The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease


1
Division of Diabetes and Endocrinology, Kobe University Hospital, 7-5-2, Kusunoki-cho, Chuo-ku, Kobe 650-0017, Japan
2
Division of Diabetes and Endocrinology, Kobe University Graduate School of Medicine, 7-5-2, Kusunoki-cho, Chuo-ku, Kobe 650-0017, Japan
3
Department of Diabetes and Endocrinology, Nara Medical University, 840 Shijo-cho, Kashihara, Nara 634-8522, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(23), 9132; https://doi.org/10.3390/ijms21239132
Submission received: 20 October 2020
/
Revised: 21 November 2020
/
Accepted: 29 November 2020
/
Published: 30 November 2020
(This article belongs to the Special Issue Molecular Biology of the Pituitary)
{kind=link}
{kind=link}
Abstract
:Cushing’s disease caused due to adrenocorticotropic hormone (ACTH)-secreting pituitary adenomas (ACTHomas) leads to hypercortisolemia, resulting in increased morbidity and mortality. Autonomous ACTH secretion is attributed to the impaired glucocorticoid negative feedback (glucocorticoid resistance) response. Interestingly, other conditions, such as ectopic ACTH syndrome (EAS) and non-neoplastic hypercortisolemia (NNH, also known as pseudo-Cushing’s syndrome) also exhibit glucocorticoid resistance. Therefore, to differentiate between these conditions, several dynamic tests, including those with desmopressin (DDAVP), corticotrophin-releasing hormone (CRH), and Dex/CRH have been developed. In normal pituitary corticotrophs, ACTH synthesis and secretion are regulated mainly by CRH and glucocorticoids, which are the ACTH secretion-stimulating and -suppressing factors, respectively. These factors regulate ACTH synthesis and secretion through genomic and non-genomic mechanisms. Conversely, glucocorticoid negative feedback is impaired in ACTHomas, which could be due to the overexpression of 11β-HSD2, HSP90, or TR4, or loss of expression of CABLES1 or nuclear BRG1 proteins. Genetic analysis has indicated the involvement of several genes in the etiology of ACTHomas, including USP8, USP48, BRAF, and TP53. However, the association between glucocorticoid resistance and these genes remains unclear. Here, we review the clinical aspects and molecular mechanisms of ACTHomas and compare them to those of other related conditions.
1. Introduction
Cushing’s disease is characterized by hypercortisolemia occurring due to autonomous secretion of adrenocorticotropic hormone (ACTH) from a pituitary tumor. However, the mechanisms underlying the impaired physiological hormonal regulation in ACTH-secreting pituitary corticotroph adenomas (ACTHomas) remain unclear. In response to diurnal variation and stress, the synthesis and secretion of ACTH in normal pituitary corticotroph cells is stimulated by the hypothalamic neuroendocrine hormones, namely corticotrophin-releasing hormone (CRH) and arginine vasopressin (AVP). Conversely, ACTH synthesis and secretion are suppressed mainly through the action of glucocorticoid derived from the adrenal cortex. This negative feedback regulation occurs not only in corticotrophs within the pituitary, but also in hypothalamic CRH-secreting neurons [1]. ACTHomas maintain high ACTH levels as they respond to CRH and AVP, which can be evaluated using the CRH and 1-deamino-8-d-arginine vasopressin (DDAVP) tests, respectively. The impaired ability of glucocorticoids to suppress ACTH secretion is clinically proven to occur through inappropriate ACTH secretion with non-suppressive cortisol secretion after dexamethasone (Dex) treatment, and this functional assay is named as the ‘Dex suppression test’ (DST). These provocative and inhibitory tests are used to screen the patients for Cushing’s syndrome and act as a diagnostic tool to differentiate it from ectopic ACTH syndrome (EAS) or non-neoplastic hypercortisolemia (NNH, also known as pseudo-Cushing’s syndrome) [2,3]. Although the clinical significance of these tests is well established, knowledge regarding the molecular mechanisms underlying the pathophysiological regulation of ACTH in these conditions is limited, which is partially attributed to the lack of availability of human ACTHoma cell lines.
In this review, we mainly focus on the mechanisms underlying ACTH synthesis and secretion in the normal pituitary, ACTHomas, and other related conditions.
2. Material and Methods
To search the bibliographic references, online databases, including PubMed, Web of Science, SCOPUS, and Google were used. The following MeSH terms and their combinations were used for searching the references: Cushing, Cushing’s disease, POMC, ACTH secretion, ACTH synthesis, USP8, pseudo Cushing syndrome, ectopic ACTH syndrome, glucocorticoid negative feedback, DDAVP, CRH, and stress.
3. Results
3.1. Physiological Regulation of ACTH
3.1.1. ACTH Synthesis and Secretion
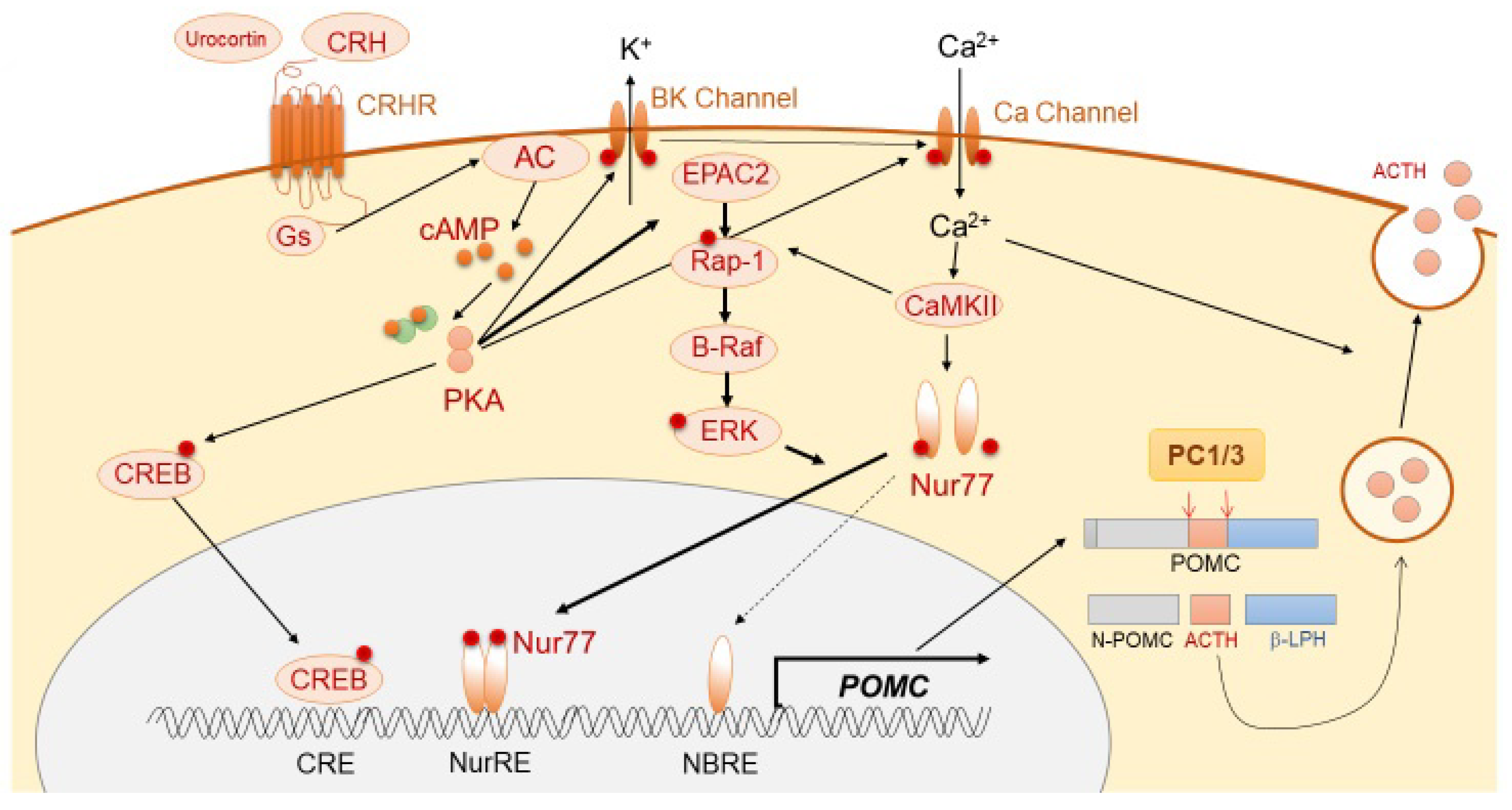
ACTH is derived mainly from the pituitary corticotroph cells, in which a 266-amino-acid precursor protein, pro-opiomelanocortin (POMC), is translated from the POMC gene located on chromosome 2q23. The 8 kb long human POMC gene comprises 3 exons. In corticotrophs, POMC is mostly encoded by exon 3 of the gene. The POMC transcriptional activity is thought to be regulated mainly via the orphan nuclear receptor, Nurr77, and also Tpit, Pitx, NeuroD1, signal transducer and activator of transcription 3 (STAT3), and ETS variant transcription factor 1 (Etv1) [4]. It is modulated by pituitary-specific enhancer, which is located at −7 kb from the POMC initiation site [5], and also epigenetic modification, such as chromatin remodeling [6]. POMC protein is thereafter sorted into the dense-core secretory granules of the regulated secretory pathway (RSP). The N-terminal of POMC (1–26 aa) possesses a sorting signal motif, which helps POMC in binding to the membrane-associated form of carboxypeptidase E (CPE), a sorting receptor of POMC, thereby leading to normal post-translational protein processing [7]. In CPE knockout mice (Cpefat/fat mice), POMC processing is reduced in both the pituitary and hypothalamus [8]. Secretogranin III (SgIII) is a sorting receptor of chromogranin A, which also binds to POMC in the pituitary [9,10]. In SgIII-deficient corticotroph cells, POMC has been shown to be accumulated in the trans-Golgi network (TGN), suggesting its important role in POMC trafficking. In the regulation of trafficking of POMC to the RSP, two proteins, including CPE and SgIII, have been shown to play a synergistic role and compensate for each other [11]. POMC is enzymatically cleaved into N-POMC (1–77), ACTH (1–39), and β-lipotropin (β-LPH) (1–89). Post-translational POMC processing by prohormone convertases (PCs) is required to produce ACTH. Corticotrophs predominantly express PC1 and PC3, which cleave POMC to generate ACTH (1–39) (Figure 1), while PC2 (expressed in melanotrophs) further cleaves ACTH into α-melanocyte-stimulating hormone (MSH) [12,13,14]. POMC processing is also modulated by other enzymes, including Yapsin A, ACTH-converting enzyme (AACE), aminopeptidases B-like (AMB), and peptidylglycine α-amidating monooxygenase (PAM) [15]. POMC also expresses in non-pituitary tissues, including hypothalamus, testis, adrenal gland, pancreas, adipose tissue, kidney, and placenta [16,17,18,19,20,21]. Following its synthesis, ACTH is stored in cytoplasmic secretory vesicles. The secretion of ACTH is predominantly mediated through non-genomic mechanisms, including calcium- and voltage-activated potassium (BK) channels [22,23], and annexin 1 (ANXA1).
3.1.2. Positive Regulation of ACTH Synthesis and Secretion
ACTH synthesis is mainly stimulated through the action of CRH, which is a trophic hormone synthesized in the hypothalamic paraventricular nucleus (PVN) and secreted into hypophyseal portal vessels at the median eminence. CRH binds to CRH receptor 1 (CRHR1), which is expressed on the cellular membrane of corticotrophs, further triggering the accumulation of cAMP, activation of protein kinase A (PKA), and subsequent POMC transcription [24,25]. Although the classical PKA pathway to Ca2+/cAMP response-element binding protein (CREB) is involved in POMC transcription [26], Nur77 (also known as NGFI-B) appears to play a dominant role [27,28,29]. Although Nur77-binding response element (NBRE) is present in the POMC promoter, another motif, Nur response element (NurRE), is also required for Nur77-dependent POMC mRNA production [30]. The homo- or hetero-dimerization of Nur77 with other Nur family members, such as Nurr1 or NOR1, is required for its translocation into the nucleus and binding to NurRE. This cascade is mainly induced through MAPK downstream of PKA [30]. PKA also induces cytosolic Ca2+ accumulation through calcium-dependent voltage channels, thereby resulting in calmodulin kinase II (CaMKII) activation. This Ca2+-dependent pathway stimulates distinct downstream signaling, including Nurr induction and MAPK-mediated activation of Nurr [31]. Conversely, during CRH-mediated POMC expression, a pituitary-specific enhancer located 7 kb upstream of the initiation site has been shown to play a pivotal role [5]. In regard to ACTH secretion, CRH rapidly stimulates the burst of Ca2+ cytosolic inclusion with large-conductance BK channels in a concentration-dependent manner [22,23,32].
AVP, a cyclic nonapeptide, is a main regulator of water homeostasis via the receptor AVPR2, located in the renal tubules. Furthermore, AVP triggers vasoconstriction via AVPR1a, which is predominantly expressed on vascular smooth muscle cells [33]. Additionally, AVP is known to be an important modulator of the HPA axis, especially during stress. AVP is synthesized in the hypothalamic PVN and supraoptic nuclei (SON) and stored in the posterior pituitary lobe. It is released into the hypophysial portal vein and can reach the anterior lobe of the pituitary [34], where it binds to AVPR1b on the pituitary corticotrophs to induce the ACTH secretion process. Intracellular signaling stimulates a biphasic release of cytosolic Ca2+ from intracellular inositol 1,4,5-triphosphate (IP3)-sensitive stores, and extracellular Ca2+ via L-type Ca2+ channels [25,35,36]. CRH, but not AVP, plays a major role in ACTH secretion under non-stress conditions. In fact, basal ACTH levels are similar in both AVP- and AVPR1b-deficient Brattleboro rats when compared to wild type [37,38]. Conversely, AVPR1b knockout mice exhibit dampened ACTH response to acute stress. Moreover, AVP storage and expression of AVPR1b receptor gradually increase during chronic stress, suggesting that AVP plays a primary role in adaptation to stress through enhancing the responses at the HPA axis [38,39,40]. Furthermore, ACTH secretion induced by AVP is mediated via BK-independent pathways [22].
Several cytokines, such as interleukin-1β (IL-1β), IL-6, and leukemia inhibitory factor (LIF) can stimulate ACTH secretion via direct or indirect mechanisms [41]. LIF and IL-6 are derived from hypothalamus and pituitary in response to inflammation, and act in an autocrine and paracrine manner on corticotrophs, respectively [42]. These proinflammatory cytokines induce STAT3 activation upon translocating to the nucleus, which is followed by binding to a STAT binding element located at position –399/–379 in the rat Pomc promoter. STAT3 activated through LIF or IL-6 stimulates co-expression of POMC with CRH in a synergistic manner [43,44]. In response to inflammation, microenvironmental protons increase and induce POMC expression through the CRHR1/CaMKII pathway. Interestingly, this induction occurs without ligand stimulation of CRHR1 [45]. Urocortin is a CRH-related peptide that binds to both type 1 and type 2 CRH receptors, leading to the synthesis and secretion of ACTH via the cAMP and protein kinase C (PKC) pathway [46,47].
3.1.3. Negative Regulation of ACTH Synthesis and Secretion
Glucocorticoid is a major factor that suppresses ACTH synthesis and secretion through its action on both the hypothalamic CRH neuron and pituitary corticotrophs. This is the predominant negative feedback system for the functioning of ACTH within the HPA axis [48], with both rapid non-genomic and delayed genomic mechanisms. In hypothalamic CRH neurons, both membrane-associated G protein-coupled glucocorticoid receptor (GRmb) and nuclear glucocorticoid receptor (GR) are expressed abundantly. As a non-genomic effect, glucocorticoid binds to GRmb, thereby leading to the release of endocannabinoid (CB) from neurons. CB binds to the cannabinoid receptor (CBR) located on the presynaptic glutamate terminal, thereby suppressing glutamine release in the PVN neuron, resulting in decreased CRH secretion [49]. GR binds directly to the negative GR element (nGRE) in DNA [50], and it also suppresses forskolin-dependent CRH activity via CRE in the CRH promoter [51]. The genomic effect of glucocorticoid in CRH neurons is still debatable. In corticotrophs, glucocorticoids rapidly suppress ACTH secretion by reducing both spontaneous and CRH-induced burst firing in a BK channel-mediated manner [52]. ACTH secretion from corticotrophs is also suppressed by Annexin 1 (ANXA1), secreted from the pituitary folliculostellate cells. In fact, glucocorticoids induce ANXA1 expression and its translocation to the outer surface of the plasma membrane of corticotrophs, suggesting that ANXA1 mediates glucocorticoid-dependent suppression of ACTH secretion [53]. As part of the genomic mechanism of glucocorticoid-dependent POMC suppression, there are several sites in the POMC promoter that might contribute to the glucocorticoid-dependent transcriptional suppression of ACTH. These sites include a negative glucocorticoid response element (nGRE), a direct GR binding site, an E-box, and a neurogenic differentiation factor 1 (NeuroD1) binding site [54,55]. However, glucocorticoid-mediated ACTH suppression is not mediated through direct binding of GR to the POMC promoter, but through its interaction with Nur factors, thereby inducing protein-protein interaction-dependent transrepression [56,57]. This process requires Brg1, the ATPase subunit of the Swi/Snf complex, which stabilizes the interaction between GR, Nur77, and HDAC2 on the POMC promoter [6] (Figure 2). The TGF-β superfamily member bone morphogenetic protein (BMP)4 has been implicated in pituitary organogenesis and differentiation. BMP4 suppresses POMC expression through preventing the binding of Tpit and Pitx1 transcription factors to the POMC promoter [58,59,60]. Furthermore, through its binding to somatostatin receptor subtype 2 (SSTR2) and subtype 5 (SSTR5), hypothalamic somatostatin (SST) suppresses the secretion of several pituitary hormones, including GH, TSH, and ACTH [61,62,63,64]. SST also suppresses intracellular cAMP levels and inhibits exocytosis through reducing cytosolic Ca2+ inclusion [65,66]. Constitutively activated SSTR5 attenuates both CRH-dependent increase in intracellular cAMP levels and ACTH secretion via posttranscriptional suppression of CRHR1 [67].
3.2. Pathological Regulation of ACTH in ACTHomas
3.2.1. ACTHomas Tumorigenesis
Pituitary adenomas, such as ACTHomas, are of monoclonal origin, suggesting that a single somatic gene abnormality might lead to their incidence [68,69,70]. Until recently, there were only few reports regarding the genetic changes in ACTHomas, except for familial Cushing’s disease, such as multiple endocrine neoplasm type 1 (MEN1), MEN4, and familial isolated pituitary adenomas (FIPAs), and the germline mutations associated with these hereditary syndromes were identified in the MEN1, CDKN1B, and AIP genes, respectively. However, these hereditary syndromes account for less than 3% of pituitary adenomas and do not have specific association with ACTHomas [71,72,73]. A major milestone in the understanding of ACTHoma pathology was achieved with the identification of somatic mutations in the ubiquitin-specific-protease 8 (USP8) gene. Mutations in this gene have been found in 20–60% of ACTHomas cases [74,75]. All point mutations are specifically located within the 14-3-3 binding motif of USP8, and lead to an increase in the deubiquitylation activity of this enzyme. The epidermal growth factor receptor (EGFR) tyrosine kinase, which is frequently overexpressed in ACTHomas, is one of the deubiquitination substrates of USP8 [76]. Moreover, corticotroph-specific EGFR overexpression leads to ACTHomas mediated via E2F1 activation [77,78]. Recently, a de novo germline USP8 mutation (c.2155T>C, p.S719P) in the 14-3-3 binding motif has also been reported in pediatric patients with Cushing’s disease [79]. Furthermore, subsequent analysis using next-generation sequencing led to the identification of somatic mutations in USP48, BRAF, and TP53 genes, which were considered to be the other causes of this disease [80,81,82]. USP48 activates NF-κB, which in turn binds to and transactivates the POMC promoter [80]. Almost 13% of ACTHomas cases have been shown to harbor specific p.Met415 mutations within the catalytic domain of USP48 [82], indicating a functional relevance of this ubiquitin-specific protease. The BRAF p.V600E mutation has been found in several cancers, including melanoma, papillary thyroid carcinomas, colon cancer, gliomas, and papillary craniopharyngiomas [83,84,85,86,87]. This activating mutation causes increased cell proliferation due to the activation of the MAPK pathway. In Cushing’s disease, MAPK activation can also induce ACTH synthesis, which leads to autonomous ACTH secretion. This mutation accounts for approximately 7% of cases with Cushing’s disease [82]. TP53 is a well-recognized tumor suppressor gene, which guards the genome and regulates apoptosis, DNA repair, and cellular senescence. Loss-of-function mutations in the TP53 gene are present in approximately 12.5% of ACTHomas cases and are associated with aggressive tumor behavior. Another gene associated with aggressive ACTHomas is CABLES1, a negative regulator of the cell cycle that interacts with cyclin-dependent kinase 3. Cables1 has been identified as a glucocorticoid-dependent cell cycle modulator in AtT20, a cell line derived from a mouse with ACTHomas. Low or undetectable expression level of CABLES1 is a distinct feature of ACTHomas in pediatric or young adult patients with Cushing’s disease, whereas the gene expresses abundantly in normal corticotrophs [88].
In pediatric patients with Cushing’s disease, mutations in DICER1, a small RNA processing endoribonuclease that cleaves precursor microRNAs (miRNAs) into mature miRNAs, lead to DICER1 syndrome [89]. This syndrome is associated with the development of pleuropulmonary blastomas, cystic nephroma, rhabdomyosarcoma, and several endocrine neoplasms, including thyroid cancer, ovarian tumors, and ACTH-secreting pituitary blastoma [90,91], thereby suggesting a pathogenic role for microRNAs in ACTHomas. Additionally, whole exome sequencing (WES) analysis revealed several other mutations associated with ACTHomas, including those in the NR3C1, DAXX, ATRX, and HCFC1 genes [82]. Somatic mutations in the MEN1, PRKAR1A, and GNAS1 genes have also been reported in ACTHomas [92,93]. Although several genes that are associated with cAMP signaling or the cell cycle have therefore been implicated in the pathogenesis of ACTHomas, the precise molecular mechanisms through which these mutations lead to tumorigenesis remain unclear.
3.2.2. ACTH Regulation in ACTHomas
Based on the response to low-dose DST, one of their most prominent characteristics of ACTHomas is the impaired ACTH suppression by glucocorticoid negative feedback [94], which further leads to ACTH-dependent hypercortisolemia. The decreased sensitivity to glucocorticoids cannot be explained by the loss-of-function mutations in GR in most cases [95]. At the intracellular pre-receptor level, glucocorticoid activity has been shown to be negatively regulated through 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which converts cortisol into its inactive form, cortisone. Increased expression of 11β-HSD2 has been observed in ACTHomas, and is in turn implicated in inducing glucocorticoid resistance [96,97]. Another mechanism possibly underlying glucocorticoid resistance is the increased expression of the chaperone protein, heat shock protein 90 (HSP90) [98,99]. GR is mainly cytoplasmic in its inactive state associated with HSP90, which regulates its intracellular trafficking, folding, maturation, and activation [100]. Silibinin, an inhibitor that binds to the C-terminal domain of HSP90, enhanced glucocorticoid sensitivity and suppressed ACTH secretion, suggesting that the increased level of HSP90 in ACTHomas plays an important role in impaired glucocorticoid negative feedback [98]. Additionally, a SWI/SNF protein, Brg1 and histone deacetylase 2 (HDAC2). are associated with the POMC promoter. Brg1 stabilizes the interaction between GR and HDAC2 to suppress POMC transcription. Loss of either nuclear Brg1 or HDAC2 can lead to glucocorticoid resistance in ACTHoma [101]. Testicular receptor 4 (TR4), an orphan nuclear receptor, interacts directly with the N-terminal domain of GR, resulting in the disruption of GR binding to the POMC promoter [102]. Therefore, an increase in the expression of TR4 is thought to contribute to glucocorticoid resistance in ACTHomas [103]. Using the murine AtT20 cell line, Cables1 was identified as a glucocorticoid-responsive cell cycle regulatory gene, as described above in the tumorigenesis of ACTHomas. In 55% of ACTHomas cases, loss of expression of CABLES1 has been reported, resulting in impaired sensitivity to glucocorticoids [104]. Conclusively, increased expression of 11β-HSD2, HSP90, or TR4, and loss of expression of BRG1 or CABLES1, contribute to the pathogenesis of ACTHomas. This mainly occurs through conceding GR function, which in turn impairs the glucocorticoid negative feedback system. However, the precise mechanism through which the expression of these molecules is altered, and the link between these molecules and other genetic changes remains unclear. In addition to the impaired glucocorticoid negative feedback, ACTHomas exhibits increased or aberrant expression of AVPR1b or AVPR2, which is associated with a paradoxical response to DDAVP [105,106]. Elevated expression levels of EGFR have been associated with high ACTH synthesis and secretion via E2F1-mediated transcriptional activity, which has been shown to be attenuated through the application of its tyrosine kinase inhibitor, gefitinib, and E2F1 inhibitor, HLM006474 [77,78]. The newly developed high-throughput “ACTH AlphaLISA assay” led to the identification a dual PI3K/HDAC inhibitor, CUDC-907, as a potential targeted therapy for ACTHomas. It was thought to be mediated via Nurr1 transcriptional activity in corticotroph adenomas, suggesting an important role of HDAC action in addition to PI3K in ACTHomas [107]. Furthermore, epigenetic-targeting compound, JQ1, which is an inhibiter of bromo and extra-terminal domain (BET), and targets bromodomain of the protein family members BRD2, BRD3, BRD4, and BRDT, has been identified to suppress POMC expression, supporting the important role of epigenetic control in ACTH synthesis in ACTHomas [108].
3.3. Pathological Regulation of Acth under Other Conditions
3.3.1. Ectopic ACTH Syndrome
Ectopic ACTH syndrome (EAS) is a rare form of ACTH-dependent Cushing’s syndrome that occurs at a frequency of approximately 12% [109,110]. Extra-pituitary ACTH hypersecretion commonly occurs in neuroendocrine tumors of various tissue types, including small-cell lung carcinomas (SCLCs), bronchial carcinoids, thymic neuroendocrine neoplasms (NENs), pheochromocytomas, and medullary thyroid carcinomas [111,112,113,114,115,116,117,118,119,120,121]. Although the mechanism of EAS tumorigenesis is not fully understood, several genetic abnormalities have been identified in thymic NEN, including HRAS, PAK1, and MEN1 using whole-exome sequencing [122]. The pathogenesis of NENs has been widely investigated, including the ones originating from lungs, gastrointestinal, and pancreatic tissues, showing genetic abnormalities. These involve DNA damage repair, chromatin remodeling, mTOR signaling, and telomere maintenance-related genes. In addition to the genetic alterations, epigenetic modification has been described, including DNA methylation and histone modifications [123,124]. However, these might be associated only with tumorigenesis but not with the incidence of autonomous hormone secretion. Regarding the molecular pathology of ectopic POMC expression in NENs, ACTH synthesis is not regulated through the action of pituitary-specific transcriptional factors, namely Pitx1 and Tpit; however, it might be enhanced by the phosphorylation-dependent DNA binding of E2F1 at the proximal region of the POMC promoter [125]. The detailed mechanisms underlying the ectopic expression of ACTH in EAS remain to be elucidated. However, in case of ACTH-secreting pheochromocytoma, a paradoxical increase in ACTH levels post-glucocorticoid administration was observed, which was found to be mediated by demethylation of the E2F-binding site in the POMC promoter [126]. In these tumors, immature ACTH precursors are frequently released in the circulation, which may contribute to the relatively high concentration of ACTH levels (100–200 pM/454.1–908.2 pg/mL), while the concentration in patients with Cushing’s disease is generally found to be less than 100 pM (454.1 pg/mL) [127,128,129]. In these NENs, the expression of POMC-processing enzymes, PC1 and PC3, is limited, probably due to impaired differentiation. For some carcinoids, ectopic PC2 expression can be explained as a reason for the secretion of corticotroph-like intermediary lobe peptide (CLIP) and β-MSH5-22 instead of ACTH [128,130,131]. As the ACTH precursors exhibit lower bioactivity, the ACTH to cortisol ratio is generally higher in patients with EAS than in those with Cushing’s disease and normal subjects. In addition to the ACTH precursors, elevated circulating levels of the hypothalamic neuropeptide agouti-related protein (AgRP) have been reported in EAS but not in Cushing’s disease. Therefore, AgRP has been suggested as a potential neuroendocrine tumor diagnostic marker [132]. In EAS, ACTH secretion is not suppressed through high-dose exogenous glucocorticoids, which is attributed to a defect in GR or GR-signaling [133]. This has been used during the diagnosis of EAS, and to differentiate it from Cushing’s disease. However, ectopic GR expression has been detected in several cases of EAS, which is a limitation of this test [134]. A dampened ACTH response to DDAVP has been observed in most EAS patients, whereas ACTH response is enhanced in Cushing’s disease, which is probably due to the ectopic AVPR1b or AVPR2 expression in these tumors [135,136]. However, in some EAS cases, AVPR1b has been reported to be expressed and respond to DDAVP [136,137,138,139]. Since the expression of CRH receptor has not been observed in most EAS cases, lack of ACTH secretion in response to CRH can be used as a reliable confirmatory test to diagnose EAS [140].
3.3.2. Non-Neoplastic Hypercortisolemia
NNH, also known as pseudo-Cushing’s syndrome, is defined as an ACTH-dependent hypercortisolism state occurring in the absence of ACTH-secreting neoplasms. It is caused due to many clinical situations, such as excess alcohol intake, chronic kidney disease, depression, obesity, and poorly controlled Type 2 diabetes mellitus [141].
In patients with alcohol-induced hypercortisolemia, elevated midnight cortisol and high urinary free cortisol (UFC) levels with either elevated or normal ACTH levels have been reported. Low-dose DST failed to suppress the cortisol levels in these patients, making it difficult to clinically differentiate the illness from neoplastic hypercortisolemia. Alcohol can stimulate hypothalamic CRH and AVP secretion and impair hepatic clearance of cortisol [142,143,144]. In these patients, it is known that glucocorticoid resistance can be normalized through implementing alcohol abstinence [145].
Depression has been associated with impaired adequate termination of stress-induced HPA axis hyperactivity [146,147]. In these patients, both late-night cortisol and UFC levels are found to be elevated, and glucocorticoid sensitivity is reduced, which is similar to the clinical presentation of patients with Cushing’s disease. Impaired cortisol suppression following Dex administration was observed in 64% of patients with active depression [148]. Moreover, CRH administration failed to increase ACTH levels in these patients, probably due to low CRHR1 expression in corticotrophs caused by chronic CRH excess [149]. Although mechanisms underlying the pathology of this disease remain unclear, FKBP5 overexpression might be associated with HPA hyperactivity [150,151]. Moreover, in patients with depression and reduced glucocorticoid sensitivity, single nucleotide polymorphisms (SNPs) in the FKBP5 gene leading to high FKBP51 expression have been identified [152].
Type 2 diabetes mellitus and obesity are associated with increased late-night salivary cortisol levels [153]. Aging, current DM, and high blood pressure have been associated with late-night salivary cortisol rather than a history of depression and current alcoholism [154]. Increased expression of 11-β-hydroxysteroid dehydrogenase 1 in adipose tissue might be a causal factor for the overproduction of cortisol in local tissue [155]. Cortisol hypersecretion is usually mild in these patients. However, it is unclear whether this cortisol elevation is a cause of metabolic syndrome rather than resulting from fat accumulation.
The DDAVP and/or combined DST/CRH tests have been used to distinguish patients with Cushing’s disease from those with non-tumoral hypercortisolism [138,156,157]. As normal corticotrophs express lower levels of AVPR1b, and intra-venous DDAVP injection does not stimulate ACTH secretion in patients who do not have ACTHomas, AVPR1b or AVPR2 has been shown to express abundantly in ACTHomas [105,106]. A combined DST/CRH test has also been applied to diagnose NNH. The rationale behind using this test is that the two conditions exhibit different sensitivity to low doses of dexamethasone and response to CRH [158]. Patients with NNH exhibit sensitivity to glucocorticoid-induced negative feedback, and therefore show a dampened response to CRH after Dex treatment. However, this test has not been useful in distinguishing alcohol-induced NNH [159]. It is therefore challenging to physiologically differentiate hyper-activity of the HPA axis from Cushing’s disease.
4. Conclusions
The molecular and genetic mechanisms underlying the pathophysiological regulation of ACTH secretion have been investigated in various clinical studies on patients with Cushing’s disease, EAS, and related conditions. Mainly, basic research has been performed using animal models and ACTH-secreting cell lines. Although there are several gaps in our understanding regarding these aspects, rapid progress due to recent technological advances, such as whole-exome sequencing, have enabled us to gain deeper insights into ACTH-related pathophysiology. However, the diagnostic methods and treatment of abnormal ACTH secretion are still limited, and thus, further investigations with a multifaceted approach are required to be performed in the future studies.
Author Contributions
Conceptualization, H.F. and Y.T.; methodology, H.F.; writing—original draft preparation, H.S.; writing—review and editing, M.Y. and H.F. supervision, Y.T.; funding acquisition, H.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology, grant number 15K09432.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| AACE | ACTH-converting enzyme |
| ACTH | Adrenocorticotropic hormone |
| ACTHomas | Adrenocorticotropic hormone (ACTH)-secreting pituitary adenomas |
| AgRP | Agouti-related protein |
| AMB | Aminopeptidases B-like |
| ANXA1 | Annexin 1 |
| AVP | Arginine vasopressin |
| β-LPH | β-lipotropin |
| BET | Bromo and extra-terminal domain |
| CaMKII | Calmodulin kinase II |
| CBR | Cannabinoid receptor |
| CPE | Carboxypeptidase E |
| CRH | Corticotrophin-releasing hormone |
| CRHR1 | CRH receptor 1 |
| DDAVP | 1-deamino-8-D-arginine vasopressin |
| Dex | Dexamethasone |
| DST | Dex suppression test |
| EAS | Ectopic ACTH syndrome |
| FIPAs | Familial isolated pituitary adenomas |
| GR | Glucocorticoid receptor |
| GRmb | G protein-coupled glucocorticoid receptor |
| HDAC2 | Histone deacetylase 2 |
| HSP90 | Heat shock protein 90 |
| MEN1 | Multiple endocrine neoplasm type 1 |
| nGRE | Negative glucocorticoid response element |
| NNH | Non-neoplastic hypercortisolemia |
| PAM | Peptidylglycine-amidating monooxygenase |
| PKC | Protein kinase C |
| POMC | Pro-opiomelanocortin |
| PVN | Paraventricular nucleus |
| SgIII | Secretogranin III |
| SON | Supraoptic nuclei |
| SSTR2 | Somatostatin receptor subtype 2 |
| TR4 | Testicular receptor 4 |
References
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, H.; Yamada, S. Cushing’s Disease. J. Clin. Med. 2019, 8, 1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, L.K.; Biller, B.M.K.; Findling, J.W.; Newell-Price, J.; Savage, M.O.; Stewart, P.M.; Montori, V.M.; Edwards, H. The diagnosis of Cushing’s syndrome: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2008, 93, 1526–1540. [Google Scholar] [CrossRef] [PubMed]
- Drouin, J. 60 Years of POMC: Transcriptional and epigenetic regulation of POMC gene expression. J. Mol. Endocrinol. 2016, 56, T99–T112. [Google Scholar] [CrossRef]
- Langlais, D.; Couture, C.; Sylvain-Drolet, G.; Drouin, J. A Pituitary-Specific Enhancer of the POMC Gene with Preferential Activity in Corticotrope Cells. Mol. Endocrinol. 2011, 25, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Bilodeau, S.; Vallette-Kasic, S.; Gauthier, Y.; Figarella-Branger, D.; Brue, T.; Berthelet, F.; Lacroix, A.; Batista, D.; Stratakis, C.; Hanson, J.; et al. Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev. 2006, 20, 2871–2886. [Google Scholar] [CrossRef] [Green Version]
- Cool, D.R.; Normant, E.; Shen, F.; Chen, H.-C.; Pannell, L.; Zhang, Y.; Loh, Y.P. Carboxypeptidase E Is a Regulated Secretory Pathway Sorting Receptor: Genetic Obliteration Leads to Endocrine Disorders in Cpefat Mice. Cell 1997, 88, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef]
- Hosaka, M.; Watanabe, T.; Sakai, Y.; Uchiyama, Y.; Takeuchi, T. Identification of a Chromogranin A Domain That Mediates Binding to Secretogranin III and Targeting to Secretory Granules in Pituitary Cells and Pancreatic β-Cells. Mol. Biol. Cell 2002, 13, 3388–3399. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Watanabe, T.; Bochimoto, H.; Sakai, Y.; Torii, S.; Takeuchi, T.; Hosaka, M. Multiple Sorting Systems for Secretory Granules Ensure the Regulated Secretion of Peptide Hormones. Traffic 2013, 14, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Cawley, N.X.; Rathod, T.; Young, S.; Lou, H.; Birch, N.; Loh, Y.P. Carboxypeptidase E and Secretogranin III Coordinately Facilitate Efficient Sorting of Proopiomelanocortin to the Regulated Secretory Pathway in AtT20 Cells. Mol. Endocrinol. 2016, 30, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateno, T.; Kato, M.; Tani, Y.; Yoshimoto, T.; Oki, Y.; Hirata, Y. Processing of high-molecular-weight form adrenocorticotropin in human adrenocorticotropin-secreting tumor cell line (DMS-79) after transfection of prohormone convertase 1/3 gene. J. Endocrinol. Investig. 2010, 33, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Marcinkiewicz, M.; Benjannet, S.; Gaspar, L.; Beaubien, G.; Mattei, M.G.; Lazure, C.; Mbikay, M.; Chrétien, M. Cloning and Primary Sequence of a Mouse Candidate Prohormone Convertase PC1 Homologous to PC2, Furin, and Kex2: Distinct Chromosomal Localization and Messenger RNA Distribution in Brain and Pituitary Compared to PC2. Mol. Endocrinol. 1991, 5, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Korner, J.; Chun, J.; Harter, D.; Axel, R. Isolation and functional expression of a mammalian prohormone processing enzyme, murine prohormone convertase 1. Proc. Natl. Acad. Sci. USA 1991, 88, 6834–6838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cawley, N.X.; Li, Z.; Loh, Y.P. 60 YEARS OF POMC: Biosynthesis, trafficking, and secretion of pro-opiomelanocortin-derived peptides. J. Mol. Endocrinol. 2016, 56, T77–T97. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.L.; Mather, J.P.; Morris, P.L.; Bardin, C.W. Expression of pro-opiomelanocortin-like gene in the testis and Leydig cell lines. Ann. N. Y. Acad. Sci. 1984, 438, 659–662. [Google Scholar] [CrossRef]
- Chen, C.L.; Chang, C.C.; Krieger, D.T.; Bardin, C.W. Expression and regulation of proopiomelanocortin-like gene in the ovary and placenta: Comparison with the testis. Endocrinology 1986, 118, 2382–2389. [Google Scholar] [CrossRef]
- Pintar, J.; Schachter, B.; Herman, A.; Durgerian, S.; Krieger, D. Characterization and localization of proopiomelanocortin messenger RNA in the adult rat testis. Science 1984, 225, 632–634. [Google Scholar] [CrossRef]
- Lolait, S.J.; Autelitano, D.J.; Markwick, A.J.; Toh, B.H.; Funder, J.W. Co-expression of vasopressin with β-endorphin and dynorphin in individual cells from the ovaries of Brattleboro and Long-Evans rats: Immunocytochemical studies. Peptides 1986, 7, 267–276. [Google Scholar] [CrossRef]
- DeBold, C.R.; Menefee, J.K.; Nicholson, W.E.; Orth, D.N. Proopiomelanocortin Gene is Expressed in Many Normal Human Tissues and in Tumors not Associated with Ectopic Adrenocorticotropin Syndrome. Mol. Endocrinol. 1988, 2, 862–870. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.L. 60 YEARS OF POMC: The proopiomelanocortin gene: Discovery, deletion and disease. J. Mol. Endocrinol. 2016, 56, T27–T37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, P.J.; Şengül, S.; Tabak, J.; Ruth, P.; Bertram, R.; Shipston, M.J. Large conductance Ca2+ -activated K+ (BK) channels promote secretagogue-induced transition from spiking to bursting in murine anterior pituitary corticotrophs. J. Physiol. 2015, 593, 1197–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsaneva-Atanasova, K.; Sherman, A.; van Goor, F.; Stojilkovic, S.S. Mechanism of Spontaneous and Receptor-Controlled Electrical Activity in Pituitary Somatotrophs: Experiments and Theory. J. Neurophysiol. 2007, 98, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Veilleux, R.; Lefevre, G.; Coy, D.H.; Sueiras-Diaz, J.; Schally, A.V. Corticotropin-releasing factor stimulates accumulation of adenosine 3′, 5′-monophosphate in rat pituitary corticotrophs. Science 1982, 216, 1007–1008. [Google Scholar] [CrossRef]
- Abou-Samra, A.B.; Harwood, J.P.; Catt, K.J.; Aguilera, G. Mechanisms of action of CRF and other regulators of ACTH release in pituitary corticotrophs. Ann. N. Y. Acad. Sci. 1987, 512, 67–84. [Google Scholar] [CrossRef]
- Boutillier, A.L.; Gaiddon, C.; Lorang, D.; Roberts, J.L.; Loeffler, J.P. Transcriptional activation of the proopiomelanocortin gene by cyclic AMP-responsive element binding protein. Pituitary 1998, 1, 33–43. [Google Scholar] [CrossRef]
- Maira, M.; Martens, C.; Batsché, E.; Gauthier, Y.; Drouin, J. Dimer-Specific Potentiation of NGFI-B (Nur77) Transcriptional Activity by the Protein Kinase A Pathway and AF-1-Dependent Coactivator Recruitment. Mol. Cell. Biol. 2003, 23, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Jenks, B.G. Regulation of Proopiomelanocortin Gene Expression. Ann. N. Y. Acad. Sci. 2009, 1163, 17–30. [Google Scholar] [CrossRef]
- Lee, A.K.; Tse, A. Mechanism underlying corticotropin-releasing hormone (CRH) triggered cytosolic Ca2+ rise in identified rat corticotrophs. J. Physiol. 1997, 504, 367–378. [Google Scholar] [CrossRef]
- Philips, A.; Lesage, S.; Gingras, R.; Maira, M.H.; Gauthier, Y.; Hugo, P.; Drouin, J. Novel dimeric Nur77 signaling mechanism in endocrine and lymphoid cells. Mol. Cell. Biol. 1997, 17, 5946–5951. [Google Scholar] [CrossRef] [Green Version]
- Kovalovsky, D.; Refojo, D.; Liberman, A.C.; Hochbaum, D.; Pereda, M.P.; Coso, O.A.; Stalla, G.K.; Holsboer, F.; Arzt, E. Activation and Induction of NUR77/NURR1 in Corticotrophs by CRH/cAMP: Involvement of Calcium, Protein Kinase A, and MAPK Pathways. Mol. Endocrinol. 2002, 16, 1638–1651. [Google Scholar] [CrossRef] [PubMed]
- Tabak, J.; Tomaiuolo, M.; Gonzalez-Iglesias, A.E.; Milescu, L.S.; Bertram, R. Fast-Activating Voltage- and Calcium-Dependent Potassium (BK) Conductance Promotes Bursting in Pituitary Cells: A Dynamic Clamp Study. J. Neurosci. 2011, 31, 16855–16863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knepper, M.A.; Kwon, T.-H.; Nielsen, S. Molecular Physiology of Water Balance. N. Engl. J. Med. 2015, 372, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Wotjak, C.T.; Kubota, M.; Kohl, G.; Landgraf, R. Release of vasopressin from supraoptic neurons within the median eminence in vivo. A combined microdialysis and push-pull perfusion study in the rat. Brain Res. 1996, 726, 237–241. [Google Scholar] [CrossRef]
- Tse, A.; Lee, A.K. Arginine Vasopressin Triggers Intracellular Calcium Release, a Calcium-Activated Potassium Current and Exocytosis in Identified Rat Corticotropes 1. Endocrinology 1998, 139, 2246–2252. [Google Scholar] [CrossRef]
- Raymond, V.; Leung, P.C.K.; Veilleux, R.; Labrie, F. Vasopressin rapidly stimulates phosphatidic acid-phosphatidylinositol turnover in rat anterior pituitary cells. FEBS Lett. 1985, 182, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Makara, G.B.; Mergl, Z.; Zelena, D. The role of vasopressin in hypothalamo-pituitary-adrenal axis activation during stress: An assessment of the evidence. Ann. N. Y. Acad. Sci. 2004, 1018, 151–161. [Google Scholar] [CrossRef]
- Roper, J.; O’Carroll, A.-M.; Young, W.; Lolait, S. The vasopressin Avpr1b receptor: Molecular and pharmacological studies. Stress 2011, 14, 98–115. [Google Scholar] [CrossRef]
- DeBold, C.R.; Sheldon, W.R.; DeCherney, G.S.; Jackson, R.V.; Alexander, A.N.; Vale, W.; Rivier, J.; Orth, D.N. Arginine vasopressin potentiates adrenocorticotropin release induced by ovine corticotropin-releasing factor. J. Clin. Investig. 1984, 73, 533–538. [Google Scholar] [CrossRef] [Green Version]
- Lamberts, S.W.; Verleun, T.; Oosterom, R.; de Jong, F.; Hackeng, W.H. Corticotropin-releasing factor (ovine) and vasopressin exert a synergistic effect on adrenocorticotropin release in man. J. Clin. Endocrinol. Metab. 1984, 58, 298–303. [Google Scholar] [CrossRef]
- Turnbull, A.V.; Rivier, C.L. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: Actions and mechanisms of action. Physiol. Rev. 1999, 79, 1–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Ren, S.G.; Melmed, S. Hypothalamic and pituitary leukemia inhibitory factor gene expression in vivo: A novel endotoxin-inducible neuro-endocrine interface. Endocrinology 1996, 137, 2947–2953. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, C.; Melmed, S. Critical Role for STAT3 in Murine Pituitary Adrenocorticotropin Hormone Leukemia Inhibitory Factor Signaling. J. Biol. Chem. 1999, 274, 10723–10730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousquet, C.; Zatelli, M.C.; Melmed, S. Direct regulation of pituitary proopiomelanocortin by STAT3 provides a novel mechanism for immuno-neuroendocrine interfacing. J. Clin. Investig. 2000, 106, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Kameda, H.; Yamamoto, M.; Tone, Y.; Tone, M.; Melmed, S. Proton Sensitivity of Corticotropin-Releasing Hormone Receptor 1 Signaling to Proopiomelanocortin in Male Mice. Endocrinology 2019, 160, 276–291. [Google Scholar] [CrossRef] [Green Version]
- Asaba, K.; Makino, S.; Hashimoto, K. Effect of urocortin on ACTH secretion from rat anterior pituitary in vitro and in vivo: Comparison with corticotropin-releasing hormone. Brain Res. 1998, 806, 95–103. [Google Scholar] [CrossRef]
- Vaughan, J.; Donaldson, C.; Bittencourt, J.; Perrin, M.H.; Lewis, K.; Sutton, S.; Chan, R.; Turnbull, A.V.; Lovejoy, D.; Rivier, C.; et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature 1995, 378, 287–292. [Google Scholar] [CrossRef]
- Spiga, F.; Walker, J.J.; Gupta, R.; Terry, J.R.; Lightman, S.L. Glucocorticoid dynamics: Insights from mathematical, experimental and clinical studies. J. Endocrinol. 2015, 226, T55–T66. [Google Scholar] [CrossRef] [Green Version]
- Di, S.; Malcher-Lopes, R.; Halmos, K.C.; Tasker, J.G. Nongenomic Glucocorticoid Inhibition via Endocannabinoid Release in the Hypothalamus: A Fast Feedback Mechanism. J. Neurosci. 2003, 23, 4850–4857. [Google Scholar] [CrossRef] [Green Version]
- Malkoski, S.P.; Dorin, R.I. Composite Glucocorticoid Regulation at a Functionally Defined Negative Glucocorticoid Response Element of the Human Corticotropin-Releasing Hormone Gene. Mol. Endocrinol. 1999, 13, 1629–1644. [Google Scholar] [CrossRef]
- Yamamori, E.; Iwasaki, Y.; Taguchi, T.; Nishiyama, M.; Yoshida, M.; Asai, M.; Oiso, Y.; Itoi, K.; Kambayashi, M.; Hashimoto, K. Molecular mechanisms for corticotropin-releasing hormone gene repression by glucocorticoid in BE(2)C neuronal cell line. Mol. Cell. Endocrinol. 2007, 264, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Duncan, P.J.; Tabak, J.; Ruth, P.; Bertram, R.; Shipston, M.J. Glucocorticoids Inhibit CRH/AVP-Evoked Bursting Activity of Male Murine Anterior Pituitary Corticotrophs. Endocrinology 2016, 157, 3108–3121. [Google Scholar] [CrossRef] [Green Version]
- Chapman, L.P.; Epton, M.J.; Buckingham, J.C.; Morris, J.F.; Christian, H.C. Evidence for a Role of the Adenosine 5′-Triphosphate-Binding Cassette Transporter A1 in the Externalization of Annexin I from Pituitary Folliculo-Stellate Cells. Endocrinology 2003, 144, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Drouin, J.; Trifiro, M.A.; Plante, R.K.; Nemer, M.; Eriksson, P.; Wrange, O. Glucocorticoid receptor binding to a specific DNA sequence is required for hormone-dependent repression of pro-opiomelanocortin gene transcription. Mol. Cell. Biol. 1989, 9, 5305–5314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvin, R.; Saito-Hakoda, A.; Shimada, H.; Shimizu, K.; Noro, E.; Iwasaki, Y.; Fujiwara, K.; Yokoyama, A.; Sugawara, A. Role of NeuroD1 on the negative regulation of Pomc expression by glucocorticoid. PLoS ONE 2017, 12, e0175435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, M.J.M.; Cidlowski, J.A. Molecular mechanisms of glucocorticoid action and resistance. J. Steroid Biochem. Mol. Biol. 2002, 83, 37–48. [Google Scholar] [CrossRef]
- Martens, C.; Bilodeau, S.; Maira, M.; Gauthier, Y.; Drouin, J. Protein-Protein Interactions and Transcriptional Antagonism between the Subfamily of NGFI-B/Nur77 Orphan Nuclear Receptors and Glucocorticoid Receptor. Mol. Endocrinol. 2005, 19, 885–897. [Google Scholar] [CrossRef]
- Giacomini, D.; Páez-Pereda, M.; Theodoropoulou, M.; Labeur, M.; Refojo, D.; Gerez, J.; Chervin, A.; Berner, S.; Losa, M.; Buchfelder, M.; et al. Bone Morphogenetic Protein-4 Inhibits Corticotroph Tumor Cells: Involvement in the Retinoic Acid Inhibitory Action. Endocrinology 2006, 147, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Nudi, M.; Ouimette, J.-F.; Drouin, J. Bone Morphogenic Protein (Smad)-Mediated Repression of Proopiomelanocortin Transcription by Interference with Pitx/Tpit Activity. Mol. Endocrinol. 2005, 19, 1329–1342. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, N.; Otsuka, F.; Ogura-Ochi, K.; Inagaki, K.; Nakamura, E.; Toma, K.; Terasaka, T.; Iwasaki, Y.; Makino, H. Melatonin receptor activation suppresses adrenocorticotropin production via BMP-4 action by pituitary AtT20 cells. Mol. Cell. Endocrinol. 2013, 375, 1–9. [Google Scholar] [CrossRef]
- Brown, M.R.; Rivier, C.; Vale, W. Central Nervous System Regulation of Adrenocorticotropin Secretion: Role of Somatostatins *. Endocrinology 1984, 114, 1546–1549. [Google Scholar] [CrossRef] [PubMed]
- Kraicer, J.; Gajewski, T.C.; Moor, B.C. Release of Pro-Opiomelanocortin-Derived Peptides from the Pars intermedia and Pars distalis of the Rat Pituitary: Effect of Corticotrophin-Releasing Factor and Somatostatin. Neuroendocrinology 1985, 41, 363–373. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, C.; Feelders, R.A.; Lamberts, S.W.J.; Hofland, L.J. Somatostatin and dopamine receptors as targets for medical treatment of Cushing’s Syndrome. Rev. Endocr. Metab. Disord. 2009, 10, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, Y.C. Somatostatin and Its Receptor Family. Front. Neuroendocrinol. 1999, 20, 157–198. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Schonbrunn, A.; Armstrong, D.L. Somatostatin stimulates Ca2+-activated K+ channels through protein dephosphorylation. Nature 1991, 351, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Draznin, B.; Dahl, R.; Sherman, N.; Sussman, K.E.; Staehelin, L.A. Exocytosis in normal anterior pituitary cells. Quantitative correlation between growth hormone release and the morphological features of exocytosis. J. Clin. Investig. 1988, 81, 1042–1050. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Ben-Shlomo, A.; Kameda, H.; Fukuoka, H.; Deng, N.; Ding, Y.; Melmed, S. Somatostatin receptor subtype 5 modifies hypothalamic-pituitary-adrenal axis stress function. JCI Insight 2018, 3, e122932. [Google Scholar] [CrossRef]
- Schulte, H.M.; Oldfield, E.H.; Allolio, B.; Katz, D.A.; Berkman, R.A.; Ali, I.U. Clonal composition of pituitary adenomas in patients with Cushing’s disease: Determination by X-chromosome inactivation analysis. J. Clin. Endocrinol. Metab. 1991, 73, 1302–1308. [Google Scholar] [CrossRef]
- Gicquel, C.; Le Bouc, Y.; Luton, J.P.; Girard, F.; Bertagna, X. Monoclonality of corticotroph macroadenomas in Cushing’s disease. J. Clin. Endocrinol. Metab. 1992, 75, 472–475. [Google Scholar]
- Biller, B.M.; Alexander, J.M.; Zervas, N.T.; Hedley-Whyte, E.T.; Arnold, A.; Klibanski, A. Clonal origins of adrenocorticotropin-secreting pituitary tissue in Cushing’s disease. J. Clin. Endocrinol. Metab. 1992, 75, 1303–1309. [Google Scholar]
- Guru, S.C.; Manickam, P.; Crabtree, J.S.; Olufemi, S.E.; Agarwal, S.K.; Debelenko, L.V. Identification and characterization of the multiple endocrine neoplasia type 1 (MEN1) gene. J. Intern. Med. 1998, 243, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Vanbellinghen, J.-F.; Khoo, S.K.; Jaffrain-Rea, M.-L.; Naves, L.A.; Guitelman, M.A.; Murat, A.; Emy, P.; Gimenez-Roqueplo, A.-P.; Tamburrano, G.; et al. Aryl Hydrocarbon Receptor-Interacting Protein Gene Mutations in Familial Isolated Pituitary Adenomas: Analysis in 73 Families. J. Clin. Endocrinol. Metab. 2007, 92, 1891–1896. [Google Scholar] [CrossRef] [PubMed]
- Igreja, S.; Chahal, H.S.; King, P.; Bolger, G.B.; Srirangalingam, U.; Guasti, L.; Chapple, J.P.; Trivellin, G.; Gueorguiev, M.; Guegan, K.; et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum. Mutat. 2010, 31, 950–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-Y.; Song, Z.-J.; Chen, J.-H.; Wang, Y.-F.; Li, S.-Q.; Zhou, L.-F.; Mao, Y.; Li, Y.-M.; Hu, R.-G.; Zhang, Z.-Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015, 25, 306–317. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Arzberger, T.; Gruebler, Y.; Jaffrain-Rea, M.L.; Schlegel, J.; Schaaf, L.; Petrangeli, E.; Losa, M.; Stalla, G.K.; Pagotto, U. Expression of epidermal growth factor receptor in neoplastic pituitary cells: Evidence for a role in corticotropinoma cells. J. Endocrinol. 2004, 183, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, H.; Cooper, O.; Ben-Shlomo, A.; Mamelak, A.; Ren, S.-G.; Bruyette, D.; Melmed, S. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J. Clin. Investig. 2011, 121, 4712–4721. [Google Scholar] [CrossRef] [Green Version]
- Araki, T.; Liu, X.; Kameda, H.; Tone, Y.; Fukuoka, H.; Tone, M.; Melmed, S. EGFR Induces E2F1-Mediated Corticotroph Tumorigenesis. J. Endocr. Soc. 2017, 1, 127–143. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Persky, R.; Stegemann, R.; Hernández-Ramírez, L.C.; Zeltser, D.; Lodish, M.B.; Chen, A.; Keil, M.F.; Tatsi, C.; Faucz, F.R.; et al. Germline USP8 Mutation Associated With Pediatric Cushing Disease and Other Clinical Features: A New Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 4676–4682. [Google Scholar] [CrossRef]
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun. 2018, 9, 3171. [Google Scholar] [CrossRef] [Green Version]
- Sbiera, S.; Perez-Rivas, L.G.; Taranets, L.; Weigand, I.; Flitsch, J.; Graf, E.; Monoranu, C.-M.; Saeger, W.; Hagel, C.; Honegger, J.; et al. Driver mutations in USP8 wild-type Cushing’s disease. Neuro. Oncol. 2019, 21, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Sbiera, S.; Kunz, M.; Weigand, I.; Deutschbein, T.; Dandekar, T.; Fassnacht, M. The New Genetic Landscape of Cushing’s Disease: Deubiquitinases in the Spotlight. Cancers 2019, 11, 1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, H.L.; Merchant, T.E.; Warmuth-Metz, M.; Martinez-Barbera, J.-P.; Puget, S. Craniopharyngioma. Nat. Rev. Dis. Prim. 2019, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Haugh, A.M.; Johnson, D.B. Management of V600E and V600K BRAF-Mutant Melanoma. Curr. Treat. Options Oncol. 2019, 20, 81. [Google Scholar] [CrossRef]
- Fugazzola, L.; Muzza, M.; Pogliaghi, G.; Vitale, M. Intratumoral Genetic Heterogeneity in Papillary Thyroid Cancer: Occurrence and Clinical Significance. Cancers 2020, 12, 383. [Google Scholar] [CrossRef] [Green Version]
- Maraka, S.; Janku, F. BRAF alterations in primary brain tumors. Discov. Med. 2018, 26, 51–60. [Google Scholar]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190.e3. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Ramírez, L.C.; Gam, R.; Valdés, N.; Lodish, M.B.; Pankratz, N.; Balsalobre, A.; Gauthier, Y.; Faucz, F.R.; Trivellin, G.; Chittiboina, P.; et al. Loss-of-function mutations in the CABLES1 gene are a novel cause of Cushing’s disease. Endocr. Relat. Cancer 2017, 24, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Solarski, M.; Rotondo, F.; Foulkes, W.D.; Priest, J.R.; Syro, L.V.; Butz, H.; Cusimano, M.D.; Kovacs, K. DICER1 gene mutations in endocrine tumors. Endocr. Relat. Cancer 2018, 25, R197–R208. [Google Scholar] [CrossRef]
- De Kock, L.; Sabbaghian, N.; Plourde, F.; Srivastava, A.; Weber, E.; Bouron-Dal Soglio, D.; Hamel, N.; Choi, J.H.; Park, S.-H.; Deal, C.L.; et al. Pituitary blastoma: A pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014, 128, 111–122. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.; Salpea, P.; Stratakis, C.A. Carney complex: An update. Eur. J. Endocrinol. 2015, 173, M85–M97. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.; Tichomirowa, M.; Boikos, S.; Azevedo, M.; Lodish, M.; Martari, M.; Verma, S.; Daly, A.; Raygada, M.; Keil, M.; et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin. Genet. 2010, 78, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drouin, J.; Bilodeau, S.; Vallette, S. Of old and new diseases: Genetics of pituitary ACTH excess (Cushing) and deficiency. Clin. Genet. 2007, 72, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, S.W. Glucocorticoid receptors and Cushing’s disease. Mol. Cell. Endocrinol. 2002, 197, 69–72. [Google Scholar] [CrossRef]
- Korbonits, M.; Bujalska, I.; Shimojo, M.; Nobes, J.; Jordan, S.; Grossman, A.B.; Stewart, P.M. Expression of 11β-hydroxysteroid dehydrogenase isoenzymes in the human pituitary: Induction of the type 2 enzyme in corticotropinomas and other pituitary tumors. J. Clin. Endocrinol. Metab. 2001, 86, 2728–2733. [Google Scholar] [CrossRef] [Green Version]
- Tateno, T.; Izumiyama, H.; Doi, M.; Yoshimoto, T.; Shichiri, M.; Inoshita, N.; Oyama, K.; Yamada, S.; Hirata, Y. Differential gene expression in ACTH -secreting and non-functioning pituitary tumors. Eur. J. Endocrinol. 2007, 157, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Riebold, M.; Kozany, C.; Freiburger, L.; Sattler, M.; Buchfelder, M.; Hausch, F.; Stalla, G.K.; Paez-Pereda, M. A C-terminal HSP90 inhibitor restores glucocorticoid sensitivity and relieves a mouse allograft model of Cushing disease. Nat. Med. 2015, 21, 276–280. [Google Scholar] [CrossRef]
- Echeverria, P.C.; Picard, D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 641–649. [Google Scholar] [CrossRef]
- Pratt, W.B.; Galigniana, M.D.; Harrell, J.M.; DeFranco, D.B. Role of hsp90 and the hsp90-binding immunophilins in signalling protein movement. Cell. Signal. 2004, 16, 857–872. [Google Scholar] [CrossRef]
- Lu, J.; Chatain, G.P.; Bugarini, A.; Wang, X.; Maric, D.; Walbridge, S.; Zhuang, Z.; Chittiboina, P. Histone deacetylase inhibitor SAHA is a promising treatment of Cushing disease. J. Clin. Endocrinol. Metab. 2017, 102, 2825–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Du, L.; Heaney, A.P. Testicular Receptor-4: Novel Regulator of Glucocorticoid Resistance. J. Clin. Endocrinol. Metab. 2016, 101, 3123–3133. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Bergsneider, M.; Mirsadraei, L.; Young, S.H.; Jonker, J.W.; Downes, M.; Yong, W.H.; Evans, R.M.; Heaney, A.P. Evidence for orphan nuclear receptor TR4 in the etiology of Cushing disease. Proc. Natl. Acad. Sci. USA 2013, 110, 8555–8560. [Google Scholar] [CrossRef] [Green Version]
- Roussel-Gervais, A.; Couture, C.; Langlais, D.; Takayasu, S.; Balsalobre, A.; Rueda, B.R.; Zukerberg, L.R.; Figarella-Branger, D.; Brue, T.; Drouin, J. The Cables1 Gene in Glucocorticoid Regulation of Pituitary Corticotrope Growth and Cushing Disease. J. Clin. Endocrinol. Metab. 2016, 101, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque, R.M.; Ibáñez-Costa, A.; López-Sánchez, L.M.; Jiménez-Reina, L.; Venegas-Moreno, E.; Gálvez, M.A.; Villa-Osaba, A.; Madrazo-Atutxa, A.M.; Japón, M.A.; de la Riva, A.; et al. A Cellular and Molecular Basis for the Selective Desmopressin-Induced ACTH Release in Cushing Disease Patients: Key Role of AVPR1b Receptor and Potential Therapeutic Implications. J. Clin. Endocrinol. Metab. 2013, 98, 4160–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.-F.; Tang, K.-T.; Yen, Y.-S.; Ho, D.M.-T.; Yang, A.-H.; Hwang, C.-I.; Lin, H.-D.; Won, J.G.S. Plasma corticotrophin response to desmopressin in patients with Cushing’s disease correlates with the expression of vasopressin receptor 2, but not with that of vasopressin receptor 1 or 3, in their pituitary tumours. Clin. Endocrinol. 2012, 76, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Damoiseaux, R.; Babayan, L.; Rivera-Meza, E.K.; Yang, Y.; Bergsneider, M.; Wang, M.B.; Yong, W.H.; Kelly, K.; Heaney, A.P. Targeting Corticotroph HDAC and PI3-Kinase in Cushing Disease. J. Clin. Endocrinol. Metab. 2020. [Google Scholar] [CrossRef]
- Lines, K.E.; Filippakopoulos, P.; Stevenson, M.; Müller, S.; Lockstone, H.E.; Wright, B.; Knapp, S.; Buck, D.; Bountra, C.; Thakker, R. V Effects of epigenetic pathway inhibitors on corticotroph tumour AtT20 cells. Endocr. Relat. Cancer 2020, 27, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, A.; Feelders, R.A.; Stratakis, C.A.; Nieman, L.K. Cushing’s syndrome. Lancet 2015, 386, 913–927. [Google Scholar] [CrossRef]
- Sharma, S.T.; Nieman, L.K.; Feelders, R.A. Cushing’s syndrome: Epidemiology and developments in disease management. Clin. Epidemiol. 2015, 7, 281–293. [Google Scholar]
- Isidori, A.M.; Kaltsas, G.A.; Pozza, C.; Frajese, V.; Newell-Price, J.; Reznek, R.H.; Jenkins, P.J.; Monson, J.P.; Grossman, A.B.; Besser, G.M. Extensive clinical experience—The ectopic adrenocorticotropin syndrome: Clinical features, diagnosis, management, and long-term follow-up. J. Clin. Endocrinol. Metab. 2006, 91, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Aniszewski, J.P.; Young, W.F.; Thompson, G.B.; Grant, C.S.; van Heerden, J.A. Gushing syndrome doe to ectopic adrenocorticotropic hormone secretion. World J. Surg. 2001, 25, 934–940. [Google Scholar] [CrossRef]
- Kent Jex, R.; van Heerden, J.A.; Carpenter, P.C.; Grant, C.S. Ectopic ACTH syndrome. Am. J. Surg. 1985, 149, 276–282. [Google Scholar] [CrossRef]
- Howlett, T.A.; Drury, P.L.; Perry, L.; Doniach, I.; Rees, L.H.; Besser, G.M. Diagnosis and management of ACTH-dependent Cushing’s syndrome: Comparison of the features in ectopic and pituitary ACTH production. Clin. Endocrinol. 1986, 24, 699–713. [Google Scholar] [CrossRef]
- Doppman, J.L.; Nieman, L.; Miller, D.L.; Pass, H.I.; Chang, R.; Cutler, G.B.; Schaaf, M.; Chrousos, G.P.; Norton, J.A.; Ziessman, H.A. Ectopic adrenocorticotropic hormone syndrome: Localization studies in 28 patients. Radiology 1989, 172, 115–124. [Google Scholar] [CrossRef]
- Tabarin, A.; Valli, N.; Chanson, P.; Bachelot, Y.; Rohmer, V.; Bex-Bachellerie, V.; Catargi, B.; Roger, P.; Laurent, F. Usefulness of Somatostatin Receptor Scintigraphy in Patients with Occult Ectopic Adrenocorticotropin Syndrome. J. Clin. Endocrinol. Metab. 1999, 84, 1193–1202. [Google Scholar] [CrossRef]
- Torpy, D.J.; Mullen, N.; Ilias, I.; Nieman, L.K. Association of hypertension and hypokalemia with Cushing’s syndrome caused by ectopic ACTH secretion: A series of 58 cases. Ann. N. Y. Acad. Sci. 2002, 970, 134–144. [Google Scholar] [CrossRef]
- Ilias, I.; Torpy, D.J.; Pacak, K.; Mullen, N.; Wesley, R.A.; Nieman, L.K. Cushing’s Syndrome Due to Ectopic Corticotropin Secretion: Twenty Years’ Experience at the National Institutes of Health. J. Clin. Endocrinol. Metab. 2005, 90, 4955–4962. [Google Scholar] [CrossRef] [Green Version]
- Hernández, I.; Espinosa-de-los-Monteros, A.L.; Mendoza, V.; Cheng, S.; Molina, M.; Sosa, E.; Mercado, M. Ectopic ACTH-Secreting Syndrome: A Single Center Experience Report with a High Prevalence of Occult Tumor. Arch. Med. Res. 2006, 37, 976–980. [Google Scholar] [CrossRef]
- Salgado, L.R.; Fragoso, M.C.B.V.; Knoepfelmacher, M.; Machado, M.C.; Domenice, S.; Pereira, M.A.A.; de Mendonça, B.B. Ectopic ACTH syndrome: Our experience with 25 cases. Eur. J. Endocrinol. 2006, 155, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Wajchenberg, B.L.; Mendonca, B.B.; Liberman, B.; Pereira, M.A.; Carneiro, P.C.; Wakamatsu, A.; Kirschner, M.A. Ectopic adrenocorticotropic hormone syndrome. Endocr. Rev. 1994, 15, 752–787. [Google Scholar] [CrossRef]
- Li, Y.; Peng, Y.; Jiang, X.; Cheng, Y.; Zhou, W.; Su, T.; Xie, J.; Zhong, X.; Song, D.; Wu, L.; et al. Whole exome sequencing of thymic neuroendocrine tumor with ectopic ACTH syndrome. Eur. J. Endocrinol. 2017, 176, 187–194. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Di Domenico, A.; Wiedmer, T.; Marinoni, I.; Perren, A. Genetic and epigenetic drivers of neuroendocrine tumours (NET). Endocr. Relat. Cancer 2017, 24, R315–R334. [Google Scholar] [CrossRef] [Green Version]
- Araki, T.; Liu, N.-A.; Tone, Y.; Cuevas-Ramos, D.; Heltsley, R.; Tone, M.; Melmed, S. E2F1-mediated human POMC expression in ectopic Cushing’s syndrome. Endocr. Relat. Cancer 2016, 23, 857–870. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, I.; Higuchi, S.; Fujimoto, M.; Takiguchi, T.; Nakayama, A.; Tamura, A.; Kohno, T.; Komai, E.; Shiga, A.; Nagano, H.; et al. Cushing Syndrome Due to ACTH-Secreting Pheochromocytoma, Aggravated by Glucocorticoid-Driven Positive-Feedback Loop. J. Clin. Endocrinol. Metab. 2016, 101, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Oliver, R.L.; Davis, J.R.E.; White, A. Characterisation of ACTH Related Peptides in Ectopic Cushing’s Syndrome. Pituitary 2003, 6, 119–126. [Google Scholar] [CrossRef]
- Raffin-Sanson, M.L.; Massias, J.F.; Dumont, C.; Raux-Demay, M.C.; Proeschel, M.F.; Luton, J.P.; Bertagna, X. High plasma proopiomelanocortin in aggressive adrenocorticotropin-secreting tumors. J. Clin. Endocrinol. Metab. 1996, 81, 4272–4277. [Google Scholar]
- Scopsi, L.; Gullo, M.; Rilke, F.; Martin, S.; Steiner, D.F. Proprotein convertases (PC1/PC3 and PC2) in normal and neoplastic human tissues: Their use as markers of neuroendocrine differentiation. J. Clin. Endocrinol. Metab. 1995, 80, 294–301. [Google Scholar]
- Vieau, D.; Seidah, N.G.; Mbikay, M.; Chrétien, M.; Bertagna, X. Expression of the prohormone convertase PC2 correlates with the presence of corticotropin-like intermediate lobe peptide in human adrenocorticotropin-secreting tumors. J. Clin. Endocrinol. Metab. 1994, 79, 1503–1506. [Google Scholar]
- Kimura, N.; Ishikawa, T.; Sasaki, Y.; Sasano, N.; Onodera, K.; Shimizu, Y.; Kimura, I.; Steiner, D.F.; Nagura, H. Expression of prohormone convertase, PC2, in adrenocorticotropin-producing thymic carcinoid with elevated plasma corticotropin-releasing hormone. J. Clin. Endocrinol. Metab. 1996, 81, 390–395. [Google Scholar] [PubMed] [Green Version]
- Page-Wilson, G.; Freda, P.U.; Jacobs, T.P.; Khandji, A.G.; Bruce, J.N.; Foo, S.T.; Meece, K.; White, A.; Wardlaw, S.L. Clinical Utility of Plasma POMC and AgRP Measurements in the Differential Diagnosis of ACTH-Dependent Cushing’s Syndrome. J. Clin. Endocrinol. Metab. 2014, 99, E1838–E1845. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.W.; Littlewood, A.C.; Clark, A.J.; Davis, J.R.; White, A. Human small cell lung cancer cell lines expressing the proopiomelanocortin gene have aberrant glucocorticoid receptor function. J. Clin. Investig. 1994, 93, 1625–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, T.; Tozawa, F.; Dobashi, I.; Horiba, N.; Ohmori, N.; Yamakado, M.; Yamada, M.; Demura, H. Corticotropin-releasing hormone, proopiomelanocortin, and glucocorticoid receptor gene expression in adrenocorticotropin-producing tumors in vitro. J. Clin. Investig. 1993, 92, 2790–2795. [Google Scholar] [CrossRef] [Green Version]
- Machado, M.C.; Valeria de Sa, S.; Correa-Giannella, M.L.; Giorgi, R.R.; Pereira, M.A.A.; Cescato, V.A.S.; Giannella-Neto, D.; Salgado, L.R. Association between tumoral GH-releasing peptide receptor type 1a mRNA expression and in vivo response to GH-releasing peptide-6 in ACTH-dependent Cushing’s syndrome patients. Eur. J. Endocrinol. 2008, 158, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Tsagarakis, S.; Tsigos, C.; Vasiliou, V.; Tsiotra, P.; Kaskarelis, J.; Sotiropoulou, C.; Raptis, S.A.; Thalassinos, N. The Desmopressin and Combined CRH-Desmopressin Tests in the Differential Diagnosis of ACTH-Dependent Cushing’s Syndrome: Constraints Imposed by the Expression of V2 Vasopressin Receptors in Tumors with Ectopic ACTH Secretion. J. Clin. Endocrinol. Metab. 2002, 87, 1646–1653. [Google Scholar]
- De Keyzer, Y.; Lenne, F.; Auzan, C.; Jégou, S.; René, P.; Vaudry, H.; Kuhn, J.M.; Luton, J.P.; Clauser, E.; Bertagna, X. The pituitary V3 vasopressin receptor and the corticotroph phenotype in ectopic ACTH syndrome. J. Clin. Investig. 1996, 97, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Dahia, P.L.; Ahmed-Shuaib, A.; Jacobs, R.A.; Chew, S.L.; Honegger, J.; Fahlbusch, R.; Besser, G.M.; Grossman, A.B. Vasopressin receptor expression and mutation analysis in corticotropin-secreting tumors. J. Clin. Endocrinol. Metab. 1996, 81, 1768–1771. [Google Scholar] [PubMed] [Green Version]
- Arlt, W.; Dahia, P.L.M.; Callies, F.; Nordmeyer, J.P.; Allolio, B.; Grossman, A.B.; Reincke, M. Ectopic ACTH production by a bronchial carcinoid tumour responsive to desmopressin in vivo and in vitro. Clin. Endocrinol. 1997, 47, 623–627. [Google Scholar] [CrossRef]
- Tani, Y.; Sugiyama, T.; Hirooka, S.; Izumiyama, H.; Hirata, Y. Ectopic ACTH syndrome caused by bronchial carcinoid tumor indistinguishable from Cushing’s disease. Endocr. J. 2010, 57, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Findling, J.W.; Raff, H. Diagnosis of Endocrine Disease: Differentiation of pathologic/neoplastic hypercortisolism (Cushing’s syndrome) from physiologic/non-neoplastic hypercortisolism (formerly known as pseudo-Cushing’s syndrome). Eur. J. Endocrinol. 2017, 176, R205–R216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wand, G.S.; Dobs, A.S. Alterations in the hypothalamic-pituitary-adrenal axis in actively drinking alcoholics. J. Clin. Endocrinol. Metab. 1991, 72, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C.; Imaki, T.; Vale, W. Prolonged exposure to alcohol: Effect on CRF mRNA levels, and CRF- and stress-induced ACTH secretion in the rat. Brain Res. 1990, 520, 1–5. [Google Scholar] [CrossRef]
- Ogilvie, K.M.; Lee, S.; Rivier, C. Role of arginine vasopressin and corticotropin-releasing factor in mediating alcohol-induced adrenocorticotropin and vasopressin secretion in male rats bearing lesions of the paraventricular nuclei. Brain Res. 1997, 744, 83–95. [Google Scholar] [CrossRef]
- Rees, L.; Besser, G.; Jeffcoate, W.; Goldie, D.; Marks, V. Alcohol-Induced Pseudo-Cushing’s Syndrome. Lancet 1977, 309, 726–728. [Google Scholar] [CrossRef]
- Jacobson, L. Hypothalamic-pituitary-adrenocortical axis: Neuropsychiatric aspects. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; Volume 4, pp. 715–738. [Google Scholar]
- Pariante, C.M.; Lightman, S.L. The HPA axis in major depression: Classical theories and new developments. Trends Neurosci. 2008, 31, 464–468. [Google Scholar] [CrossRef]
- Nelson, J.C.; Davis, J.M. DST Studies in Psychotic Depression: A Meta-Analysis. Am. J. Psychiatry 1997, 154, 1497–1503. [Google Scholar] [CrossRef]
- Holsboer, F. How can we realize the promise of personalized antidepressant medicines? Nat. Rev. Neurosci. 2008, 9, 638–646. [Google Scholar] [CrossRef]
- Reynolds, P.D.; Ruan, Y.; Smith, D.F.; Scammell, J.G. Glucocorticoid Resistance in the Squirrel Monkey Is Associated with Overexpression of the Immunophilin FKBP51 1. J. Clin. Endocrinol. Metab. 1999, 84, 663–669. [Google Scholar] [CrossRef]
- Scammell, J.G.; Denny, W.B.; Valentine, D.L.; Smith, D.F. Overexpression of the FK506-Binding Immunophilin FKBP51 Is the Common Cause of Glucocorticoid Resistance in Three New World Primates. Gen. Comp. Endocrinol. 2001, 124, 152–165. [Google Scholar] [CrossRef]
- Binder, E.B.; Salyakina, D.; Lichtner, P.; Wochnik, G.M.; Ising, M.; Pütz, B.; Papiol, S.; Seaman, S.; Lucae, S.; Kohli, M.A.; et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat. Genet. 2004, 36, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Hackett, R.A.; Steptoe, A.; Kumari, M. Association of Diurnal Patterns in Salivary Cortisol with Type 2 Diabetes in the Whitehall II Study. J. Clin. Endocrinol. Metab. 2014, 99, 4625–4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Bravata, D.M.; Cabaccan, J.; Raff, H.; Ryzen, E. Elevated late-night salivary cortisol levels in elderly male type 2 diabetic veterans. Clin. Endocrinol. 2005, 63, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Constantinopoulos, P.; Michalaki, M.; Kottorou, A.; Habeos, I.; Psyrogiannis, A.; Kalfarentzos, F.; Kyriazopoulou, V. Cortisol in tissue and systemic level as a contributing factor to the development of metabolic syndrome in severely obese patients. Eur. J. Endocrinol. 2015, 172, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Rabadan-Diehl, C.; Makara, G.; Kiss, A.; Lolait, S.; Zelena, D.; Ochedalski, T.; Aguilera, G. Regulation of Pituitary V1b Vasopressin Receptor Messenger Ribonucleic Acid by Adrenalectomy and Glucocorticoid Administration. Endocrinology 1997, 138, 5189–5194. [Google Scholar] [CrossRef]
- Dahia, P.L.M.; Grossman, A.B. The Molecular Pathogenesis of Corticotroph Tumors. Endocr. Rev. 1999, 20, 136–155. [Google Scholar] [CrossRef]
- Pecori Giraldi, F.; Pivonello, R.; Ambrogio, A.G.; De Martino, M.C.; De Martin, M.; Scacchi, M.; Colao, A.; Toja, P.M.; Lombardi, G.; Cavagnini, F. The dexamethasone-suppressed corticotropin-releasing hormone stimulation test and the desmopressin test to distinguish Cushing’s syndrome from pseudo-Cushing’s states. Clin. Endocrinol. 2007, 66, 251–257. [Google Scholar] [CrossRef]
- Hundt, W.; Zimmermann, U.; Pöttig, M.; Spring, K.; Holsboer, F. The combined dexamethasone-suppression/CRH-stimulation test in alcoholics during and after acute withdrawal. Alcohol. Clin. Exp. Res. 2001, 25, 687–691. [Google Scholar] [CrossRef]
Figure 1.
Schema of physiological ACTH synthesis and secretion.

Figure 2.
Schematic representation of glucocorticoid negative-feedback in corticotrophs.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fukuoka, H.; Shichi, H.; Yamamoto, M.; Takahashi, Y. The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease. Int. J. Mol. Sci. 2020, 21, 9132. https://doi.org/10.3390/ijms21239132
AMA Style
Fukuoka H, Shichi H, Yamamoto M, Takahashi Y. The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease. International Journal of Molecular Sciences. 2020; 21(23):9132. https://doi.org/10.3390/ijms21239132
Chicago/Turabian StyleFukuoka, Hidenori, Hiroki Shichi, Masaaki Yamamoto, and Yutaka Takahashi. 2020. "The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease" International Journal of Molecular Sciences 21, no. 23: 9132. https://doi.org/10.3390/ijms21239132
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.