Density Functional Theory, Chemical Reactivity, Pharmacological Potential and Molecular Docking of Dihydrothiouracil-Indenopyridopyrimidines with Human-DNA Topoisomerase II



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Methods Benchmark
2.2. Comparison of Atomic Charges Methods
2.3. Determination of The Chemical Reactivity of TUDHIPP Derivatives in the Gas and Aqueous Phases
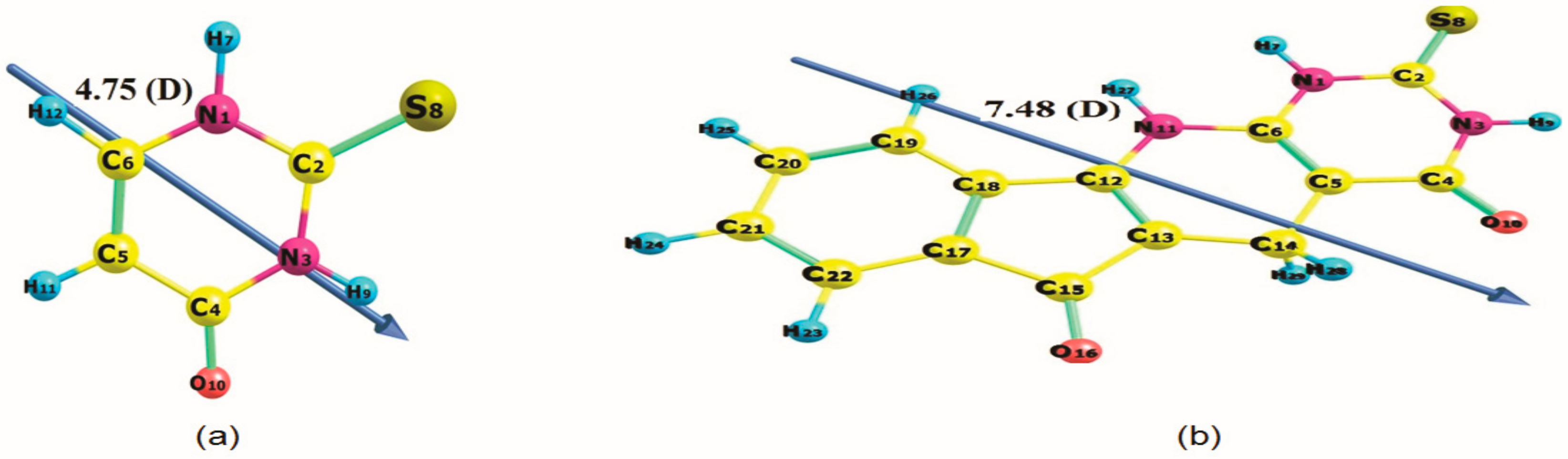
2.3.1. Effect of Solvent on Energy and Dipole Moment
2.3.2. Global Reactivity Descriptors
2.3.3. Local Reactivity Descriptors
2.4. Molecular Electrostatic Potential Surface (MEPS) of TUDHIPP
2.5. Molecular Docking Simulations
2.5.1. TUDHIPP Docked with Human DNA Topoisomerase II alpha (II α)
2.5.2. TUDHIPP docked with human DNA topoisomerase II beta (IIβ)
2.6. Drug Likeness Screening
2.7. Structural Activity/Property Relationship
3. Computational Details
3.1. Geometry Optimization
3.2. Molecular Docking Simulation
3.2.1. Ligand Structure Preparation
3.2.2. Protein Structure Preparation
3.2.3. Molecular Docking
3.2.4. Molecular Docking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khamouli, S.; Belaidi, S.; Medjahed, S.; Belaidi, H. Property/Activity Relationships and Drug Likeness for Pyrimidine Derivatives as Serine/Threonine Protein Kinase B Inhibitors. J. Bionanoscience 2017, 11, 301–309. [Google Scholar] [CrossRef]
- Romagnoli, C.; Zonefrati, R.; Puppi, D.; Rosati, C.; Aldinucci, A.; Palmini, G.; Galli, G.; Chiellini, F.; Martelli, F.S.; Tanini, A. Human Adipose Tissue-Derived Stem Cells and a Poly (ε-Caprolactone) Scaffold Produced by Computer-Aided Wet Spinning for Bone Tissue Engineering. J. Biomater. Tissue Eng. 2017, 7, 622–633. [Google Scholar] [CrossRef]
- Straka, F.; Schornik, D.; Masin, J.; Filova, E.; Mirejovsky, T.; Burdikova, Z.; Svindrych, Z.; Chlup, H.; Horny, L.; Vesely, J. A new approach to heart valve tissue engineering based on modifying autologous human pericardium by 3D cellular mechanotransduction. J. Biomater. Tissue Eng. 2017, 7, 527–543. [Google Scholar] [CrossRef]
- Harkati, D.; Belaidi, S.; Kerassa, A.; Gherraf, N. Molecular Structure, Substituent Effect and Physical-Chemistry Property Relationship of Indole Derivatives. Quantum Matter 2016, 5, 36–44. [Google Scholar] [CrossRef]
- Dua, R.; Shrivastava, S.; Sonwane, S.K.; Srivastava, S.K. Pharmacological significance of synthetic heterocycles scaffold: A review. Adv. Biol. Res. 2011, 5, 120–144. [Google Scholar]
- Builla, J.A.; Vaquero, J.J.; Barluenga, J. Modern Heterocyclic Chemistry; Wiley-Blackwell: Hoboken, NJ, USA, 2011. [Google Scholar]
- Bouchlaleg, L.; Belaidi, S.; Salah, T.; Alafeefy, A.M. Quantitative Structure Activity Relationship Study for Development of Plasmin Inhibitors Controlled by the Spacer Hydantoin. J. Comput. Theore. Nanosci. 2015, 12, 3949–3955. [Google Scholar] [CrossRef]
- Dermeche, K.; Tchouar, N.; Belaidi, S.; Salah, T. Qualitative Structure-Activity Relationships and 2D-QSAR Modeling of TNF-a Inhibition by Thalidomide Derivatives. J. Bionanoscience 2015, 9, 395–400. [Google Scholar] [CrossRef]
- Belaidi, S.; Youcef, O.; Salah, T.; Lanez, T. In Silico Approach for Conformational Analysis, Drug-Likeness Properties and Structure Activity Relationships of 12-Membered Macrolides. J. Comput. Theore. Nanosci. 2015, 12, 4855–4861. [Google Scholar] [CrossRef]
- Cecil, R. Intramolecular bonds in proteins I. The role of sulfur in proteins; Academic Press: New York, NY, USA, 1963; Volume 1. [Google Scholar]
- Evdokimov, N.M.; Van Slambrouck, S.; Heffeter, P.; Tu, L.; Le Calve, B.; Lamoral-Theys, D.; Hooten, C.J.; Uglinskii, P.Y.; Rogelj, S.; Kiss, R.; et al. Structural simplification of bioactive natural products with multicomponent synthesis. 3. Fused uracil-containing heterocycles as novel topoisomerase-targeting agents. J. Med. Chem. 2011, 54, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Hassaneen, H.M.E. A novel one-pot three-components reaction: Synthesis of indeno [2’, 1’: 5, 6] pyrido [2, 3: 4’’, 5’’] pyrimido [2’’, 1’’-c] triazole-5, 7-dione. A new ring system. Arkivoc 2007, 154–163. [Google Scholar]
- Word Health Organization. Cancer Key Facts. 2018. Available online: https://www.who.int/cancer/about/facts/en/ (accessed on 4 January 2019).
- Zheng, H.; Jie, J.; Zheng, Y. Multi-swarm chaotic particle swarm optimization for protein folding. J. Bionanoscience 2013, 7, 643–648. [Google Scholar] [CrossRef]
- Xu, H.; Fang, G.; Gou, J.; Wang, S.; Yao, Q.; Zhang, Y.; Tang, X.; Zhao, Y. Lyophilization of self-assembled polymeric nanoparticles without compromising their microstructure and their in vivo evaluation: Pharmacokinetics, tissue distribution and toxicity. J. Biomater. Tissue Eng. 2015, 5, 919–929. [Google Scholar] [CrossRef]
- Li, X.; Rong, Q.; Chen, S.-L. The Innate Osteogenic Potential of the Canine Maxillary Sinus Membrane: An In Vitro and In Vivo Study. J. Biomater. Tissue Eng. 2015, 5, 445–451. [Google Scholar] [CrossRef]
- Guo, H.-G.; Guo, H.-S.; Yao, F.-L.; Yang, S.-G.; Chen, Z.; Wang, T. In Vitro Study on Biological Potential of Tissue-Engineered Cage by Using Surface-Modified Technique: A Preliminary Evaluation. J. Biomater. Tissue Eng. 2016, 6, 114–121. [Google Scholar] [CrossRef]
- Liu, Y.; Hsing, I. Others Cancer specific targeting by glucosamine coated gold nanoparticles in vitro and in vivo. J. Biomater. Tissue Eng. 2015, 5, 687–696. [Google Scholar] [CrossRef]
- Kim, K.; Kim, D.Y.; Baek, J.H.; Kim, J.H.; Park, Y.H.; Kim, Y.J.; Min, B.H.; Kim, M.S. Comparison of the In Vivo Bioactivity of Electrospun Poly (D, L-lactic-co-glycolic acid) and Poly (L-lactide) Fibrous Scaffolds. J. Biomater. Tissue Eng. 2015, 5, 372–377. [Google Scholar] [CrossRef]
- Jeyapragas, R.; Poovi, G.; Rao, M.S.; Gopal, R.; Sivabalan, M. Formulation, In-Vitro Characterization, Dissolution and Stability Enhancement of Oral Delivery of Nano Self-Emulsifying Powder for Poorly Water Soluble Drug. J. Bionanoscience 2015, 9, 465–474. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. Quantitative prediction of antitarget interaction profiles for chemical compounds. Chem. Res. Toxicol. 2012, 25, 2378–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, K.; Bai, X.-Z.; Li, X.-Q.; Zhang, Y.; Shi, J.-H.; Tang, C.-W.; Hu, D.-H. In vitro induction of adipose-derived mesenchymal stem cells differentiated into sweat gland epithelial cells. J. Comput.Thero. Nanosci. 2014, 11, 1785–1789. [Google Scholar] [CrossRef]
- Campos, J.; Jiménez, C.; Trigo, C.; Ibarra, P.; Rana, D.; Thiruganesh, R.; Ramalingam, M.; Haidar, Z.S. Quartz crystal microbalance with dissipation monitoring: A powerful tool for bionanoscience and drug discovery. J. Bionanoscience 2015, 9, 249–260. [Google Scholar] [CrossRef]
- D Segall, M. Multi-parameter optimization: Identifying high quality compounds with a balance of properties. Curr. Pharm. Des. 2012, 18, 1292–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medjahed, S.; Belaidi, S.; Djekhaba, S.; Tchouar, N.; Kerassa, A. Computational Study of Molecular Electrostatic Potential, Drug Likeness Screening and Structure-Activity/Property Relationships of Thiazolidine-2, 4-Dione Derivatives. J. Bionanoscience 2016, 10, 118–126. [Google Scholar] [CrossRef]
- Soualmia, F.; Belaidi, S.; Belaidi, H.; Tchouar, N.; Almi, Z. Quantitative Structure Anti-Proliferative Activity Against HEPG2 and SW1116 Relationships in a Series of Pyrazine Derivatives. J. Bionanoscience 2017, 11, 584–591. [Google Scholar] [CrossRef]
- Belaidi, S.; Melkemi, N.; Bouzidi, D. Molecular geometry and structure-property relationships for 1, 2-dithiole-3-thione derivatives. Int. J. Chem. Res. 2012, 4, 134–139. [Google Scholar]
- Yam, C.-Y.; Zheng, X.; Chen, G. Some recent progresses in density-functional theory: Efficiency, accuracy, and applicability. J. Comput. Theor. Nanosci. 2006, 3, 857–863. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, A.K.; Karuppayil, S.M. Molecular docking studies on thirteen fluoroquinolines with human topoisomerase II a and b. In Silico Pharmacol. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.H.S.; Oliveira, A.R.S.; Pereira, M.J.S.; Figueiredo, A.F.; Ferreira, J.E.V.; Miranda, R.M. Quantum Chemistry, Quantitative Structure-Activity Relationship and Molecular Docking Study on Fenarimol Derivatives with Biological Activity Against Chagas Disease. J. Comput. Theor. Nanosci. 2015, 12, 3309–3321. [Google Scholar] [CrossRef]
- Langueur, H.; Kassali, K.; Lebgaa, N. Density Functional Study of Structural, Mechanic, Thermodynamic and Dynamic Properties of SiGe Alloys. J. Comput. Theor. Nanosci. 2013, 10, 86–94. [Google Scholar] [CrossRef]
- Melkemi, N.; Belaidi, S. Structure-property relationships and quantitative structure-activity relationship modeling of detoxication properties of some 1, 2-dithiole-3-thione derivatives. J. Comput. Theor. Nanosci. 2014, 11, 801–806. [Google Scholar] [CrossRef]
- Mellaoui, M.; Belaidi, S.; Bouzidi, D.; Gherraf, N. Electronic Structure and Physical-Chemistry Property Relationship for Cephalosporin Derivatives. Quantum Matter 2014, 3, 435–441. [Google Scholar] [CrossRef]
- Belaidi, S.; Belaidi, H.; Bouzidi, D. Computational Methods Applied in Physical-Chemistry Property Relationships of Thiophene Derivatives. J. Comput. Theor. Nanosci. 2015, 12, 1737–1745. [Google Scholar] [CrossRef]
- Belaidi, S.; Mazri, R.; Belaidi, H.; Lanez, T.; Bouzidi, D. Electronic Structure and Physico-Chemical Property Relationship for Thiazole Derivatives. Asian J. Chem. 2013, 25. [Google Scholar] [CrossRef]
- Belaidi, S.; Belaidi, H.; Kerassa, A.; Saoula, M.; Bouzidi, D. Predictive Qualitative Structure-Property/Activity Relationships for Drug Design in Some of Antimycobacterial Pyrrole Derivatives. Quantum Matter 2016, 5, 798–805. [Google Scholar] [CrossRef]
- Esposito, E.X.; Hopfinger, A.J.; Madura, J.D. Methods for applying the quantitative structure-activity relationship paradigm. In Chemoinformatics; Springer: Berlin/Heidelberg, Germany, 2004; pp. 131–213. [Google Scholar]
- Poovi, G. Bio-Physicochemical, Pharmacological Challenges, and Opportunities in the Design of Polymeric Nanoparticles. J. Bionanoscience 2017, 11, 87–104. [Google Scholar] [CrossRef]
- Roy, S. Green Synthesized Gold Nanoparticles: Study of Antimicrobial Activity. J. Bionanoscience 2017, 11, 131–135. [Google Scholar] [CrossRef]
- Bradbury, S.P. Quantitative structure-activity relationships and ecological risk assessment: An overview of predictive aquatic toxicology research. Toxicol. Lett. 1995, 79, 229–237. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Hoekman, D.H. Exploring QSAR: Fundamentals and applications in chemistry and biology; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Hansen, C.; Telzer, B.R.; Zhang, L. Comparative QSAR in toxicology: Examples from teratology and cancer chemotherapy of aniline mustards. Crit. Rev. Toxicol. 1995, 25, 67–89. [Google Scholar] [CrossRef]
- Chen, J.-Z.; Han, X.-W.; Liu, Q.; Makriyannis, A.; Wang, J.; Xie, X.-Q. 3D-QSAR studies of arylpyrazole antagonists of cannabinoid receptor subtypes CB1 and CB2. A combined NMR and CoMFA approach. J. Med. Chem. 2006, 49, 625–636. [Google Scholar] [CrossRef]
- Perkins, R.; Fang, H.; Tong, W.; Welsh, W.J. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Env. Toxicol. Chem. 2003, 22, 1666–1679. [Google Scholar] [CrossRef]
- Salum, L.B.; Andricopulo, A.D. Fragment-based QSAR: Perspectives in drug design. Mol. Divers. 2009, 13, 277–285. [Google Scholar] [CrossRef] [Green Version]
- McGinnity, D.F.; Collington, J.; Austin, R.P.; Riley, R.J. Evaluation of human pharmacokinetics, therapeutic dose and exposure predictions using marketed oral drugs. Curr. Drug Metab. 2007, 8, 463–479. [Google Scholar] [CrossRef] [PubMed]
- Nithiyanantham, S.; Palaniappan, L.; Lenin, M. Physico-Chemical and Bio-Chemical Nature of Pectin with Amylase. J. Bionanoscience 2015, 9, 359–362. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Kubsik, M.; Kaminski, R.; Wozniak, K.; Dominiak, P.M. From a single molecule to molecular crystal architectures: Structural and energetic studies of selected uracil derivatives. Cryst. Growth Des. 2012, 12, 2508–2524. [Google Scholar] [CrossRef]
- Cookney, J.; Light, M.E.; Benes, N.E.; Fila, V. CCDC 1483947: Experimental Crystal Structure Determination. Available online: https://research.utwente.nl/en/publications/ccdc-1483947-experimental-crystal-structure-determination-2 (accessed on 13 February 2020).
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Cryst. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Zolghadr, A.R.; Ghatee, M.H.; Moosavi, F. The effect of various quantum mechanically derived partial atomic charges on the bulk properties of chloride-based ionic liquids. Chem. Phys. 2016, 475, 23–31. [Google Scholar] [CrossRef]
- Defranceschi, M.; Le Bris, C. Mathematical Models and Methods for ab Initio Quantum Chemistry; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Mendoza-Huizar, L.H.; Rios-Reyes, C.H. Chemical reactivity of atrazine employing the Fukui function. J. Mex. Chem. Soc. 2011, 55, 142–147. [Google Scholar]
- Parr, R.G. Density-Functional Theory of Atoms and Molecules. Int. Academy Quantum Mole. Sci. 1980, 3, 5–15. [Google Scholar]
- Wu, R.; Zhang, T.; Qiao, X.; Zhang, J.; Yang, L.; Yu, W. Synthesis and crystal structure of 7-nitro-5-sulfo-napthalene-1,4-dicarboxylate acid. Chin. J. Struct. Chem. 2006, 7, 849–853. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Roy, D.R. Update 1 of: Electrophilicity index. Chem. Rev. 2007, 107, PR46–PR74. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Giri, S. Stability, reactivity, and aromaticity of compounds of a multivalent superatom. J. Phys. Chem. A 2007, 111, 11116–11121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, R.R.; Fuentealba, P.; Galvan, M.; Perez, P. A direct evaluation of regional Fukui functions in molecules. Chem. Phys. Lett. 1999, 304, 405–413. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbe, A. New dual descriptor for chemical reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Sajith, P.K.; Suresh, C.H. Mechanisms of reductive eliminations in square planar Pd (II) complexes: Nature of eliminated bonds and role of trans influence. Inorg. Chem. 2011, 50, 8085–8093. [Google Scholar] [CrossRef]
- Vinod, K.S.; Periandy, S.; Govindarajan, M. Spectroscopic analysis of cinnamic acid using quantum chemical calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 136, 808–817. [Google Scholar] [CrossRef]
- Luque, F.J.; Lopez, J.M.; Orozco, M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects.”. Theor. Chem. Acc. 2000, 103, 343–345. [Google Scholar]
- Molecular electrostatic potentials for large systems. In Molecular Electrostatic Potentials Concepts and Applications; Murray, J.S.; Sen, K. (Eds.) Theoretical and Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 1996. [Google Scholar]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.L.; Baranowski, J.; Pernet, A.G. Mechanism of inhibition of DNA gyrase by quinolone antibacterials: Specificity and cooperativity of drug binding to DNA. Biochemistry 1989, 28, 3879–3885. [Google Scholar] [CrossRef]
- Wilson, C.O.; Beale, J.M.; Block, J.H. Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry; Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011. [Google Scholar]
- Wei, B.; Shi, Z.; Xiao, J.; Xu, Y.; Lv, L. In vivo and in vitro antibacterial effect of nano-structured titanium coating incorporated with silver oxide nanoparticles. J. Biomater. Tissue Eng. 2017, 7, 418–425. [Google Scholar] [CrossRef]
- Khojasteh, S.C.; Wong, H.; Hop, C.E.C.A. Drug Metabolism and Pharmacokinetics Quick Guide; Springer: New York, NY, USA, 2011. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Schultes, S.; de Graaf, C.; Haaksma, E.E.J.; de Esch, I.J.P.; Leurs, R.; Krämer, O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discov. Today Technol. 2010, 7, e157–e162. [Google Scholar] [CrossRef] [Green Version]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Singh, T.; Adekoya, O.A.; Jayaram, B. Understanding the binding of inhibitors of matrix metalloproteinases by molecular docking, quantum mechanical calculations, molecular dynamics simulations, and a MMGBSA/MMBappl study. Mol. Biosyst. 2015, 11, 1041–1051. [Google Scholar] [CrossRef] [Green Version]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Viswanadhan, V.N.; Ghose, A.K.; Revankar, G.R.; Robins, R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain. J. Chem. Inf. Comput. Sci. 1989, 29, 163–172. [Google Scholar]
- Johnson, T.W.; Dress, K.R.; Edwards, M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 2009, 19, 5560–5564. [Google Scholar] [CrossRef]
- Wang, J.; Xie, X.-Q.; Hou, T.; Xu, X. Fast approaches for molecular polarizability calculations. J. Phys. Chem. A 2007, 111, 4443–4448. [Google Scholar] [CrossRef] [Green Version]
- Kapupara, P.P.; Matholiya, C.R.; Dedakiya, A.S.; Desai, T.R. 2D-QSAR Study on 1-Acetyl-3-Aryl-5-(4-Methoxyphenyl) Pyrazole Analogues As an Antifungal Agents. Int. Bull. Drug Res. 2011, 1, 1–10. [Google Scholar]
- Andrasi, M.; Buglyo, P.; Zekany, L.; Gaspar, A. A comparative study of capillary zone electrophoresis and pH-potentiometry for determination of dissociation constants. J. Pharm. Biomed. Anal. 2007, 44, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revis. C. 01 2010. Available online: https://gaussian.com/g09citation/ (accessed on 3 March 2018).
- Roothaan, C.C.J. New developments in molecular orbital theory. Rev. Mod. Phys. 1951, 23, 69. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.G.; Frisch, M.J. Analytic second derivatives of the gradient-corrected density functional energy. Effect of quadrature weight derivatives. Chem. Phys. Lett. 1993, 216, 133–140. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Ulic, S.E.; Vedova, C.O.D.; Hermann, A.; Mack, H.-G.; Oberhammer, H. Preparation and Properties of Trifluorothioacetic Acid-S-(trifluoromethyl) ester, CF 3C (O) SCF 3. J. Phys. Chem. A 2008, 112, 6211–6216. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree—Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009; Available online: https://www.who.int/cancer/about/facts/en/ (accessed on 4 January 2019).
- Andrienko, G.A. Chemcraf Program. 2018. Available online: https://www.chemcraftprog.com/about.html (accessed on 13 February 2020).
- Froimowitz, M. HyperChem: A software package for computational chemistry and molecular modeling. Biotechniques 1993, 14, 1010. [Google Scholar]
- MarvinSketch Software. Version 18. ChemAxon Company, 2015. Available online: http://www.chemaxon.com (accessed on 13 February 2020).
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Bora, U. Molecular docking studies of curcumin natural derivatives with DNA topoisomerase I and II-DNA complexes. Interdiscip. Sci. Comput. Life Sci. 2014, 6, 285–291. [Google Scholar] [CrossRef]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase II alpha: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Li, T.-K.; Farh, L.; Lin, L.-Y.; Lin, T.-S.; Yu, Y.-J.; Yen, T.-J.; Chiang, C.-W.; Chan, N.-L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand—Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Exp.* | 6-311++G (d, p) | CC-PVDZ | ||||
|---|---|---|---|---|---|---|---|
| ab initio/HF | ab initio/MP2 | ab initio/B3LYP | ab initio/HF | ab initio/MP2 | ab initio/B3LYP | ||
| R(N1,C2) | 1.351 | 1.353 | 1.380 | 1.378 | 1.355 | 1.382 | 1.380 |
| R(N1,C6) | 1.371 | 1.374 | 1.376 | 1.374 | 1.374 | 1.377 | 1.375 |
| R(N1,H7) | 1.030 | 0.994 | 1.011 | 1.010 | 0.998 | 1.016 | 1.014 |
| R(C2,N3) | 1.357 | 1.350 | 1.376 | 1.370 | 1.351 | 1.376 | 1.370 |
| R(C2,S8) | 1.683 | 1.661 | 1.643 | 1.662 | 1.665 | 1.658 | 1.668 |
| R(N3,C4) | 1.394 | 1.398 | 1.414 | 1.417 | 1.398 | 1.418 | 1.419 |
| R(N3,H9) | 1.030 | 0.998 | 1.015 | 1.013 | 1.002 | 1.021 | 1.018 |
| R(C4,C5) | 1.442 | 1.460 | 1.458 | 1.456 | 1.462 | 1.463 | 1.460 |
| R(C4,O10) | 1.235 | 1.187 | 1.220 | 1.214 | 1.190 | 1.222 | 1.217 |
| R(C5,C6) | 1.354 | 1.328 | 1.356 | 1.348 | 1.332 | 1.362 | 1.353 |
| R(C5,H11) | 1.080 | 1.070 | 1.082 | 1.079 | 1.077 | 1.090 | 1.088 |
| R(C6,H12) | 1.080 | 1.073 | 1.085 | 1.083 | 1.080 | 1.093 | 1.091 |
| A% | 1.386 | 1.200 | 1.102 | 1.192 | 1.257 | 1.105 | |
| B3lyp | Atoms | 6−31G | 6−311G | 6−31+G (d) | 6−311+G (d) | 6−31++G (d,p) | 6−311++G (d,p) | Stander Error Mean |
|---|---|---|---|---|---|---|---|---|
| Mulliken | N1 | −0.742 | −0.774 | −0.572 | −0.377 | −0.393 | −0.175 | 0.095 |
| C2 | 0.215 | 0.160 | 0.251 | 0.528 | 0.174 | 0.476 | 0.065 | |
| N3 | −0.674 | −0.703 | −0.612 | −0.525 | −0.393 | −0.330 | 0.062 | |
| C4 | 0.462 | 0.554 | 0.919 | 0.699 | 0.766 | 0.383 | 0.082 | |
| C5 | 0.064 | −0.165 | 0.293 | 0.872 | 0.308 | 1.170 | 0.205 | |
| C6 | 0.628 | 0.811 | 0.078 | −0.334 | −0.378 | −0.809 | 0.256 | |
| S8 | −0.064 | −0.058 | −0.068 | −0.580 | 0.011 | −0.600 | 0.116 | |
| O10 | −0.427 | −0.378 | −0.513 | −0.327 | −0.487 | −0.296 | 0.035 | |
| N11 | −0.844 | −0.916 | −0.599 | −0.328 | −0.320 | −0.036 | 0.139 | |
| C12 | 0.250 | 0.286 | −0.318 | −0.871 | −0.560 | −1.235 | 0.248 | |
| C13 | 0.072 | −0.007 | 1.429 | 1.339 | 0.737 | 1.004 | 0.252 | |
| C14 | −0.309 | −0.414 | −0.594 | −1.241 | −0.549 | −0.765 | 0.135 | |
| C15 | 0.190 | 0.234 | −0.651 | −1.284 | −0.320 | −1.004 | 0.254 | |
| O16 | −0.387 | −0.317 | −0.479 | −0.247 | −0.468 | −0.239 | 0.043 | |
| NBO | N1 | −0.587 | −0.573 | −0.608 | −0.583 | −0.607 | −0.587 | 0.006 |
| C2 | 0.269 | 0.277 | 0.239 | 0.269 | 0.239 | 0.269 | 0.007 | |
| N3 | −0.613 | −0.588 | −0.634 | −0.608 | −0.633 | −0.613 | 0.007 | |
| C4 | 0.654 | 0.619 | 0.654 | 0.654 | 0.654 | 0.654 | 0.006 | |
| C5 | −0.195 | −0.165 | −0.205 | −0.193 | −0.202 | −0.195 | 0.006 | |
| C6 | 0.429 | 0.417 | 0.420 | 0.430 | 0.420 | 0.429 | 0.002 | |
| S8 | −0.177 | −0.224 | −0.169 | −0.180 | −0.170 | −0.177 | 0.008 | |
| O10 | −0.592 | −0.584 | −0.596 | −0.593 | −0.599 | −0.592 | 0.002 | |
| N11 | −0.576 | −0.564 | −0.596 | −0.574 | −0.595 | −0.576 | 0.005 | |
| C12 | 0.225 | 0.220 | 0.236 | 0.236 | 0.233 | 0.225 | 0.003 | |
| C13 | −0.144 | −0.113 | −0.162 | −0.144 | −0.154 | −0.144 | 0.007 | |
| C14 | −0.392 | −0.410 | −0.478 | −0.397 | −0.462 | −0.392 | 0.016 | |
| C15 | 0.540 | 0.498 | 0.550 | 0.531 | 0.537 | 0.540 | 0.007 | |
| O16 | −0.523 | −0.512 | −0.526 | −0.524 | −0.526 | −0.523 | 0.002 | |
| ChelpG | N1 | −0.395 | −0.423 | −0.370 | −0.386 | −0.370 | −0.362 | 0.009 |
| C2 | 0.305 | 0.321 | 0.286 | 0.308 | 0.284 | 0.297 | 0.006 | |
| N3 | −0.377 | −0.400 | −0.354 | −0.384 | −0.350 | −0.362 | 0.008 | |
| C4 | 0.661 | 0.709 | 0.670 | 0.700 | 0.672 | 0.688 | 0.008 | |
| C5 | −0.447 | −0.487 | −0.447 | −0.476 | −0.461 | −0.478 | 0.007 | |
| C6 | 0.520 | 0.561 | 0.511 | 0.509 | 0.548 | 0.503 | 0.010 | |
| S8 | −0.413 | −0.407 | −0.380 | −0.381 | −0.379 | −0.379 | 0.007 | |
| O10 | −0.555 | −0.578 | −0.568 | −0.571 | −0.570 | −0.569 | 0.003 | |
| N11 | −0.598 | −0.619 | −0.601 | −0.572 | −0.648 | −0.575 | 0.012 | |
| C12 | 0.214 | 0.213 | 0.207 | 0.202 | 0.247 | 0.228 | 0.007 | |
| C13 | −0.398 | −0.422 | −0.368 | −0.368 | −0.383 | −0.388 | 0.008 | |
| C14 | 0.539 | 0.578 | 0.558 | 0.555 | 0.538 | 0.574 | 0.007 | |
| C15 | 0.559 | 0.587 | 0.565 | 0.560 | 0.566 | 0.562 | 0.004 | |
| O16 | −0.489 | −0.506 | −0.508 | −0.504 | −0.508 | −0.503 | 0.003 |
| Compound | Gas Phase | Water Phase | ||
|---|---|---|---|---|
| ET, au | μ, D | ET, au | μ, D | |
| 1H | −1252.08722 | 7.48 | −1252.11530 | 11.01 |
| 2H | −1483.19061 | 6.96 | −1483.22089 | 11.02 |
| 3H | −1942.81366 | 7.99 | −1942.84419 | 12.23 |
| 4H | −1582.45943 | 7.86 | −1582.49025 | 12.04 |
| 5H | −4056.73331 | 8.01 | −4056.76391 | 12.24 |
| 6H | −1687.75484 | 10.68 | −1687.79006 | 15.26 |
| 7H | −1597.74680 | 7.78 | −1597.77917 | 12.19 |
| 8H | −1538.56733 | 6.98 | −1538.60157 | 11.09 |
| 9H | −1617.19402 | 6.59 | −1617.22616 | 10.44 |
| Comp. | Phases | EHOMO, au | ELUMO, au | Eg, eV | I, eV | A, eV | χ, eV | η, eV | S, eV−1 | V, eV | ω, eV | N, eV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1H | Gas phase | −0.23393 | −0.11200 | 3.32 | 6.37 | 3.05 | 4.71 | 1.66 | 0.30 | −4.71 | 6.68 | −2.15 |
| Aqueous phase | −0.22618 | −0.10754 | 3.23 | 6.15 | 2.93 | 4.54 | 1.61 | 0.31 | −4.54 | 6.39 | −1.94 | |
| 2H | Gas phase | −0.23738 | −0.11034 | 3.46 | 6.46 | 3.00 | 4.73 | 1.73 | 0.29 | −4.73 | 6.47 | −2.25 |
| Aqueous phase | −0.23132 | −0.10799 | 3.36 | 6.29 | 2.94 | 4.62 | 1.68 | 0.30 | −4.62 | 6.35 | −2.08 | |
| 3H | Gas phase | −0.24158 | −0.11415 | 3.47 | 6.57 | 3.11 | 4.84 | 1.73 | 0.29 | −4.84 | 6.76 | −2.36 |
| Aqueous phase | −0.23243 | −0.10867 | 3.37 | 6.32 | 2.96 | 4.64 | 1.68 | 0.30 | −4.64 | 6.40 | −2.11 | |
| 4H | Gas phase | −0.24081 | −0.11339 | 3.47 | 6.55 | 3.09 | 4.82 | 1.73 | 0.29 | −4.82 | 6.70 | −2.34 |
| Aqueous phase | −0.23216 | −0.10839 | 3.37 | 6.32 | 2.95 | 4.63 | 1.68 | 0.30 | −4.63 | 6.37 | −2.11 | |
| 5H | Gas phase | −0.24176 | −0.11435 | 3.47 | 6.58 | 3.11 | 4.85 | 1.73 | 0.29 | −4.85 | 6.77 | −2.37 |
| Aqueous phase | −0.23248 | −0.10874 | 3.37 | 6.33 | 2.96 | 4.64 | 1.68 | 0.30 | −4.64 | 6.40 | −2.12 | |
| 6H | Gas phase | −0.24851 | −0.12131 | 3.46 | 6.76 | 3.30 | 5.03 | 1.73 | 0.29 | −5.03 | 7.31 | −2.55 |
| Aqueous phase | −0.23470 | −0.11624 | 3.22 | 6.39 | 3.16 | 4.77 | 1.61 | 0.31 | −4.77 | 7.07 | −2.18 | |
| 7H | Gas phase | −0.22399 | −0.10882 | 3.13 | 6.09 | 2.96 | 4.53 | 1.57 | 0.32 | −4.53 | 6.54 | −1.88 |
| Aqueous phase | −0.22796 | −0.10734 | 3.28 | 6.20 | 2.92 | 4.56 | 1.64 | 0.30 | −4.56 | 6.34 | −1.99 | |
| 8H | Gas phase | −0.20975 | −0.10691 | 2.80 | 5.71 | 2.91 | 4.31 | 1.40 | 0.36 | −4.31 | 6.63 | −1.50 |
| Aqueous phase | −0.21423 | −0.10680 | 2.92 | 5.83 | 2.91 | 4.37 | 1.46 | 0.34 | −4.37 | 6.53 | −1.62 | |
| 9H | Gas phase | −0.19625 | −0.10520 | 2.48 | 5.34 | 2.86 | 4.10 | 1.24 | 0.40 | −4.10 | 6.79 | −1.13 |
| Aqueous phase | −0.20005 | −0.10646 | 2.55 | 5.44 | 2.90 | 4.17 | 1.27 | 0.39 | −4.17 | 6.83 | −1.23 |
| Atoms | Gas Phase | Water | ||||
|---|---|---|---|---|---|---|
| f(−) | f(+) | Δf | f(−) | f(+) | Δf | |
| N1 | 0.019 | 0.006 | −0.013 | 0.005 | 0.007 | 0.002 |
| C2 | 0.006 | 0.004 | −0.002 | 0.001 | 0.006 | 0.005 |
| N3 | 0.018 | 0.009 | −0.010 | 0.010 | 0.012 | 0.002 |
| C4 | 0.009 | 0.011 | 0.001 | 0.012 | 0.015 | 0.003 |
| C5 | 0.165 | 0.041 | −0.124 | 0.129 | 0.044 | −0.085 |
| C6 | 0.041 | 0.035 | −0.005 | 0.025 | 0.043 | 0.017 |
| H7 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| S8 | 0.120 | 0.005 | −0.115 | 0.051 | 0.005 | −0.046 |
| H9 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| O10 | 0.021 | 0.011 | −0.010 | 0.022 | 0.012 | −0.010 |
| N11 | 0.107 | 0.015 | −0.092 | 0.118 | 0.017 | −0.101 |
| C12 | 0.060 | 0.155 | 0.095 | 0.063 | 0.168 | 0.105 |
| C13 | 0.178 | 0.077 | −0.101 | 0.241 | 0.058 | −0.183 |
| C14 | 0.031 | 0.001 | −0.030 | 0.029 | 0.001 | −0.028 |
| C15 | 0.008 | 0.148 | 0.141 | 0.014 | 0.166 | 0.152 |
| O16 | 0.021 | 0.154 | 0.133 | 0.030 | 0.143 | 0.113 |
| C17 | 0.026 | 0.090 | 0.064 | 0.038 | 0.078 | 0.040 |
| C18 | 0.024 | 0.069 | 0.045 | 0.036 | 0.061 | 0.025 |
| C19 | 0.011 | 0.024 | 0.012 | 0.013 | 0.025 | 0.012 |
| C20 | 0.023 | 0.058 | 0.035 | 0.034 | 0.055 | 0.021 |
| C21 | 0.032 | 0.061 | 0.029 | 0.045 | 0.053 | 0.008 |
| C22 | 0.004 | 0.025 | 0.021 | 0.005 | 0.030 | 0.025 |
| H23 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| H24 | 0.000 | 0.001 | 0.000 | 0.000 | 0.001 | 0.000 |
| H25 | 0.000 | 0.001 | 0.000 | 0.000 | 0.001 | 0.000 |
| H26 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| H27 | 0.002 | 0.000 | −0.002 | 0.002 | 0.000 | −0.002 |
| H28 | 0.039 | 0.000 | −0.039 | 0.038 | 0.000 | −0.038 |
| H29 | 0.036 | 0.000 | −0.035 | 0.038 | 0.000 | −0.038 |
| Ligand | Run no. | Interaction Residue in Human Topo II | Interaction Atoms (Amino Acid…Ligand) HB Length (Ao) | H Bonds Formed | Binding Energy Kcal/mol | Inhibition Constant Ki, nM |
|---|---|---|---|---|---|---|
| 1H | 22 | ASN780.A SER891.A | ND2……….O16 (3.06) OG…………H9N3 (2.71) | 2 | −7.9 | 1.61, µM |
| 2H | 18 | GLN773.A ASN770.A SER800.A DC9.C | OE1………..H27N11 (3.11) OD1………..H7N1 (2.6), OD1…………H27N11 (3.15) OG…………S8 (3.78) O3…………O16 (3.58) | 5 | −9.29 | 155.7 |
| 3H | 46 | GLN773.A ASN770.A ASN779.A TYR892.A DG10.C | OE1………...H7N1 (3.7) O…………...H7N1 (3.72) ND2………..H27N11 (3.17) OH…………Cl39 (3.16) OP2………..N3H9 (2.65) | 5 | −8.61 | 485.91 |
| 4H | 62 | SER778.A GLU854.A ARG929.A ASN779.A TYR892.A | OG…………H7N1 (3.19) N…………...O16 (2.98) NH2………..O10 (2.85) OD1………..H27N11 (2.62) OH…………F39 (2.66) | 5 | −8.85 | 327.53 |
| 5H | 98 | GLN773.A ASN779.A DG10.C | OE1………..H7N1 (3.69) ND2……….H27N11 (3.2) OP2………..H9N3 (2.62) | 3 | −8.74 | 392.18 |
| 6H | 3 | DG10.C ARG929.A TYR892.A ASN779.A | OP2………..N3H9 (2.67) NE…………O40 (3.37), NH2…………O40 (2.81) OH…………O40 (2.65) ND2...............H27N11 (3.2) | 5 | −9.17 | 189.18 |
| 7H | 48 | SER778.A GLU854.A TYR892.A ARG929.A ASN779.A | OG…………H7N1 (3.18) N…………...O16 (2.99) OH…………O39 (2.69) NH2………..O10 (3.17) OD1………..H27N11 (2.6) | 5 | −9.23 | 170.11 |
| 8H | 43 | SER778.A GLU854.A TYR892.A ILE864.A ARG929.A | OG…………H7N1 (3.02) N…………...O16 (3.02) OH…………N39 (2.65) O…………...H40N39 (2.81) NH2………..O10 (3.23) | 5 | −9.06 | 227.858 |
| 9H | 93 | TYR892.A GLU854.A | OH…………S8 (3.121) N…………...O10 (3.05) | 2 | −8.53 | 555.3 |
| Etoposide | 43 | GLN773.A LYS798.A SER800.A DC9.C DG10.C | NE2………..O1 (2.85) O………….. O9 (2.75) O…………...O9 (2.81) O3/…………O11 (3.1) O5/…………O11 (2.64) | 5 | −8.39 | 708.84 |
| Ligand | Run no. | Interaction residue in human Topo IIα | Interaction Atoms (Amino Acid …Ligand) HB Length (Ao) | H Bonds Formed | Binding Energy Kcal/mol | Inhibition Constant Ki, nM |
|---|---|---|---|---|---|---|
| 1H | 56 | DC8.C DG10.D DT9.D | O3…………O16 (3.74) O4/…………H7N1 (3.05) O3/…………H27N11 (2.89) | 3 | −7.25 | 4.88, µM |
| 2H | 4 | ASP479 LYS456 DG10.D | OD1………..H7N1 (3.05), OD1…H27N11 (3.13) NZ…………S8 (3.17) O3/…………S8 (3.19) | 4 | −9.29 | 154.3 |
| 3H | 31 | DC8.C DT9.D | O3/…………S8 (3.13) OP2………...H9N3 (2.71) | 2 | −9.28 | 157.67 |
| 4H | 71 | DC8.C DT9.D | O3/…………S8 (3.31) OP2………...H9N3 (2.66) | 2 | −9.04 | 235.79 |
| 5H | 75 | ASP479.A DC8.C DT9.D | OD1……….H7N1 (2.81), OD1….H27N11 (3.15) O3/………...O16 (3.11) O3/………...H27N11 (3.3) | 4 | −9.5 | 108.42 |
| 6H | 28 | ASP479 DC11.D DT9.D | OD1……….H9N3 (2.8), N…S8 (3.71) N1…………O41 (3.81) O3/…………S8 (3.26) | 4 | −10.07 | 41.62 |
| 7H | 75 | GLN778.A DC8.C | N…………...O10 (3.51) O3/…………S8 (3.58) | 2 | −9.34 | 142.45 |
| 8H | 28 | ASP479.A DC8.C | OD1………..N39 (2.64), N…N39 (3.12) O3/…………O10 (3.39), N1…O10 (3.46) | 4 | −9.56 | 97.78 |
| 9H | 49 | ASP479.A DT9.D | OD1……….H7N1 (3.27), OD1…H27N11 (3.04) O3/…………H27N11 (3.42) | 3 | −9.16 | 193.43 |
| Etoposide | 19 | ASP479.A DC8.C DT9.D | N…………...O9 (3.08), OD1….O9 (2.78) O3/…………O8 (2.87) O3/…………O9 (3.37) | 4 | −11.59 | 3.17 |
| Compounds | Lipinski Rules | Veber Rules | Log D (7.4) | |||||
|---|---|---|---|---|---|---|---|---|
| Log P | MW (DA) | HBD | HBA | Lipinski Score of 5 | nrotb | PSA | ||
| 1H | −0.16 | 283.3 | 3 | 4 | 4 | 0 | 70.23 | 0.893 |
| 2H | 1.39 | 359.4 | 3 | 4 | 4 | 1 | 70.23 | 2.310 |
| 3H | 1.91 | 393.85 | 3 | 5 | 4 | 1 | 70.23 | 2.914 |
| 4H | 1.53 | 377.39 | 3 | 5 | 4 | 1 | 70.23 | 2.452 |
| 5H | 2.18 | 438.3 | 3 | 5 | 4 | 1 | 70.23 | 3.078 |
| 6H | −2.52 | 404.4 | 3 | 6 | 4 | 2 | 113.37 | 2.250 |
| 7H | 1.14 | 389.43 | 3 | 5 | 4 | 2 | 79.46 | 2.152 |
| 8H | 0.61 | 374.42 | 4 | 5 | 4 | 1 | 96.25 | 1.480 |
| 9H | 1.66 | 402.47 | 3 | 5 | 4 | 2 | 73.47 | 2.416 |
| Etoposide | 1.27 | 588.56 | 3 | 13 | 2 | 5 | 160.83 | 1.15 |
| Etoposide * | 1.16 | 588.56 | 3 | 13 | 2 | 5 | 160.83 | |
| Etoposide # | 588.56 | 3 | 13 | 2 | 0.74 | |||
| Comp. | Polarizability (A3) | Refractivity (A3) | Vol (A3) | Surface Area (Grid) A2 | HE (kcal/mol) |
|---|---|---|---|---|---|
| 1H | 31.22 | 77.23 | 737.5 | 454.55 | −10.87 |
| 2H | 40.88 | 101.87 | 739.85 | 554.14 | −11.73 |
| 3H | 42.81 | 106.68 | 982.07 | 584.73 | −11.43 |
| 4H | 40.79 | 102.09 | 946.24 | 557.2 | −11.47 |
| 5H | 43.51 | 109.49 | 1000.77 | 590.22 | −11.42 |
| 6H | 42.72 | 108.18 | 998.3 | 586.26 | −16.69 |
| 7H | 43.35 | 108.33 | 1014.72 | 598.73 | −13.42 |
| 8H | 42.23 | 106.57 | 973.55 | 571.36 | −15.84 |
| 9H | 45.9 | 116.3 | 1070.91 | 621.57 | −10.22 |
| Etoposide | 55.15 | 138.73 | 1448.08 | 808.26 | −25.39 |
| Etoposide * | 58.77 | 139.02 | |||
| Etoposide #,+,† | 55.5 | 140.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elshakre, M.E.; Noamaan, M.A.; Moustafa, H.; Butt, H. Density Functional Theory, Chemical Reactivity, Pharmacological Potential and Molecular Docking of Dihydrothiouracil-Indenopyridopyrimidines with Human-DNA Topoisomerase II. Int. J. Mol. Sci. 2020, 21, 1253. https://doi.org/10.3390/ijms21041253
Elshakre ME, Noamaan MA, Moustafa H, Butt H. Density Functional Theory, Chemical Reactivity, Pharmacological Potential and Molecular Docking of Dihydrothiouracil-Indenopyridopyrimidines with Human-DNA Topoisomerase II. International Journal of Molecular Sciences. 2020; 21(4):1253. https://doi.org/10.3390/ijms21041253
Chicago/Turabian StyleElshakre, Mohamed E., Mahmoud A. Noamaan, Hussein Moustafa, and Haider Butt. 2020. "Density Functional Theory, Chemical Reactivity, Pharmacological Potential and Molecular Docking of Dihydrothiouracil-Indenopyridopyrimidines with Human-DNA Topoisomerase II" International Journal of Molecular Sciences 21, no. 4: 1253. https://doi.org/10.3390/ijms21041253