RNA Editing as a Therapeutic Approach for Retinal Gene Therapy Requiring Long Coding Sequences

 , , and
, , and
Abstract
:1. Introduction
2. ADARs: Adenosine Deaminase Acting on RNA
2.1. ADAR Expression, Structure and Function
2.2. Engineering ADARs for RNA Editing
2.2.1. A-I Editors
2.2.2. C-U Editors
3. RNA Editing with ADARs
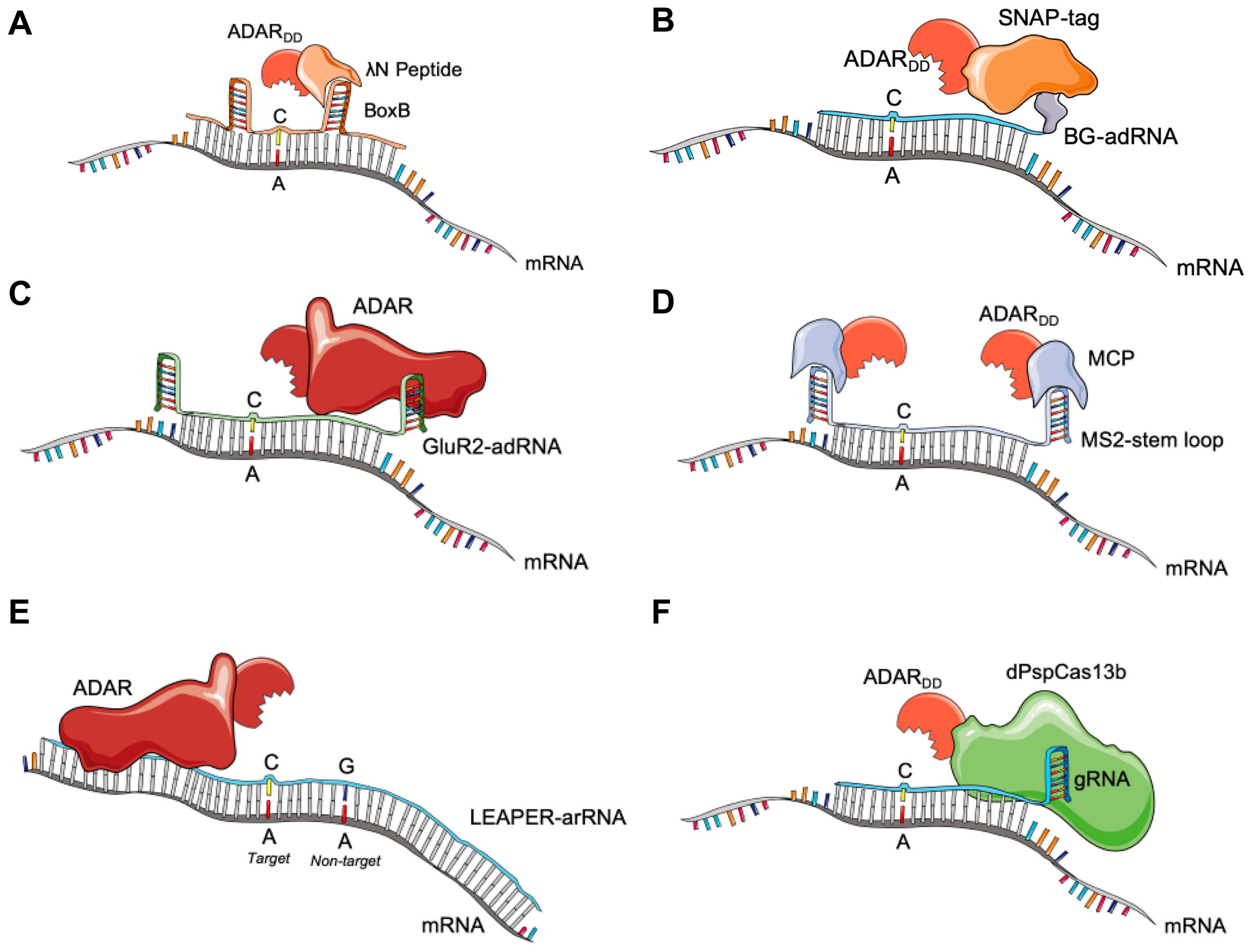
3.1. BoxB-λN-ADAR
3.2. SNAP-ADAR
3.3. GluR2-ADAR
3.4. MS2-MCP-ADAR
3.5. Endogenous ADAR Approaches
3.6. CRISPR-Cas13 for RNA Editing
3.6.1. A to I Editing
3.6.2. C to U Editing
3.6.3. Synthetic CRISPR-Like RNA Editors
4. RNA Editing for Large Inherited Retinal Genes
4.1. Gene Targets for RNA Editing in Inherited Retinal Degeneration
4.2. Distribution of Targetable Mutations with RNA Editing
4.3. A Case Study of RNA Editing for Inherited Retinal Disease
4.4. Towards Clinical use of RNA Editing
4.4.1. Clinical Considerations of RNA Editing
4.4.2. Endogenous and Exogenous ADAR Strategies
4.4.3. Delivery Challenges
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| ADAR | Adenosine deaminase acting on RNA |
| ADARDD | Deaminase domain of ADAR |
| BoxB-λN | BoxB RNA hairpin-Lambda N protein |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeat |
| Cas | CRISPR-associated genes |
| CIRTS | CRISPR-Cas-inspired RNA targeting system |
| dsRBD | Double-stranded RNA-binding domain |
| HEK | Human embryonic kidney |
| HEPN | Higher eukaryotes and prokaryotes nucleotide-binding domain |
| HUVEC | Human umbilical vein endothelial cells |
| LEAPER | Leveraging Endogenous ADAR for Programmable Editing of RNA |
| MS2-MCP | MS2 bacteriophage coat protein |
| NLS | Nuclear localization signal |
| NES | Nuclear export signal |
| PAM | Protospacer adjacent motif |
| PFS | Protospacer flanking sequences |
| REPAIR | RNA Editing for Programmable A to I Replacement |
| RESCUE | RNA Editing for Specific C to U Exchange |
| RESTORE | Recruiting Endogenous ADAR to Specific Transcripts for Oligonucleotide-mediated RNA Editing |
| RNA | Ribonucleic acid |
| UTR | Untranslated region |
References
- Zhang, F. Development of CRISPR-Cas systems for genome editing and beyond. Q. Rev. Biophys. 2019, 52, e6. [Google Scholar] [CrossRef] [Green Version]
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef]
- Lee, J.H.; Wang, J.-H.; Chen, J.; Li, F.; Edwards, T.L.; Hewitt, A.W.; Liu, G.-S. Gene therapy for visual loss: Opportunities and concerns. Prog. Retin. Eye Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65 -mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- McClements, M.E.; Maclaren, R.E. Adeno-associated virus (AAV) dual vector strategies for gene therapy encoding large transgenes. Yale J. Biol. Med. 2017, 90, 611–623. [Google Scholar]
- Charbel Issa, P.; MacLaren, R.E. Non-viral retinal gene therapy: A review. Clin. Exp. Ophthalmol. 2012, 40, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Burnight, E.R.; Wiley, L.A.; Drack, A.V.; Braun, T.A.; Anfinson, K.R.; Kaalberg, E.E.; Halder, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 2014, 21, 662–672. [Google Scholar] [CrossRef] [Green Version]
- Olsson, J.E.; Gordon, J.W.; Pawlyk, B.S.; Roof, D.; Hayes, A.; Molday, R.S.; Mukai, S.; Cowley, G.S.; Berson, E.L.; Dryja, T.P. Transgenic mice with a rhodopsin mutation (Pro23His): A mouse model of autosomal dominant retinitis pigmentosa. Neuron 1992, 9, 815–830. [Google Scholar] [CrossRef]
- Seo, S.; Mullins, R.F.; Dumitrescu, A.V.; Bhattarai, S.; Gratie, D.; Wang, K.; Stone, E.M.; Sheffield, V.; Drack, A.V. Subretinal Gene Therapy of Mice with Bardet-Biedl Syndrome Type 1. Investig. Opthalmol. Vis. Sci. 2013, 54, 6118. [Google Scholar] [CrossRef]
- Tan, E.; Wang, Q.; Quiambao, A.B.; Xu, X.; Qtaishat, N.M.; Peachey, N.S.; Lem, J.; Fliesler, S.J.; Pepperberg, D.R.; Naash, M.I.; et al. The relationship between opsin overexpression and photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2001, 42, 589–600. [Google Scholar]
- Tsai, Y.T.; Wu, W.H.; Lee, T.T.; Wu, W.P.; Xu, C.L.; Park, K.S.; Cui, X.; Justus, S.; Lin, C.S.; Jauregui, R.; et al. Clustered Regularly Interspaced Short Palindromic Repeats-Based Genome Surgery for the Treatment of Autosomal Dominant Retinitis Pigmentosa. Ophthalmology 2018, 125, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, S.G.; Luoni, M.; Castoldi, V.; Massimino, L.; Cabassi, T.; Angeloni, D.; Demontis, G.C.; Leocani, L.; Andreazzoli, M.; Broccoli, V. Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum. Mol. Genet. 2018, 27, 761–779. [Google Scholar] [CrossRef] [PubMed]
- Bakondi, B.; Lv, W.; Lu, B.; Jones, M.K.; Tsai, Y.; Kim, K.J.; Levy, R.; Akhtar, A.A.; Breunig, J.J.; Svendsen, C.N.; et al. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. 2016, 24, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Latella, M.C.; Di Salvo, M.T.; Cocchiarella, F.; Benati, D.; Grisendi, G.; Comitato, A.; Marigo, V.; Recchia, A. In vivo Editing of the Human Mutant Rhodopsin Gene by Electroporation of Plasmid-based CRISPR/Cas9 in the Mouse Retina. Mol. Ther. Nucleic Acids 2016, 5, e389. [Google Scholar] [CrossRef]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.-W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef] [Green Version]
- Jo, D.H.; Koo, T.; Cho, C.S.; Kim, J.H.; Kim, J.-S.; Kim, J.H. Long-term effects of in vivo genome editing in the mouse retina using Campylobacter jejuni Cas9 expressed via adeno-associated virus. Mol. Ther. 2018. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Cai, Y.; Cheng, T.; Yao, Y.; Li, X.; Ma, Y.; Li, L.; Zhao, H.; Bao, J.; Zhang, M.; Qiu, Z.; et al. In vivo genome editing rescues photoreceptor degeneration via a Cas9/RecA-mediated homology-directed repair pathway. Sci. Adv. 2019, 5, eaav3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, T.; Nakade, S.; Sakane, Y.; Suzuki, K.-I.T.; Yamamoto, T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2016, 11, 118–133. [Google Scholar] [CrossRef]
- Rees, H.A.; Wilson, C.; Doman, J.L.; Liu, D.R. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv. 2019, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019. [Google Scholar] [CrossRef]
- Carroll, D. Collateral damage: Benchmarking off-target effects in genome editing. Genome Biol. 2019, 20, 114. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [Green Version]
- George, C.X.; Samuel, C.E. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl. Acad. Sci. USA 1999, 96, 4621–4626. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Maas, S.; Gommans, W.M. Identification of a selective nuclear import signal in adenosine deaminases acting on RNA. Nucleic Acids Res. 2009, 37, 5822–5829. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.L.; Carroll, R.C.; Nawy, S. Down-regulation of the RNA editing enzyme ADAR2 contributes to RGC death in a mouse model of glaucoma. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Single, F.N.; Köhler, M.; Sommer, B.; Sprengel, R.; Seeburg, P.H. RNA editing of AMPA receptor subunit GluR-B: A base-paired intron-exon structure determines position and efficiency. Cell 1993, 75, 1361–1370. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Vukic, D.; Michalík, D.; O’Connell, M.A.; Keegan, L.P. ADAR RNA editing in human disease; more to it than meets the I. Hum. Genet. 2017, 136, 1265–1278. [Google Scholar] [CrossRef]
- Chalk, A.M.; Taylor, S.; Heraud-farlow, J.E.; Walkley, C.R. The majority of A-to-I RNA editing is not required for mammalian homeostasis. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Matthews, M.M.; Thomas, J.M.; Zheng, Y.; Tran, K.; Phelps, K.J.; Scott, A.I.; Havel, J.; Fisher, A.J.; Beal, P.A. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016, 23, 426–433. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.M.; Beal, P.A. How do ADARs bind RNA? New protein-RNA structures illuminate substrate recognition by the RNA editing ADARs. BioEssays 2017, 39, 1–8. [Google Scholar] [CrossRef]
- Wong, S.K.; Sato, S.; Lazinski, D.W. Substrate recognition by ADAR1 and ADAR2. Rna 2001, 7, 846–858. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.; Yi, Z.; Zhu, S.; Wang, C.; Cao, Z.; Zhou, Z.; Yuan, P.; Yu, Y.; Tian, F.; Liu, Z.; et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019, 37, 1059–1069. [Google Scholar] [CrossRef]
- Wang, Y.; Havel, J.; Beal, P.A. A Phenotypic Screen for Functional Mutants of Human Adenosine Deaminase Acting on RNA 1. ACS Chem. Biol. 2015, 10, 2512–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katrekar, D.; Chen, G.; Meluzzi, D.; Ganesh, A.; Worlikar, A.; Shih, Y.-R.R.; Varghese, S.; Mali, P. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 2019, 16, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A cytosine deaminase for programmable single-base RNA editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Woolf, T.M.; Chase, J.M.; Stinchcomb, D.T. Toward the therapeutic editing of mutated RNA sequences. Proc. Natl. Acad. Sci. USA 1995, 92, 8298–8302. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Garcia-Mena, J.; DeVito, J.; Wolska, K.; Das, A. Bipartite function of a small RNA hairpin in transcription antitermination in bacteriophage lambda. Proc. Natl. Acad. Sci. USA 1995, 92, 4061–4065. [Google Scholar] [CrossRef] [Green Version]
- Montiel-Gonzalez, M.F.; Vallecillo-Viejo, I.; Yudowski, G.A.; Rosenthal, J.J.C. Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl. Acad. Sci. USA 2013, 110, 18285–18290. [Google Scholar] [CrossRef] [Green Version]
- Montiel-Gonźalez, M.F.; Vallecillo-Viejo, I.C.; Rosenthal, J.J.C. An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 2016, 44, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sinnamon, J.R.; Kim, S.Y.; Corson, G.M.; Song, Z.; Nakai, H.; Adelman, J.P.; Mandel, G. Site-directed RNA repair of endogenous Mecp2 RNA in neurons. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [Green Version]
- Vallecillo-Viejo, I.C.; Liscovitch-Brauer, N.; Montiel-Gonzalez, M.F.; Eisenberg, E.; Rosenthal, J.J.C. Abundant off-target edits from site-directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol. 2018, 15, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Stafforst, T.; Schneider, M.F. An RNA-deaminase conjugate selectively repairs point mutations. Angew. Chem. Int. Ed. 2012, 51, 11166–11169. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Schneider, M.F.; Wettengel, J.; Stafforst, T. Improving site-directed RNA editing in vitro and in cell culture by chemical modification of the guideRNA. Angew. Chem. Int. Ed. 2014, 53, 6267–6271. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Moschref, M.; Li, Q.; Merkle, T.; Selvasaravanan, K.D.; Li, J.B.; Stafforst, T. Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat. Methods 2018, 15, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Wettengel, J.; Reautschnig, P.; Geisler, S.; Kahle, P.J.; Stafforst, T. Harnessing human ADAR2 for RNA repair—Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 2017, 45, 2797–2808. [Google Scholar] [CrossRef] [Green Version]
- Azad, T.A.; Qulsum, U.; Tsukahara, T. Comparative Activity of Adenosine Deaminase Acting on RNA (ADARs) Isoforms for Correction of Genetic Code in Gene Therapy. Curr. Gene Ther. 2018, 18, 31–39. [Google Scholar] [CrossRef]
- Fukuda, M.; Umeno, H.; Nose, K.; Nishitarumizu, A.; Noguchi, R.; Nakagawa, H. Construction of a guide-RNA for site-directed RNA mutagenesis utilising intracellular A-To-I RNA editing. Sci. Rep. 2017, 7, 8–19. [Google Scholar] [CrossRef]
- Merkle, T.; Merz, S.; Reautschnig, P.; Blaha, A.; Li, Q.; Vogel, P.; Wettengel, J.; Li, J.B.; Stafforst, T. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019, 37, 133–138. [Google Scholar] [CrossRef]
- East-Seletsky, A.; O’Connell, M.R.; Knight, S.C.; Burstein, D.; Cate, J.H.D.; Tjian, R.; Doudna, J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016, 538, 270–273. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.X.; Chong, S.; Zhang, H.; Makarova, K.S.; Koonin, E.V.; Cheng, D.R.; Scott, D.A. Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol. Cell 2018, 70, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tambe, A.; East-Seletsky, A.; Knott, G.J.; Doudna, J.A.; O’Connell, M.R. RNA Binding and HEPN-Nuclease Activation Are Decoupled in CRISPR-Cas13a. Cell Rep. 2018, 24, 1025–1036. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Li, X.; Wang, J.; Wang, M.; Chen, P.; Yin, M.; Li, J.; Sheng, G.; Wang, Y. Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell 2017, 168, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Li, X.; Ma, J.; Li, Z.; You, L.; Wang, J.; Wang, M.; Zhang, X.; Wang, Y. The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell 2017, 170, 714–726. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Mesa, P.; Kellner, M.J.; Kannan, S.; Brignole, E.; Koob, J.; Feliciano, P.R.; Stella, S.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. High-Resolution Structure of Cas13b and Biochemical Characterization of RNA Targeting and Cleavage. Cell Rep. 2019, 26, 3741–3751. [Google Scholar] [CrossRef] [Green Version]
- Smargon, A.A.; Cox, D.B.T.; Pyzocha, N.K.; Zheng, K.; Slaymaker, I.M.; Gootenberg, J.S.; Abudayyeh, O.A.; Essletzbichler, P.; Shmakov, S.; Makarova, K.S.; et al. Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol. Cell 2017, 65, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef]
- O’Connell, M.R. Molecular Mechanisms of RNA Targeting by Cas13-containing Type VI CRISPR–Cas Systems. J. Mol. Biol. 2019, 431, 66–87. [Google Scholar] [CrossRef]
- Rauch, S.; He, E.; Srienc, M.; Zhou, H.; Zhang, Z.; Dickinson, B.C. Programmable RNA-Guided RNA Effector Proteins Built from Human Parts. Cell 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stoltzfus, A.; Norris, R.W. On the Causes of Evolutionary Transition: Transversion Bias. Mol. Biol. Evol. 2016, 33, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, L.; Yi, Z.; Zhu, S.; Wang, C.; Cao, Z.; Zhou, Z.; Yuan, P.; Yu, Y.; Tian, F.; Liu, Z.; et al. Leveraging Endogenous ADAR for Programmable Editing on RNA. BioRxiv 2019, 605972. [Google Scholar] [CrossRef]
- Ring, H.; Boije, H.; Daniel, C.; Ohlson, J.; Öhman, M.; Hallböök, F. Increased A-to-I RNA editing of the transcript for GABA A receptor subunit α3 during chick retinal development. Vis. Neurosci. 2010, 27, 149–157. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Anderson, K.M.; Poosala, P.; Lindley, S.R.; Anderson, D.M. Targeted Cleavage and Polyadenylation of RNA by CRISPR-Cas13. BioRxiv 2019. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Immortalized Cells | Primary Cells | In Vivo | Ref | |
|---|---|---|---|---|---|
| Exogenous ADAR | |||||
| BoxB-λN-ADAR | Gene | Mecp2 | Mecp2 | Not tested | [47,49,50] |
| Model | N2A | Murine neuron | |||
| Efficiency | 25–50% | 72% | |||
| Delivery | Plasmid | AAV | |||
| SNAP-ADAR | Gene | Many targets | Not tested | Not tested | [53] |
| Model | HEK293 | ||||
| Efficiency | Up to 90% | ||||
| Delivery | Guide transfection | ||||
| MS2-MCP-ADAR | Gene | Many targets | Not tested | Dmd | [42,55] |
| Model | HEK293T | mdx mouse | |||
| Efficiency | 10–80% | AAV | |||
| Delivery | Plasmid transfection | 2% | |||
| GluR2-ADAR | Gene | PINK1 | Dmd, Otc | [42,54] | |
| Model | HELA HEK293T | Not tested | mdx mouse model spfash mouse model | ||
| Efficiency | 10–40% | 0.8% (mdx) 4.6–8.2% (spfash) | |||
| Delivery | Plasmid transfection | AAV | |||
| REPAIR: CRISPR-Cas13b-ADAR (A-I) | Gene | KRAS, PPIB | Not tested | Not tested | [43] |
| Model | HEK293FT | ||||
| Efficiency | 28% | ||||
| Delivery | Plasmid | ||||
| RESCUE: CRISPR-Cas13b-ADAR (C-T) | Gene | Multiple | CTNNB1 | Not tested | [44] |
| Model | HEK293FT | HUVEC cells | |||
| Efficiency | 3–42% in 5% when multiplexed | Not reported | |||
| Delivery | Plasmid transfection | Plasmid transfection | |||
| Endogenous ADAR | |||||
| LEAPER (long gRNA) | Gene | Many targets | Many targets | Not tested | [40] |
| Model | Multiple lines | Multiple lines | |||
| Efficiency | Up to 50% (plasmid) 6% (lentivirus) | 30–80% | |||
| Delivery | Plasmid Lentivirus | Plasmid electroporation | |||
| RESTORE (ASO, chemical modification) | Gene | Many targets | GAPDH, STAT1, SERPINA1 | Not tested | [57] |
| Model | Many cell types | Multiple lines | |||
| Efficiency | 3–34% | Up to 27% | |||
| Delivery | ASO Transfection | ASO Transfection | |||
| GluR2 | Gene | Not tested | Not tested | Otc | [42] |
| Model | spfash mouse model | ||||
| Efficiency | 0.6% | ||||
| Delivery | AAV | ||||
| Gene | Frequency (%) # | Coding Sequence Length (kb) |
|---|---|---|
| ABCA4 | 17.3 | 6.81 |
| USH2A | 7.6 | 15.6 |
| CEP290 | 1.8 | 7.44 |
| MYO7A | 0.8 | 6.65 |
| EYS | 0.6 | 9.43 |
| CDH23 | 0.4 | 10.1 |
| Total | 28.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fry, L.E.; Peddle, C.F.; Barnard, A.R.; McClements, M.E.; MacLaren, R.E. RNA Editing as a Therapeutic Approach for Retinal Gene Therapy Requiring Long Coding Sequences. Int. J. Mol. Sci. 2020, 21, 777. https://doi.org/10.3390/ijms21030777
Fry LE, Peddle CF, Barnard AR, McClements ME, MacLaren RE. RNA Editing as a Therapeutic Approach for Retinal Gene Therapy Requiring Long Coding Sequences. International Journal of Molecular Sciences. 2020; 21(3):777. https://doi.org/10.3390/ijms21030777
Chicago/Turabian StyleFry, Lewis E., Caroline F. Peddle, Alun R. Barnard, Michelle E. McClements, and Robert E. MacLaren. 2020. "RNA Editing as a Therapeutic Approach for Retinal Gene Therapy Requiring Long Coding Sequences" International Journal of Molecular Sciences 21, no. 3: 777. https://doi.org/10.3390/ijms21030777
APA StyleFry, L. E., Peddle, C. F., Barnard, A. R., McClements, M. E., & MacLaren, R. E. (2020). RNA Editing as a Therapeutic Approach for Retinal Gene Therapy Requiring Long Coding Sequences. International Journal of Molecular Sciences, 21(3), 777. https://doi.org/10.3390/ijms21030777