1. Introduction
Glioblastomas are intra-axial tumors that originate from neuroglial cells of the central nervous system (CNS) [
1,
2]. Glioblastomas are known to display cellular heterogeneity with stem-like glioblastoma stem cells at the apex [
3]. Glioblastoma multiforme (GBM) is the most common and aggressive malignant primary brain tumor in adults and is a major cause of morbidity and mortality among neurosurgical patients, with only 12% surviving beyond 36 months (long-term survivors) [
4]. The current standard treatment strategy for glioblastoma is multimodal, involving maximal surgical resection followed by radiotherapy with concomitant and adjuvant temozolomide (TMZ) [
2]. Although therapeutic advances in diagnosis have been made, glioblastoma is resistant to current treatments, such as chemotherapy and radiation, and effective treatment options are lacking. Recent insights into the biology of glioblastoma, including those from The Cancer Genome Atlas, have revealed important genetic events in human glioblastomas, such as gene amplification, mutation, and deletion [
1,
5]. The most frequent alterations in gliomas include dysregulation of growth factor signaling via amplification and mutational activation of receptor tyrosine kinase genes such as
EGFR. Phosphatidylinositol-3-kinase (PI3K) signaling pathways are also activated due to genetic alteration in the phosphatase and tensin homolog (
PTEN) tumor suppressor gene on 10q23.3 at the level of loss of heterozygosity, mutation, and methylation in at least 60% of gliomas [
6].
PTEN, a tumor suppressor, is a lipid phosphatase that hydrolyses phosphate in position 3′ from phosphoinositide, thereby opposing mitogenic signaling mediated by PI3K [
7]. The loss of PTEN function via its mutation or deletion leads to hyperactivation of PI3K signaling [
7]. The loss of PTEN function has been linked to tumor malignancy, including metastasis and resistance to radiotherapy and chemotherapy in brain and breast cancer patients [
8,
9,
10]. The major downstream effector of PI3K signaling is the pro-survival serine/threonine kinase, Akt [
7]. Akt is activated by phosphorylation at Thr 308 (T308) by PDK1 and is fully activated by further phosphorylation at Ser 473 (S473) by mTORC2 [
11]. Activated Akt phosphorylates numbers of substrates, and the majority of these substrates are involved in cell survival, proliferation, and metabolism. Uncontrolled Akt activation occurs through either overexpression and mutation, a loss of negative regulator PTEN, or the activation of PI3K pathways. These outcomes have been reported in a number of cancers including glioblastoma, breast, and prostate cancers [
1,
7,
12]. Therefore, Akt is an attractive target for the control of cancers where the PI3K–Akt pathway is upregulated, such as glioblastoma.
Arsenic-based drugs have been used for centuries. Arsenic trioxide (As
2O
3), an arsenic derivative, exerts potent anti-tumor effects in vitro and in vivo [
13]. As
2O
3 is known to be an effective inducer of apoptosis in patients with relapsed acute promyelocytic leukemia (APL) [
13,
14]. Because it also has a potent effect in vitro against different types of malignant leukemia as well as APL, there is increasing interest in the therapeutic development of arsenic compounds for the treatment of various solid tumors. Arsenic compounds are known to induce cell death via multiple pathways, such as reactive oxygen species (ROS) generation, JNK activation, and the activation of pro-apoptotic proteins such as Bax [
13,
15]. Recently, As
2O
3 was reported to decrease Akt protein levels in a caspase-dependent manner [
16]. Akt is highly activated in glioblastomas when PTEN function is lost, so it is possible that an arsenic compound could control glioblastomas via Akt downregulation. As
2O
3 is poorly water-soluble and it must be dissolved with sodium hydroxide and then adjusted to physiologic pH, yielding s odium meta-arsenite (NaAsO
2) [
17]. NaAsO
2, KML001, is a water soluble, and thus orally bioavailable arsenic compound that has been reported to have anti-tumor effects on several tumors, including prostate cancer and glioma cells [
17,
18]. KML001 is known to exerts its cytotoxic effects via its binding to telomeric sequences, which leads to shortened telomeres in prostate cancer [
17,
18]. KML001 has anti-tumoral effects on non-Hodgkin lymphoma cells via inhibiting cell signaling including PI3K/Akt and MAPK [
13,
14]. Recently, KML001 exhibits anti-tumoral effects in multiple myeloma cells via Akt inactivation and PTEN activation [
19]. In addition, KML001 has entered Phase I/II clinical trials for treatment of prostate cancer [
20] and Phase I clinical trials for advanced non-small-cell lung cancer as well as other platinum-responsive malignancies (
https://clinicaltrials.gov). Because KML001 induces Akt inactivation and PTEN activation [
19] and because Akt activation with PTEN mutation is associated with glioblastoma, in this study, we evaluated the anti-tumor effects of KML001 using PTEN-negative human glioblastoma U87-MG cells in vitro and in vivo, and investigated the possible mechanisms involved in this process.
3. Discussion
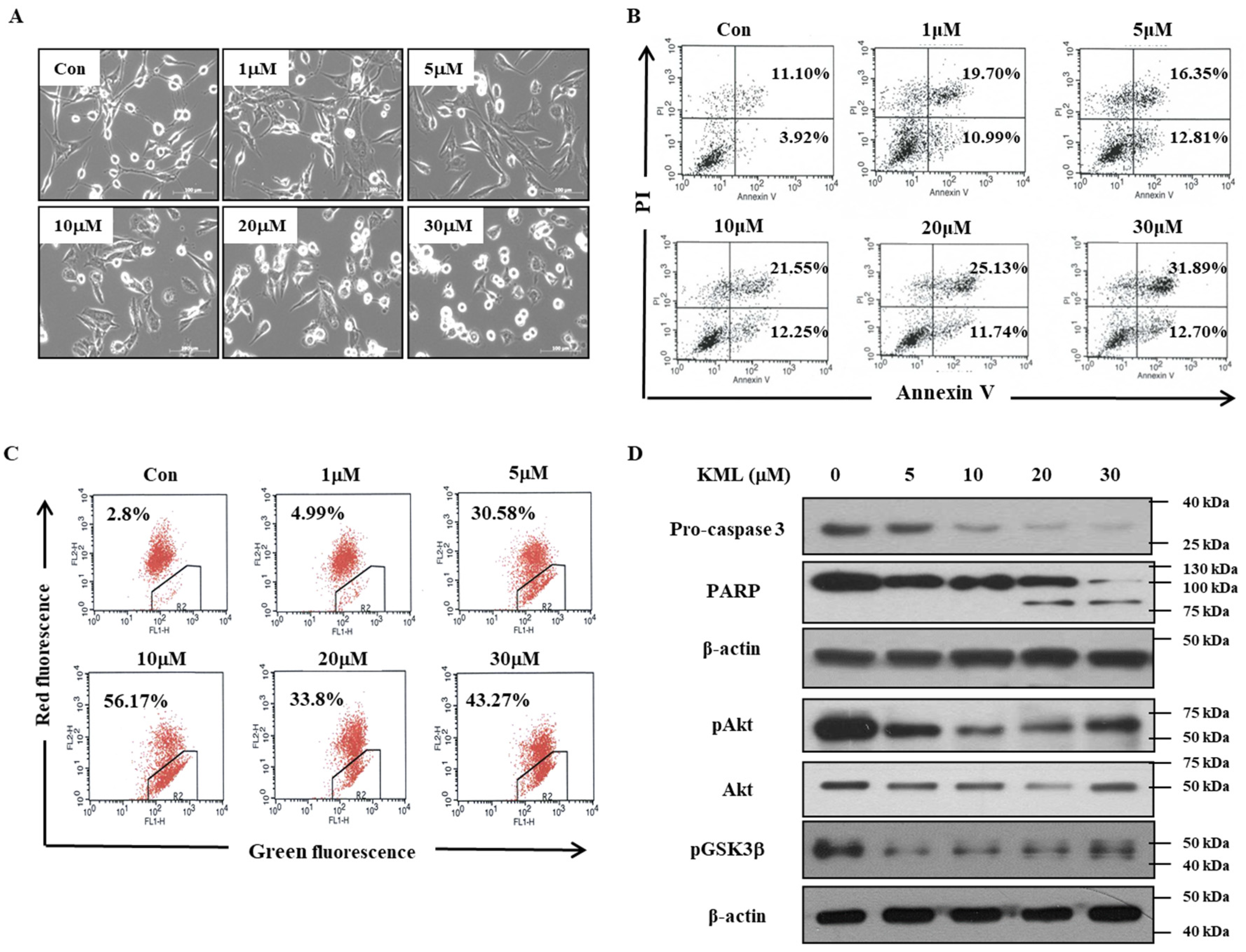
We evaluated the effects of KML001 on the growth inhibition of glioma cells, which express different levels of active-Akt depending on expression levels and/or status of PTEN, which is a negative regulator for Akt activation. We demonstrated that KML001 exhibited anti-tumor effects via the induction of glioma cell death, with a greater effect in cells with higher Akt activation. KML001 exerts its growth inhibition through Akt downregulation via the proteasomal degradation pathway. KML001 also reduced tumor growth in an orthotopic xenograft mouse model.
Glioblastoma is the most common malignant type of primary brain tumor and is classified as a Grade IV astrocytoma by the World Health Organization, with a median survival of approximately 15 months following intensive therapy including surgery, chemotherapy, and radiotherapy [
27]. The standard therapy options available to patients are minimally effective; thus, development of a novel treatment regimen is necessary to improve the survival rate among glioblastoma patients. A number of genetic and epigenetic alterations have been identified in glioblastoma that lead to the dysregulation of signaling pathways, including activation of the receptor tyrosine kinase pathway, PTEN–PI3K–Akt activation, and inhibition of p53 [
28]. Therefore, drugs targeting these commonly observed alterations have been investigated as potential targeted therapies for glioblastoma [
28]. The PI3K–Akt signaling pathway is activated in most glioblastoma via either growth factor receptor activation, such as EGFR, or through loss of PTEN, a negative regulator of Akt activation [
28]. Therefore, Akt inhibitors may be beneficial for control of glioblastoma growth. In our study, we showed that sodium meta-arsenite (KML001, NaAsO
2) inhibited Akt activation via its downregulation and induced apoptosis of glioblastoma cells.
Arsenic has been used to treat diseases for centuries. Among the various arsenicals, arsenic trioxide (ATO, As
2O
3), an FDA-approved drug, is a highly effective drug for treating acute promyelocytic leukemia (APL) with low toxicity [
29]. ATO induces apoptosis of various human solid tumor cell lines, including lung cancer, breast cancer, and glioblastoma cells [
15,
18,
30,
31]. ATO is known to exert its anti-tumor effects via multiple pathways, including the mitochondrial aggregation pathway, autophagic cell death pathway, and inhibition of telomerase activity and Notch pathway [
18,
32,
33,
34]. ATO has been administered via intravenous injection for APL treatment. KML001, an ATO derivative, is water-soluble and orally bioavailable, and expresses cytotoxic activity in various cancer cells, including prostate cancer and AML [
17,
35]. KML001 has been demonstrated to bind to telomeres and to cause telomerase erosion in prostate cancer cells [
17]. In addition, KML001 inhibits the proliferation of non-Hodgkin’s lymphoma (NHL) cell lines, while ATO is not effective. The anti-tumor effect of KML001 in NHL is mediated via the inhibition of cell signaling pathways, including STAT, PI3K–Akt, MAPK, and NF-kB signaling [
14]. It has been reported that KML001 makes glioblastoma cells more sensitive to temozolomide chemotherapy and radiotherapy by enhancing DNA damage [
18]. However, the anti-tumor effects of KML001 and the mechanism of this effect as related to glioblastoma have not been investigated. In this study, we found that KML001 induced cell growth inhibition to a greater extent in PTEN-negative glioblastoma cells than in PTEN-positive cells.
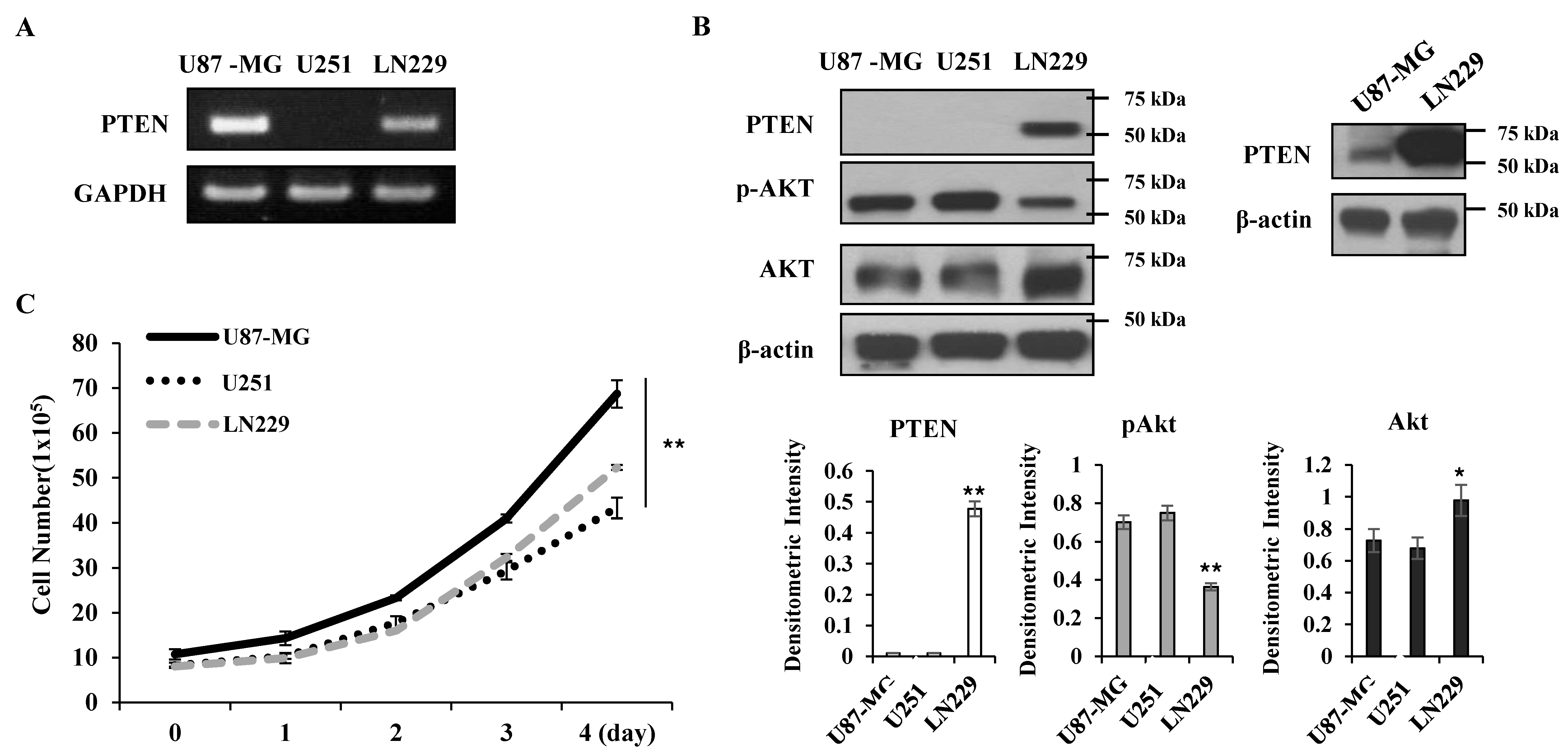
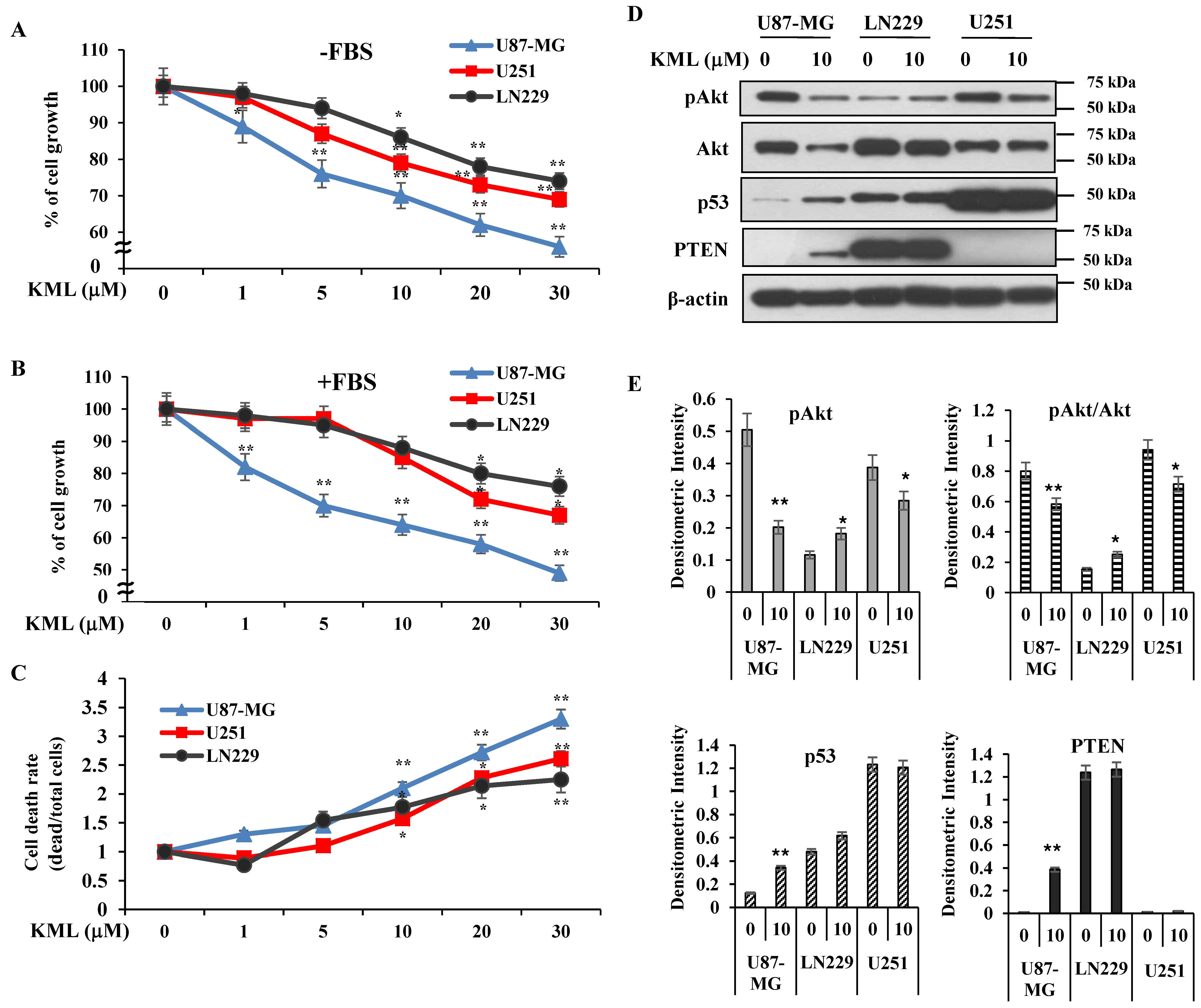
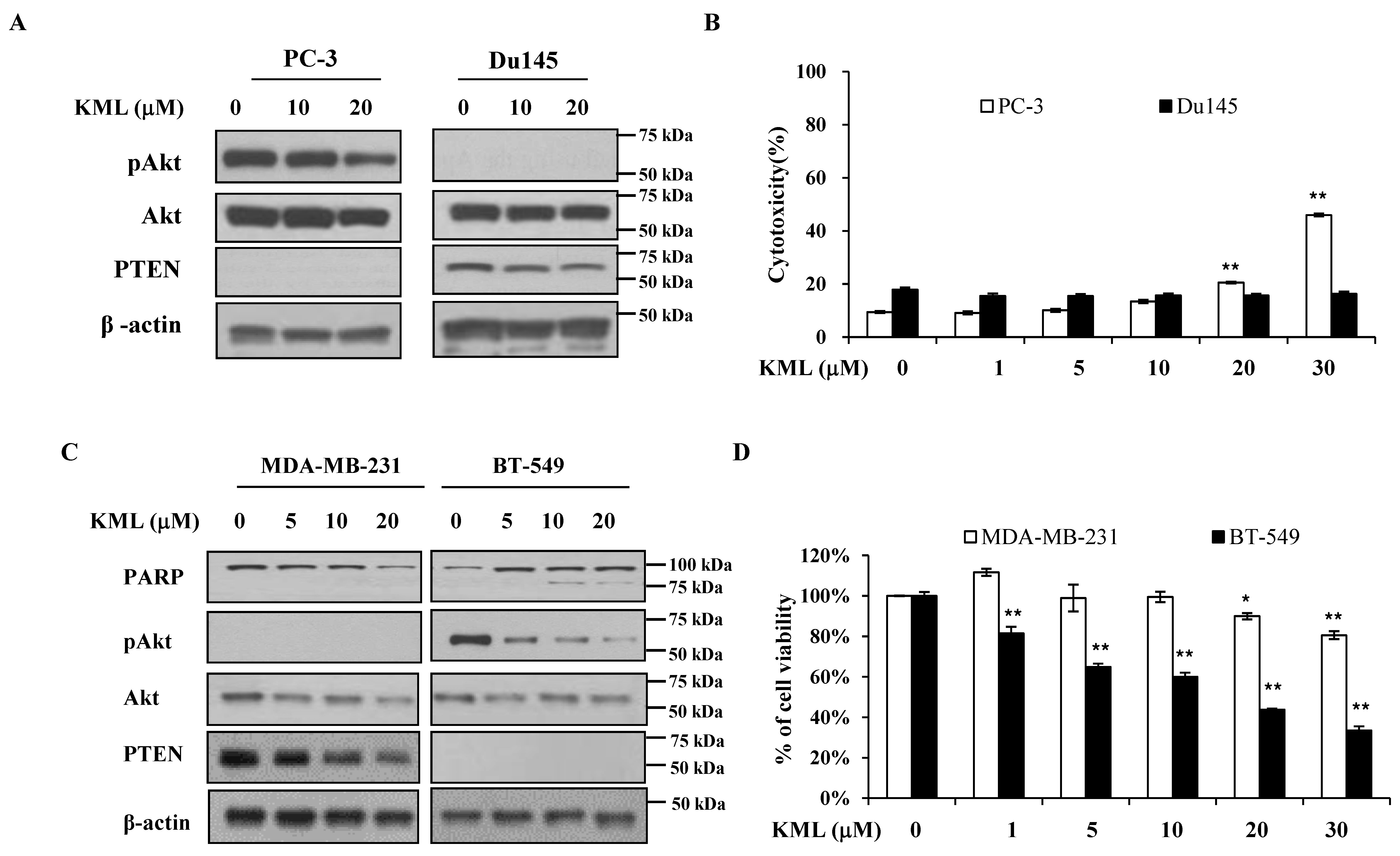
PTEN is a negative regulator for Akt [
12]; thus, Akt activity was higher in the PTEN-negative U87-MG and U251 cells than the PTEN-positive LN229 cells (
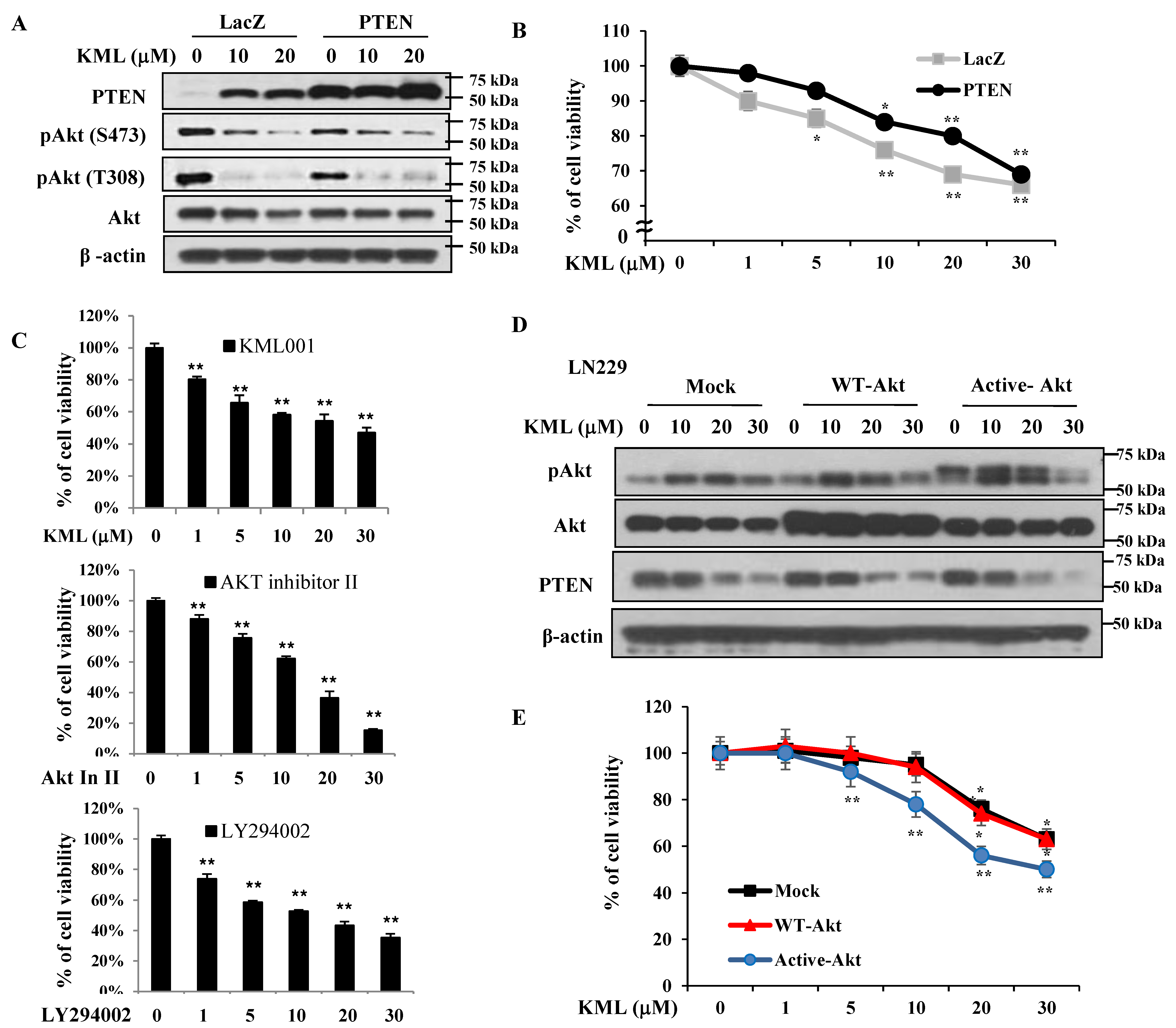
Figure 1A,B). KML001 induced growth inhibition in glioma cells, but the degree of inhibition was more prominent in the U87-MG and U251 cells. When PTEN was overexpressed in U87-MG cells, KML001-induced cell death was attenuated (
Figure 5A,B). PTEN-dependent KML001 sensitivity appears not to be limited to glioma cells, as KML001 sensitized cell growth inhibition in other PTEN-negative cells (PC-3 and BT549) when compared with PTEN-positive cells (DU145 and MDA-MB-231) (
Figure 6). In addition, when WT-Akt or active-Akt were overexpressed, KML001 induced cell death to a greater extent in the active-Akt-expressing cells than in WT-Akt-expressing cells (
Figure 5D,E). These data suggest that KML001 might efficiently target cancers with Akt activation either due to PTEN deletion or PI3K activation.
KML001 treatment decreased protein levels of Akt with little change in its mRNA levels (
Figure 4). Proteasomal inhibitors, namely MG132 and lactacyctin, attenuated KML001-induced Akt downregulation, but a lysosomal inhibitor, chloroquine, did not (
Figure 4C,D,
Supplementary Figure S4), indicating that KML001 negatively regulates Akt via its proteasomal degradation. HSP90 is a molecular chaperon for oncogenic proteins such as EGFR, Akt, and Src [
36]. Although levels of HSP90 were not changed (
Supplementary Figure S3), we cannot rule out the possibility that HSP90 inactivation is involved in Akt downregulation, because KML001 is known to deactivate HSP90 [
37]. U87-MG cells expressed a mutant form of PTEN [
23], which was expressed at the mRNA level, but hardly detected at the protein level (
Figure 1). However, KML001 increased the amount of mutant PTEN at the protein level in U87-MG cells, whereas it decreased WT-PTEN levels in LN229 cells (
Figure 4A). This mutant PTEN is stabilized by MG132, a proteasomal inhibitor, even without KML001 treatment (
Figure 4F), indicating that Exon 3 of PTEN might be involved in its stability.
PTEN and p53 are very well-known tumor suppressors and their dysregulations are often associated with tumor progression [
38]. PTEN is known to regulate p53 protein levels and activities through both its phosphatase-dependent and phosphatase-independent mechanisms. The onset of tumor development in
p53−/−PTEN−/− mice is similar to that of
p53−/− animals, and p53 protein levels are dramatically reduced in
PTEN−/− cells and tissues. Reintroducing wild-type or phosphatase-dead PTEN mutants leads to a significant increase in p53 stability [
38]. PTEN-deficient U87-MG cells possess WT-p53 but express very low levels of p53. PTEN-deficient U251 cells expressed high levels of mutant p53 initially and regardless of KML001 treatment (
Figure 2D). KML001 treatment led to increases in both mutant PTEN (
Figure 4) and p53 levels (
Figure 2D and
Supplementary Figure S8). KML001 also increased phosphorylation of p53 at Ser-46 (
Supplementary Figure S8A), which is mainly involved in apoptosis [
39], and induced p53 localization in the nucleus (
Supplementary Figure S8B). Accordingly, p53-responsible pro-apoptotic proteins such as PUMA and Bax [
40] were upregulated by KML001 (
Supplementary Figure S8A). It is possible that the restoration of mutant PTEN levels might stabilize and activate WT-P53, which facilitates KML001-induced apoptosis.
ATO has been used with other agents as a combinational therapy to increase anti-tumor effects. For example, a combination of ATO with the natural anti-cancer agent gossypol synergistically targeted glioma stem-like cells through Hedgehog and Notch signaling [
34]. KML001 has been combined with doxercalciferol and gemcitabine for synergistic anti-cancer effect in acute lymphoid leukemia and pancreatic cancer cells, respectively [
35,
41]. KML001 has also been reported to sensitize glioblastoma when combined with temozolomide or radiation by causing increased DNA damage and apoptosis [
18], although the mechanisms by which KML001 exerts its synergistic effect have not been explored. In our study, we demonstrated that KML001 alone is enough to reduce the growth of glioblastoma in vitro and in vivo, probably through Akt downregulation. However, we cannot rule out other mechanisms involved in KML001-induced cell death. Reactive oxygen species (ROS) are involved in apoptosis induced by ATO [
42] and KML001 induced autophagic cell death in prostate cancer cells via ROS [
43]. ROS were increased by KML001 in U87-MG cells (
Supplementary Figure S9A) but N-acetyl cysteine (NAC), an antioxidant, did not reduce KML001-induced cell death (
Supplementary Figure S9B). NAC did not affect the signaling pathways induced by KML001, such as downregulation and inactivation of Akt or PTEN upregulation (
Supplementary Figure S9C). Signal transducer and activator of transcription 3 (STAT3) is highly expressed and overactivated in many tumors, including glioblastoma [
44]; thus, targeting STAT3 could be a promising strategy for glioblastoma therapy [
45]. ATO is known to inactivate STAT3 in gastric cancer cells through SHP-1 induction [
46]. KML001 treatment also decreased STAT3 activation, which was not reversed by NAC treatment (
Supplementary Figure S9C). How KML001 inactivates STAT3 remains to be elucidated.
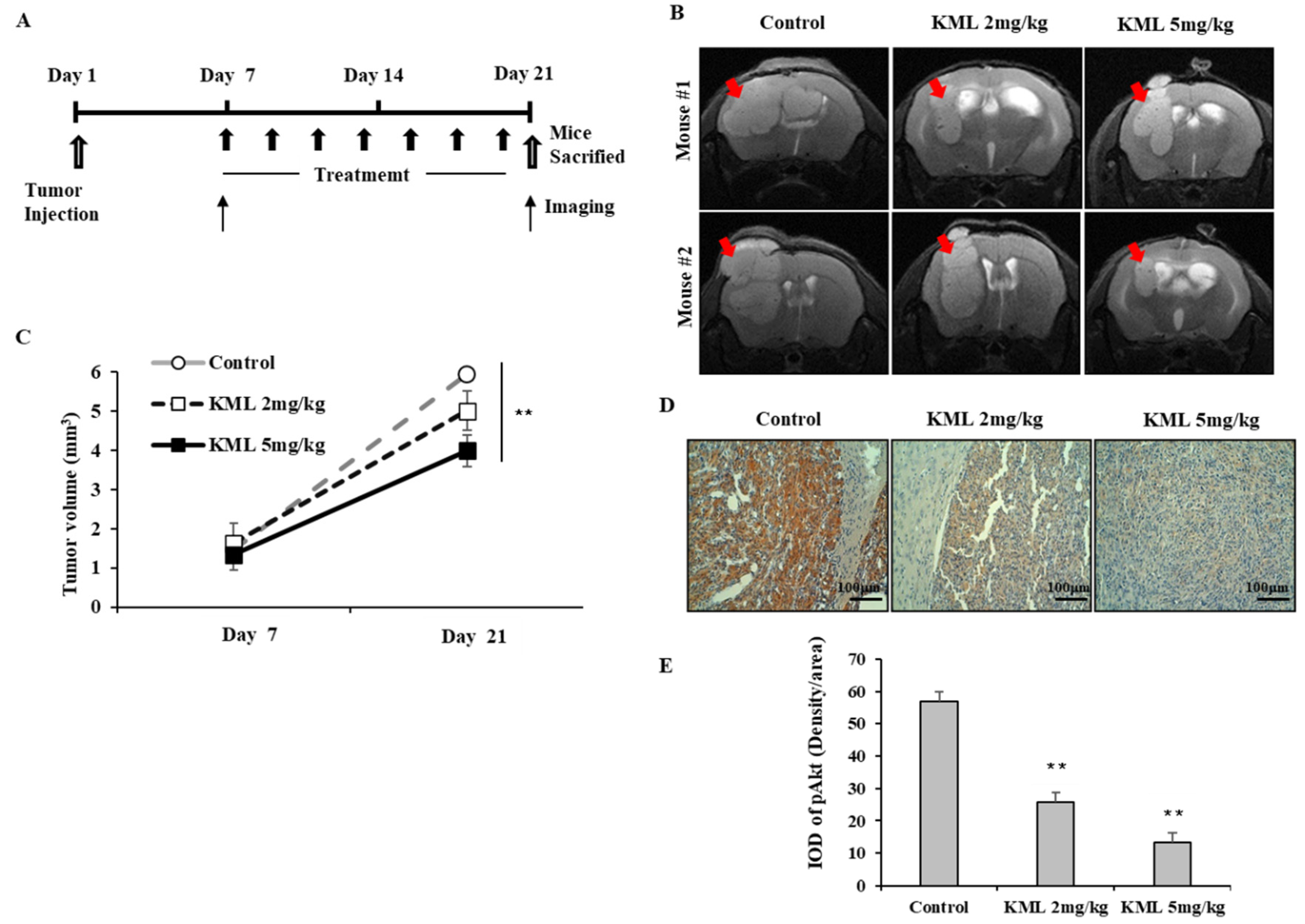
To evaluate the anti-tumor effects of KML001, seven days after tumor injection, U87-MG orthotopic xenograft mice were treated with 2 mg/kg or 5 mg/kg KML001 every other day for 14 days. KML001 treatment resulted in a significant reduction of tumor growth with decreased Akt activation (
Figure 7B,D). The doses of KML001 used in our study have been reported not to be toxic in vivo when treatment is carried out every day for 20 days, as determined by body weight change and levels of AST and ALT (K-9). We also observed little body weight reduction when treated with KML001 at a two-day interval for 14 days (data not shown). These data indicate that KML001 has anti-tumor effect on glioma in mouse xenograft model, too. In conclusion, our data support the notion that KML001 has therapeutic potential for the treatment of glioma, especially when Akt is overactivated by either PTEN deletion or mutation.
4. Materials and Methods
4.1. Cell Culture
Human glioblastoma cell lines, U87-MG, LN229 and human glioma cell line, U251, human prostate cancer cell lines, Du145 and PC3, human breast cancer cell lines, MDA-MB231 and BT-549 were obtained from the American Type Culture Collection (Rockville, MD, USA). Roswell Park Memorial Institute (RPMI), Dulbecco’s modified Eagles medium (DMEM), Fetal bovine serum (FBS), antibiotic-antimycotic (100X) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). U87-MG, U251, and LN229 cell lines were cultured in DMEM, supplemented with 10% FBS and antibiotic-antimycotic (1X). Du145, PC3, MDA-MB231, and BT-549 were cultured in RPMI, supplemented with 10% FBS and antibiotic-antimycotic (1X).
4.2. Antibodies and Reagents
KML001 was a kind gift from Komipharm International (Seoul, Korea). Anti-P53, Horseradish Peroxidase (HRP)-conjugated goat anti-mouse IgG and goat anti-rabbit IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-Akt (Ser473), anti-Akt, anti-phospho-GSK3β, anti-PTEN, anti-caspase-3, anti-PARP, anti-β-actin were from cell signaling technology (Beverly, MA, USA). FITC annexin V apoptosis detection kit was obtained from BD Pharmingen (San Jose, CA, USA). JC-1 assay kit from Molecular Probes (Eugene, OR, USA). Immobilion-P polyvinylidene difluoride (PVDF) membranes (0.45 µm) were from Millipore (Bedford, MA, USA). Micro-BCA protein assay reagents and Chemiluminescent reagents were from Pierce (Rockford, IL, USA). MG132, Pan-caspase inhibitor, Puromycin, Akt inhibitor II were obtained from Calbiochem (La Jolla, CA, USA). Chloroquine was purchased from Sigma-Aldrich. LY294002 was purchased from LC Laboratories (Woburn, MA, USA).
4.3. Drug Treatment
Cells were grown to approximately 70% confluence and then serum-starved in RPMI containing 0.1% bovine serum albumin (BSA) prior to treatment. Cells were treated with the indicated concentrations of reagents in the same media.
4.4. Cell Viability and Cell Proliferation Assay
For the cell proliferation assay, the cells were plated in 96-well cell culture plate (5 × 103 cells) and cultured overnight prior to treatment with KML001. The effects of the treatments on cell growth were determined with CellTiter 96 aqueous nonradioactive cell proliferation assay kit (MTS; 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxyme-thoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, Promega, Madison, WI, USA) as described in the manufacturer’s instruction. Absorbance was measured at 490 nm with a PowerWave HT Spectrophotometer (Biotek instruments, Winooski, VT, USA). For trypan blue exclusion assay, cells were plated in 6-well plates (1.5 × 105 cells/well) for 24 h, followed by KML001 treatment for 24 h. The cells were trypsinized, stained with trypan blue, and counted under the microscope as viable cells. Alternatively, after staining with trypan blue, cells were counted by countessTM automated cell counter from Invitrogen (Carlsbad, CA, USA). Each experiment was performed in triplicate.
4.5. Immunoblotting Analysis
After washing with ice-cold phosphate-buffered saline (PBS; 10 mM Na2HPO4, pH 7.4, 145 mM NaCl, and 2.7 mM KCl), cells were lysed with 2X SDS-PAGE sample buffer (20 mM Tris, pH 8.0, 2% SDS, 2 mM DTT, 1 mM Na3VO4, 2 mM EDTA, 20% glycerol) and boiled for 5 min. Protein concentration of each sample was determined using a Micro-BCA protein assay reagent as described by the manufacturer. In all, 30 ug of total cellular protein was separated by 8%–12% SDS-PAGE and then transferred to PVDF membranes. The membranes were blocked 1 hr at room temperature in TBST (20 mM Tris, pH 8.0, 150 mM NaCl, and 0.05% Tween 20) containing either 5% BSA (for immunoblotting with anti-phospho-Akt antibody) or 5% non-fat dried milk (for immunoblotting with other antibodies). The membranes were then incubated with the primary antibody overnight at 4 °C, washed three times with TBST, incubated with HRP-conjugated goat anti-mouse IgG or goat anti-rabbit IgG secondary antibodies for 1 h at room temperature, and then washed with TBST three times. The labeled proteins were visualized using the enhanced chemi-luminescence method. In the instances in which the same membrane was reprobed with a different primary antibody, the membrane was incubated in a stripping buffer (62.5 mM Tris, pH6.8, 2% SDS and 0.75% β-mercaptoethanol) at 37 °C for 15 min, washed extensively, reblocked with 5% non-fat milk, and then reprobed with another antibody as described above.
4.6. Flow Cytometer Analysis
Cells were trypsinized and suspended in PBS containing 2.5 mM EDTA, 2.5 mM EGTA and 1% BSA. For the measurement of mitochondrial membrane potential changes, cells were incubated with 20 nM 2 µM JC-1 in PBS for 15–30 min at 37 °C, followed by flow cytometer analysis (FACSCalibur; Becton Dickinson Bioscience, San Jose, CA, USA). JC-1 is a lipophillic dye and it may be the most sensitive indicator for mitochondrial membrane potential [
24]. At low membrane potential, JC-1 occurs as a monomer that emits green fluorescent, and at higher membrane potential JC-1 forms aggregates that emit red fluorescence. The data were analyzed with Cell Quest Software (BD bioscience, San Jose, CA, USA).
4.7. Annexin V/PI Staining
For detection of apoptotic cells, cells were harvested and incubated with fluorescein isothiocyanate (FITC)-conjugated annexin V reagent and propidium iodide (PI) according to the manufacturer’s instructions for the FITC annexin V apoptosis detection kit I (BD Biosciences, San Jose, CA, USA). Cells were then analyzed using flow cytometry. The data were analyzed with Cell Quest Software (BD Biosciences).
4.8. Reverse Transcription—Polymerase Chain Reaction
Cellular RNA was extracted from glioblastoma cell lines using the Trizol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacterer’s instructions. cDNA fragments were amplified with the following primer pairs: AKT 5′- ATCCGCTGCCTGCAGTGGACC -3′ (sense), 5′- TTGTCCAGCATGAGGTTCTCC-3′ (antisense) PTEN 5′-ACCGCCAAATTTAATTGCAG-3′(sense), 5′-GGGTCCTGAATTGGAGGAAT-3′ (anti-sense). β-actin 5′-ACAACGGCTCCGGCATGTGCAA-3′ (sense) 5′-CGGTTGGCCTTGGGGTTCAG-3′ (antisense). The condition of PCR was as follows: (AKT, EGFR) 94 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, and (PTEN) 94 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min for 30 cycles. Products were analyzed on a 1% agarose gel.
4.9. Establishment of the Stable Cell Lines Overexpressing Wild Type-Akt and Dominant-Active Akt
Constructs for wild type-
AKT/pCMV5 and dominant active-
AKT/pCMV5 were a kind gift from Dr. Jong-Sun Park (Chung-nam national university, Daejeon, Korea). Dominant active Akt is a membrane-targeted form of wild type-Akt as described before [
47]. pLL3.7 vector was obtained from Addgene Inc. (Cambridge, MA, USA). Its U6 promoter was digested with restriction enzymes
XbaI/
HpaI and then the corresponding sites were replaced by EF1α promoter/enhancer as described by Invitrogen (San Diego, CA, USA) to over-express a gene of interest. The XbaI/BglII fragment digested from either wt-
AKT/pCMV5 or active-
AKT/pCMV5 vector was inserted into pLL3.7 vector. The resultant wt-
AKT/pLL3.7 or active-
AKT/pLL3.7 constructs also co-expressed enhanced green fluorescent protein (EGFP) as a reporter gene. Recombinant lentiviruses were produced according to the manufacturer’s protocol (Invitrogen). In brief, we co-transfected pLL3.7 (for mock, wt-Akt, and active-Akt) vector and viral packing mix (Invitrogen) into 293FT cells and harvested the resulting supernatant after 48 h. Media containing live lentiviruses were collected, spun to remove cell debris, filtered through a Millex-HV syringe filter (0.45 μm, Millipore, Bedford, MA, USA), and stored at −80°C before used. The glioblastoma cells, U87-MG, LN229, cells were infected with each lentivirus supernatant and 2 μg/mL polybrene (hexadimethrine bromide). After 24 h, media was changed and then cells were incubated for 48 h before the aspiration of media. The GFP expression after cell sorting was greater than 80–90%.
4.10. Establishment of the Stable Cell Lines Expressing PTEN
Based on the gene sequences of PTEN (GenBank AF067844), the primer sequences were designed according to procedure of the pLenti6/V5-D-TOPO® Cloning Kit (Invitrogen, Carlsbad, CA, USA): Forward primer: 5′-CACCATGACA GCCATCATCAAA-3′, which contains a Kozak translation initiation sequence (underlined) with an ATG initiation codon (italicized), Reverse primer: 5′- GACTTTTGTAATTTGTGTATG-3′ (synthesized by Bioneer Corporation, Daejeon, Korea). The PCR was carried out using an AccuPower TLA PCR PreMix (Bioneer Corporation, Daejeon, Korea) and PTEN/pCDNA3 as a template, which was kindly provided by Dr. Jong Bae, Park (National Cancer Center, Goyang, Korea). The condition of PCR was as follows: 30 cycles of 94 ℃ for 30 s, 54 ℃ for 30 s, and 72 ℃ for 1 min. The gel-purified PCR products were ligated into the pLenti6/V5-D-TOPO® expression vector, resulting in the plasmid of pLenti6/V5-PTEN. The PTEN cDNA cloned in pLenti6/V5-D-TOPO vector was confirmed by restriction enzyme digestion and by DNA sequencing analysis (Macrogen, Seuoul, Korea). Lentivirus production was done with ViraPower Lentivirus Expression Systems (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. In brief, the ViraPower™ Packaging Mix and pLenti6/V5-PTEN were cotransfected into 293FT cells (6 × 106 total cells), using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA), and harvested the resulting supernatant after 72 h. The glioblastoma cells, U87-MG was infected with equivalent titers of control, PTEN viruses and were incubated for at least 48 h. Stable cell lines of PTEN were selected with fresh media containing blasticidin.
4.11. Establishment of Orthotropic Xenograft
U87-MG cells were orthotopically transplanted following washing and re-suspension in PBS (1 × 10
5 cells per mouse). Cells were injected stereotactically into the left caudate-putamen region of 6-week-old female Balb/c nude mice (n = 5 in each group) as described before [
48]. The injection coordinates were 2.2 mm to the left of the midline and 0.2 mm posterior to the bregma at a depth of 3.5 mm. After 1-week injection, mice were treated either without (control) or with KML001 for 2 weeks (two-day intervals) orally. After 2 weeks of treatment the brain of each mouse was harvested and fixed in 4% paraformaldehyde. All animal research was conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee at the National Cancer Center, Republic of Korea (NCC-10-005D, approved on 2 March 2010).
4.12. In Vivo Imaging and Analysis In Vivo MR Imaging
Mice were imaged with a 7.0-T MR imaging system (BioSpec 70/20 USR; Bruker Biospin, Ettlingen, Germany). Mice were anesthetized with 2% isoflurane gas, and their respiration rate was monitored during examinations. Anatomic images were acquired by using a rapid acquisition with relaxation enhancement (RARE) sequence and the following parameters: repetition time msec/echo time msec, 2600/30; number of averages, two; RARE factor, four; matrix, 256 × 256; slice thickness, 0.7 mm; slice distance, 1 mm; and field of view, 2.0 × 2.0 cm. the arbitrary shape region of interest (ROI) spanning a whole tumor region were drawn on the MR images. The tumor volumes were calculated by multiplying the area of the ROI’s and the slice distance (mm3).
4.13. Immunohistochemical Staining
The brains were fixed with 4% paraformaldehyde for 24 h at 4 °C. For immunostaining, after the antigen retrieval process with citrate buffer (pH 6.0) and endogenous peroxidase blocking with 3% hydrogen peroxide, tissue sections were incubated in 1% BSA blocking solution (v/v) for 30 min at room temperature, then in primary antibody overnight at 4 °C in a humidified chamber. For primary antibodies, we used rabbit antibody to pAKT (Cell Signaling Technology, 1:500). Sections were rinsed three times with a washing buffer (1% BSA, 0.1% cold fish skin gelatin, 0.5% Triton X-100, and 0.01 M PBS) and then incubated biotinylated secondary antibody (diluted 1:200, Vector Laboratories; Burlingame, USA) overnight at 4 °C. An avidin–biotin peroxidase enzyme complex was prepared and applied according to manufacturer’s instructions (Vectastain Elite ABC kit). Finally, sections were incubated for 5 min in a DAB/hydrogen peroxide substrate solution (prepared according to manufacturer’s instructions, Vectastain DAB substrate kit, Vector Laboratories). Sections were mounted in an aqueous mountant (Vectashield, Vector Laboratories) and observed under a microscope (Olympus, Tokyo, Japan). For quantification of IHC staining, three-staining fields were selected randomly from each section. The average IOD (Integrated Optical Density) levels between treated groups were then compared. Image-J software was used for analysis.
4.14. Immunofluorescence Analysis
Cells were fixed with 1% paraformaldehyde in PBS at 4 °C for 20 min, permeabilized and blocked in 3% BSA in PBS for 1 h at room temperature. Cells were then incubated with anti-p53 antibody for 4 °C overnight, and then incubated with Alexa 488-conjugated antibody for 1 h at room temperature. For nucleus staining, cells were incubated with DAPI in PBS. Cells were examined using confocal microscopy (Zeiss, Jena, Germany).
4.15. Statistical Analysis
Comparison between two groups were performed using Student’s t-test. Multiple group comparisons were made parametric one-way ANOVA followed post hoc test (Bonferroni correction). Data represent average values and standard deviations (error bars) obtained from three independent experiments.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






