, 909–923. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, T.; Knuesting, J.; Berndt, C.; Morgan, B.; Scheibe, R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 2015, 396, 523–537. [Google Scholar] [CrossRef]
- Lopez-Grueso, M.J.; Gonzalez-Ojeda, R.; Requejo-Aguilar, R.; McDonagh, B.; Fuentes-Almagro, C.A.; Muntane, J.; Barcena, J.A.; Padilla, C.A. Thioredoxin and glutaredoxin regulate metabolism through different multiplex thiol switches. Redox Biol. 2019, 21, 101049. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Lu, J.; Holmgren, A. Selenocysteine in mammalian thioredoxin reductase and application of ebselen as a therapeutic. Free Radic. Biol. Med. 2018, 127, 238–247. [Google Scholar] [CrossRef]
- Camacho, E.T.; Brager, D.; Elachouri, G.; Korneyeva, T.; Millet-Puel, G.; Sahel, J.A.; Leveillard, T. A Mathematical Analysis of Aerobic Glycolysis Triggered by Glucose Uptake in Cones. Sci. Rep. 2019, 9, 4162. [Google Scholar] [CrossRef]
- Leveillard, T.; Sahel, J.A. Metabolic and redox signaling in the retina. Cell. Mol. Life Sci. CMLS 2017, 74, 3649–3665. [Google Scholar] [CrossRef] [Green Version]
- Leveillard, T.; Ait-Ali, N. Cell Signaling with Extracellular Thioredoxin and Thioredoxin-Like Proteins: Insight into Their Mechanisms of Action. Oxidative Med. Cell. Longev. 2017, 2017, 8475125. [Google Scholar] [CrossRef] [Green Version]
- Domenger, C.; Grimm, D. Next-generation AAV vectors-do not judge a virus (only) by its cover. Hum. Mol. Genet. 2019, 28, R3–R14. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Fernandez De La Camara, C.; Cehajic-Kapetanovic, J.; MacLaren, R.E. RPGR gene therapy presents challenges in cloning the coding sequence. Expert Opin. Biol. Ther. 2019, 9, 63–71. [Google Scholar] [CrossRef]
- Bell, P.; Wang, L.; Chen, S.J.; Yu, H.; Zhu, Y.; Nayal, M.; He, Z.; White, J.; Lebel-Hagan, D.; Wilson, J.M. Effects of Self-Complementarity, Codon Optimization, Transgene, and Dose on Liver Transduction with AAV8. Hum. Gene Ther. Methods 2016, 27, 228–237. [Google Scholar] [CrossRef]
- Dai, Y.; Roman, M.; Naviaux, R.K.; Verma, I.M. Gene therapy via primary myoblasts: Long-term expression of factor IX protein following transplantation in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 10892–10895. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, J.; Takaki, S.; Araki, K.; Tashiro, F.; Tominaga, A.; Takatsu, K.; Yamamura, K. Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene 1989, 79, 269–277. [Google Scholar]
- Elachouri, G.; Lee-Rivera, I.; Clerin, E.; Argentini, M.; Fridlich, R.; Blond, F.; Ferracane, V.; Yang, Y.; Raffelsberger, W.; Wan, J.; et al. Thioredoxin rod-derived cone viability factor protects against photooxidative retinal damage. Free Radic. Biol. Med. 2015, 81, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Zack, D.J.; Bennett, J.; Wang, Y.; Davenport, C.; Klaunberg, B.; Gearhart, J.; Nathans, J. Unusual topography of bovine rhodopsin promoter-lacZ fusion gene expression in transgenic mouse retinas. Neuron 1991, 6, 187–199. [Google Scholar] [CrossRef]
- Mancuso, K.; Hauswirth, W.W.; Li, Q.; Connor, T.B.; Kuchenbecker, J.A.; Mauck, M.C.; Neitz, J.; Neitz, M. Gene therapy for red-green colour blindness in adult primates. Nature 2009, 461, 784–787. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Andreadis, S.T. Independent and high-level dual-gene expression in adult stem-progenitor cells from a single lentiviral vector. Gene Ther. 2009, 16, 874–884. [Google Scholar] [CrossRef] [Green Version]
- Nojima, T.; Dienstbier, M.; Murphy, S.; Proudfoot, N.J.; Dye, M.J. Definition of RNA polymerase II CoTC terminator elements in the human genome. Cell Rep. 2013, 3, 1080–1092. [Google Scholar] [CrossRef] [Green Version]
- Parrish, C.; Berns, K. Parvoviridae. In Fields Virology, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2437–2477. [Google Scholar]
- Dong, J.Y.; Fan, P.D.; Frizzell, R.A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 1996, 7, 2101–2112. [Google Scholar] [CrossRef]
- Grieger, J.C.; Samulski, R.J. Packaging capacity of adeno-associated virus serotypes: Impact of larger genomes on infectivity and postentry steps. J. Virol. 2005, 79, 9933–9944. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, H.; Colosi, P. Effect of genome size on AAV vector packaging. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Bennicelli, J.; Wright, J.F.; Komaromy, A.; Jacobs, J.B.; Hauck, B.; Zelenaia, O.; Mingozzi, F.; Hui, D.; Chung, D.; Rex, T.S.; et al. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 458–465. [Google Scholar] [CrossRef]
- Cheng, S.W.; Court, D.L.; Friedman, D.I. Transcription termination signals in the nin region of bacteriophage lambda: Identification of Rho-dependent termination regions. Genetics 1995, 140, 875–887. [Google Scholar]
- Chadeuf, G.; Ciron, C.; Moullier, P.; Salvetti, A. Evidence for encapsidation of prokaryotic sequences during recombinant adeno-associated virus production and their in vivo persistence after vector delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 744–753. [Google Scholar] [CrossRef]
- Wang, X.S.; Qing, K.; Ponnazhagan, S.; Srivastava, A. Adeno-associated virus type 2 DNA replication in vivo: Mutation analyses of the D sequence in viral inverted terminal repeats. J. Virol. 1997, 71, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Kole, C.; Klipfel, L.; Yang, Y.; Ferracane, V.; Blond, F.; Reichman, S.; Millet-Puel, G.; Clerin, E.; Ait-Ali, N.; Pagan, D.; et al. Otx2-Genetically Modified Retinal Pigment Epithelial Cells Rescue Photoreceptors after Transplantation. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 219–237. [Google Scholar] [CrossRef] [Green Version]
- Schmeer, M.; Buchholz, T.; Schleef, M. Plasmid DNA Manufacturing for Indirect and Direct Clinical Applications. Hum. Gene Ther. 2017, 28, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Penaud-Budloo, M.; Francois, A.; Clement, N.; Ayuso, E. Pharmacology of Recombinant Adeno-associated Virus Production. Mol. Ther. Methods Clin. Dev. 2018, 8, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebherz, C.; Maguire, A.; Tang, W.; Bennett, J.; Wilson, J.M. Novel AAV serotypes for improved ocular gene transfer. J. Gene Med. 2008, 10, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. Biodrugs Clin. Immunother. Biopharm. Gene Ther. 2017, 31, 317–334. [Google Scholar]
- Vandenberghe, L.H.; Bell, P.; Maguire, A.M.; Xiao, R.; Hopkins, T.B.; Grant, R.; Bennett, J.; Wilson, J.M. AAV9 targets cone photoreceptors in the nonhuman primate retina. PLoS ONE 2013, 8, e53463. [Google Scholar] [CrossRef]
- Boye, S.E.; Alexander, J.J.; Boye, S.L.; Witherspoon, C.D.; Sandefer, K.J.; Conlon, T.J.; Erger, K.; Sun, J.; Ryals, R.; Chiodo, V.A.; et al. The human rhodopsin kinase promoter in an AAV5 vector confers rod- and cone-specific expression in the primate retina. Hum. Gene Ther. 2012, 23, 1101–1115. [Google Scholar] [CrossRef] [Green Version]
- Virag, T.; Cecchini, S.; Kotin, R.M. Producing recombinant adeno-associated virus in foster cells: Overcoming production limitations using a baculovirus-insect cell expression strategy. Hum. Gene Ther. 2009, 20, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Kotin, R.M.; Snyder, R.O. Manufacturing Clinical Grade Recombinant Adeno-Associated Virus Using Invertebrate Cell Lines. Hum. Gene Ther. 2017, 28, 350–360. [Google Scholar] [CrossRef]
- Kondratov, O.; Marsic, D.; Crosson, S.M.; Mendez-Gomez, H.R.; Moskalenko, O.; Mietzsch, M.; Heilbronn, R.; Allison, J.R.; Green, K.B.; Agbandje-McKenna, M.; et al. Direct Head-to-Head Evaluation of Recombinant Adeno-associated Viral Vectors Manufactured in Human versus Insect Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2661–2675. [Google Scholar] [CrossRef] [Green Version]
- Robert, M.A.; Chahal, P.S.; Audy, A.; Kamen, A.; Gilbert, R.; Gaillet, B. Manufacturing of recombinant adeno-associated viruses using mammalian expression platforms. Biotechnol. J. 2017, 12, 1600193. [Google Scholar] [CrossRef]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Egan, C.; Bayley, S.T.; Branton, P.E. Binding of the Rb1 protein to E1A products is required for adenovirus transformation. Oncogene 1989, 4, 383–388. [Google Scholar] [PubMed]
- Hacker, D.L.; Bertschinger, M.; Baldi, L.; Wurm, F.M. Reduction of adenovirus E1A mRNA by RNAi results in enhanced recombinant protein expression in transiently transfected HEK293 cells. Gene 2004, 341, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Pear, W.S.; Nolan, G.P.; Scott, M.L.; Baltimore, D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 1993, 90, 8392–8396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.Y.; Simmons, D.T. The ability of large T antigen to complex with p53 is necessary for the increased life span and partial transformation of human cells by simian virus 40. J. Virol. 1991, 65, 6447–6453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmerling, V.V.; Pegel, A.; Milian, E.G.; Venereo-Sanchez, A.; Kunz, M.; Wegele, J.; Kamen, A.A.; Kochanek, S.; Hoerer, M. Rational plasmid design and bioprocess optimization to enhance recombinant adeno-associated virus (AAV) productivity in mammalian cells. Biotechnol. J. 2016, 11, 290–297. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Guidelines on Tissue Infectivity Distribution in Transmissible Spongiform Encephalopathies; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Strobel, B.; Miller, F.D.; Rist, W.; Lamla, T. Comparative Analysis of Cesium Chloride- and Iodixanol-Based Purification of Recombinant Adeno-Associated Viral Vectors for Preclinical Applications. Hum. Gene Ther. Methods 2015, 26, 147–157. [Google Scholar] [CrossRef]
- Nass, S.A.; Mattingly, M.A.; Woodcock, D.A.; Burnham, B.L.; Ardinger, J.A.; Osmond, S.E.; Frederick, A.M.; Scaria, A.; Cheng, S.H.; O’Riordan, C.R. Universal Method for the Purification of Recombinant AAV Vectors of Differing Serotypes. Mol. Ther. Methods Clin. Dev. 2018, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Mulagapati, S.H.R.; Chen, Z.; Du, J.; Zhao, X.; Xi, G.; Chen, L.; Linke, T.; Gao, C.; Schmelzer, A.E.; et al. Developing an Anion Exchange Chromatography Assay for Determining Empty and Full Capsid Contents in AAV6.2. Mol. Ther. Methods Clin. Dev. 2019, 15, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Burnham, B.; Nass, S.; Kong, E.; Mattingly, M.; Woodcock, D.; Song, A.; Wadsworth, S.; Cheng, S.H.; Scaria, A.; O’Riordan, C.R. Analytical Ultracentrifugation as an Approach to Characterize Recombinant Adeno-Associated Viral Vectors. Hum. Gene Ther. Methods 2015, 26, 228–242. [Google Scholar] [CrossRef]
- D’Costa, S.; Blouin, V.; Broucque, F.; Penaud-Budloo, M.; Francois, A.; Perez, I.C.; Le Bec, C.; Moullier, P.; Snyder, R.O.; Ayuso, E. Practical utilization of recombinant AAV vector reference standards: Focus on vector genomes titration by free ITR qPCR. Mol. Ther. Methods Clin. Dev. 2016, 5, 16019. [Google Scholar] [CrossRef]
- Lock, M.; Alvira, M.R.; Chen, S.J.; Wilson, J.M. Absolute determination of single-stranded and self-complementary adeno-associated viral vector genome titers by droplet digital PCR. Hum. Gene Ther. Methods 2014, 25, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strain, M.C.; Lada, S.M.; Luong, T.; Rought, S.E.; Gianella, S.; Terry, V.H.; Spina, C.A.; Woelk, C.H.; Richman, D.D. Highly precise measurement of HIV DNA by droplet digital PCR. PLoS ONE 2013, 8, e55943. [Google Scholar] [CrossRef] [PubMed]
- Clement, N.; Grieger, J.C. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Methods Clin. Dev. 2016, 3, 16002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Yue, Y.; Duan, D. Evidence for the failure of adeno-associated virus serotype 5 to package a viral genome > or = 8.2 kb. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Prusky, G.T.; Alam, N.M.; Beekman, S.; Douglas, R.M. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4611–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clerin, E.; Wicker, N.; Mohand-Said, S.; Poch, O.; Sahel, J.A.; Leveillard, T. e-conome: An automated tissue counting platform of cone photoreceptors for rodent models of retinitis pigmentosa. BMC Ophthalmol. 2011, 11, 38. [Google Scholar] [CrossRef]
- Cronin, T.; Lyubarsky, A.; Bennett, J. Dark-rearing the rd10 mouse: Implications for therapy. Adv. Exp. Med. Biol. 2012, 723, 129–136. [Google Scholar]
- Barone, I.; Novelli, E.; Piano, I.; Gargini, C.; Strettoi, E. Environmental enrichment extends photoreceptor survival and visual function in a mouse model of retinitis pigmentosa. PLoS ONE 2012, 7, e50726. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Gografe, S.; Munchow, A.; Lopez-Toledano, M.; Pan, Z.H.; Shen, W. Sex-related differences in the progressive retinal degeneration of the rd10 mouse. Exp. Eye Res. 2019, 187, 107773. [Google Scholar] [CrossRef]
- Narayan, D.S.; Chidlow, G.; Wood, J.P.M.; Casson, R.J. Investigations into Bioenergetic Neuroprotection of Cone Photoreceptors: Relevance to Retinitis Pigmentosa. Front. Neurosci. 2019, 13, 1234. [Google Scholar] [CrossRef]
- The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Guideline ICH. Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals M3 (R2). In International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; ICH: Geneva, Switzerland, 2009. [Google Scholar]
- European Medicines Agency. Guideline on the Non-Clinical Studies Required Before First Clinical Use of Gene Therapy Medicinal Products; EMA: London, UK, 2008. [Google Scholar]
- The Food and Drug Administration. Gene Therapy Guidance Document: Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products (2013); The Food and Drug Administration: White Oak, MD, USA, 2018. [Google Scholar]
- Weed, L.; Ammar, M.J.; Zhou, S.; Wei, Z.; Serrano, L.W.; Sun, J.; Lee, V.; Maguire, A.M.; Bennett, J.; Aleman, T.S. Safety of Same-Eye Subretinal Sequential Readministration of AAV2-hRPE65v2 in Non-human Primates. Mol. Ther. Methods Clin. Dev. 2019, 15, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, K.; Shen, J.; Hafiz, Z.; Hackett, S.F.; Silva, R.L.E.; Khan, M.; Lorenc, V.E.; Chen, D.; Chadha, R.; Zhang, M.; et al. AAV8-vectored suprachoroidal gene transfer produces widespread ocular transgene expression. J. Clin. Investig. 2019, 130, 4901–4911. [Google Scholar] [CrossRef] [PubMed]
- Bruewer, A.R.; Mowat, F.M.; Bartoe, J.T.; Boye, S.L.; Hauswirth, W.W.; Petersen-Jones, S.M. Evaluation of lateral spread of transgene expression following subretinal AAV-mediated gene delivery in dogs. PLoS ONE 2013, 8, e60218. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.P.; Wang, S.; Stachelek, K.; Lee, S.; Reid, M.W.; Thornton, M.E.; Craft, C.M.; Grubbs, B.H.; Cobrinik, D. Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human but not mouse cone precursors. Proc. Natl. Acad. Sci. USA 2018, 115, E9391–E9400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauregui, R.; Park, K.S.; Duong, J.K.; Mahajan, V.B.; Tsang, S.H. Quantitative progression of retinitis pigmentosa by optical coherence tomography angiography. Sci. Rep. 2018, 8, 13130. [Google Scholar] [CrossRef] [PubMed]
- Cabral, T.; Sengillo, J.D.; Duong, J.K.; Justus, S.; Boudreault, K.; Schuerch, K.; Belfort, R., Jr.; Mahajan, V.B.; Sparrow, J.R.; Tsang, S.H. Retrospective Analysis of Structural Disease Progression in Retinitis Pigmentosa Utilizing Multimodal Imaging. Sci. Rep. 2017, 7, 10347. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, I.H.; Birtel, J.; Shanks, M.E.; Clouston, P.; Downes, S.M.; Charbel Issa, P.; MacLaren, R.E. Clinical Characterization of Retinitis Pigmentosa Associated With Variants in SNRNP200. JAMA Ophthalmol. 2019, 137, 1295–1300. [Google Scholar] [CrossRef]
- Fujiwara, K.; Ikeda, Y.; Murakami, Y.; Tachibana, T.; Funatsu, J.; Koyanagi, Y.; Nakatake, S.; Yoshida, N.; Nakao, S.; Hisatomi, T.; et al. Assessment of Central Visual Function in Patients with Retinitis Pigmentosa. Sci. Rep. 2018, 8, 8070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comander, J.; Weigel-DiFranco, C.; Sandberg, M.A.; Berson, E.L. Visual Function in Carriers of X-Linked Retinitis Pigmentosa. Ophthalmology 2015, 122, 1899–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tee, J.J.L.; Yang, Y.; Kalitzeos, A.; Webster, A.; Bainbridge, J.; Michaelides, M. Natural History Study of Retinal Structure, Progression, and Symmetry Using Ellipzoid Zone Metrics in RPGR-Associated Retinopathy. Am. J. Ophthalmol. 2019, 198, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cideciyan, A.V.; Charng, J.; Roman, A.J.; Sheplock, R.; Garafalo, A.V.; Heon, E.; Jacobson, S.G. Progression in X-linked Retinitis Pigmentosa Due to ORF15-RPGR Mutations: Assessment of Localized Vision Changes Over 2 Years. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4558–4566. [Google Scholar] [CrossRef] [Green Version]
- Chaumet-Riffaud, A.E.; Chaumet-Riffaud, P.; Cariou, A.; Devisme, C.; Audo, I.; Sahel, J.A.; Mohand-Said, S. Impact of Retinitis Pigmentosa on Quality of Life, Mental Health, and Employment Among Young Adults. Am. J. Ophthalmol. 2017, 177, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, G. Topography of the Layer of Rods and Cones in the Human Retina; Levin & Munksgaard: Copenhagen, Denmark, 1935. [Google Scholar]
- Staurenghi, G.; Sadda, S.; Chakravarthy, U.; Spaide, R.F. International Nomenclature for Optical Coherence Tomography. Proposed lexicon for anatomic landmarks in normal posterior segment spectral-domain optical coherence tomography: The IN*OCT consensus. Ophthalmology 2014, 121, 1572–1578. [Google Scholar]
- Smith, T.B.; Parker, M.A.; Steinkamp, P.N.; Romo, A.; Erker, L.R.; Lujan, B.J.; Smith, N. Reliability of Spectral-Domain OCT Ellipsoid Zone Area and Shape Measurements in Retinitis Pigmentosa. Transl. Vis. Sci. Technol. 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Nowomiejska, K.; Brzozowska, A.; Koss, M.J.; Weleber, R.G.; Schiefer, U.; Rejdak, K.; Juenemann, A.G.; Maciejewski, R.; Rejdak, R. Quantification of the Visual Field Loss in Retinitis Pigmentosa Using Semi-Automated Kinetic Perimetry. Curr. Eye Res. 2016, 41, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Schuerch, K.; Woods, R.L.; Lee, W.; Duncker, T.; Delori, F.C.; Allikmets, R.; Tsang, S.H.; Sparrow, J.R. Quantifying Fundus Autofluorescence in Patients with Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1843–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, M.A.; Weigel-DiFranco, C.; Dryja, T.P.; Berson, E.L. Clinical expression correlates with location of rhodopsin mutation in dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1934–1942. [Google Scholar]
- Gerkema, M.P.; Davies, W.I.; Foster, R.G.; Menaker, M.; Hut, R.A. The nocturnal bottleneck and the evolution of activity patterns in mammals. Proc. Biol. Sci. R. Soc. 2013, 280, 20130508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillard, C.; Mouret, A.; Niepon, M.L.; Clerin, E.; Yang, Y.; Lee-Rivera, I.; Ait-Ali, N.; Millet-Puel, G.; Cronin, T.; Sedmak, T.; et al. Nxnl2 splicing results in dual functions in neuronal cell survival and maintenance of cell integrity. Hum. Mol. Genet. 2012, 21, 2298–2311. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clérin, E.; Marussig, M.; Sahel, J.-A.; Léveillard, T. Metabolic and Redox Signaling of the Nucleoredoxin-Like-1 Gene for the Treatment of Genetic Retinal Diseases. Int. J. Mol. Sci. 2020, 21, 1625. https://doi.org/10.3390/ijms21051625
Clérin E, Marussig M, Sahel J-A, Léveillard T. Metabolic and Redox Signaling of the Nucleoredoxin-Like-1 Gene for the Treatment of Genetic Retinal Diseases. International Journal of Molecular Sciences. 2020; 21(5):1625. https://doi.org/10.3390/ijms21051625
Chicago/Turabian StyleClérin, Emmanuelle, Myriam Marussig, José-Alain Sahel, and Thierry Léveillard. 2020. "Metabolic and Redox Signaling of the Nucleoredoxin-Like-1 Gene for the Treatment of Genetic Retinal Diseases" International Journal of Molecular Sciences 21, no. 5: 1625. https://doi.org/10.3390/ijms21051625